
N-Acetylcysteine and Aripiprazole Improve Social Behavior and Cognition and Modulate Brain BDNF Levels in a Rat Model of Schizophrenia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Chronic Treatment of Adult Sprague-Dawley Rats with ARI or NAC Reverses the Social Deficits Induced by BSO in Early Postnatal Development
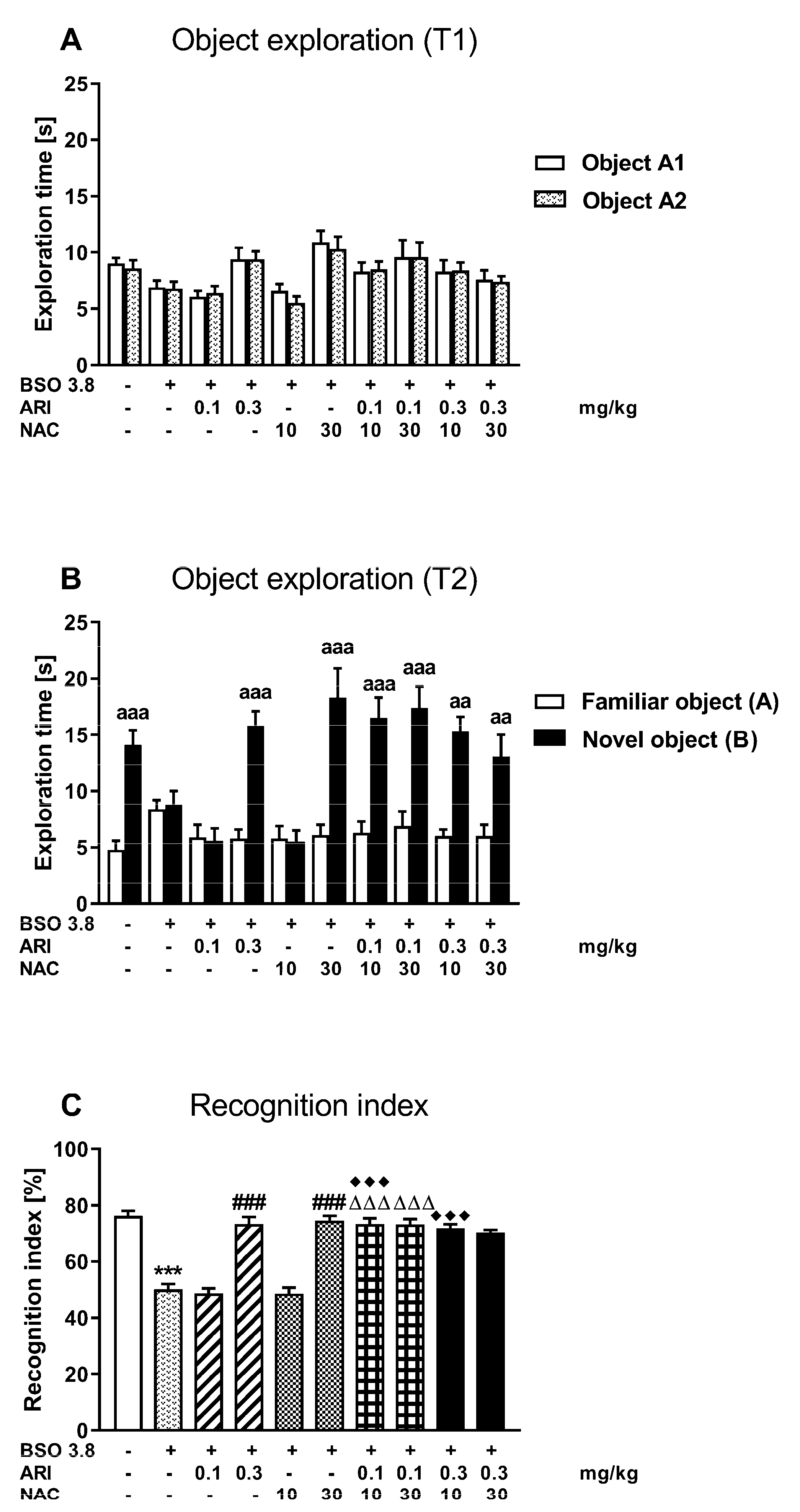
2.2. Chronic Treatment of Adult Sprague-Dawley Rats with ARI or NAC Reverses the Cognitive Deficits Induced by BSO in Early Postnatal Development
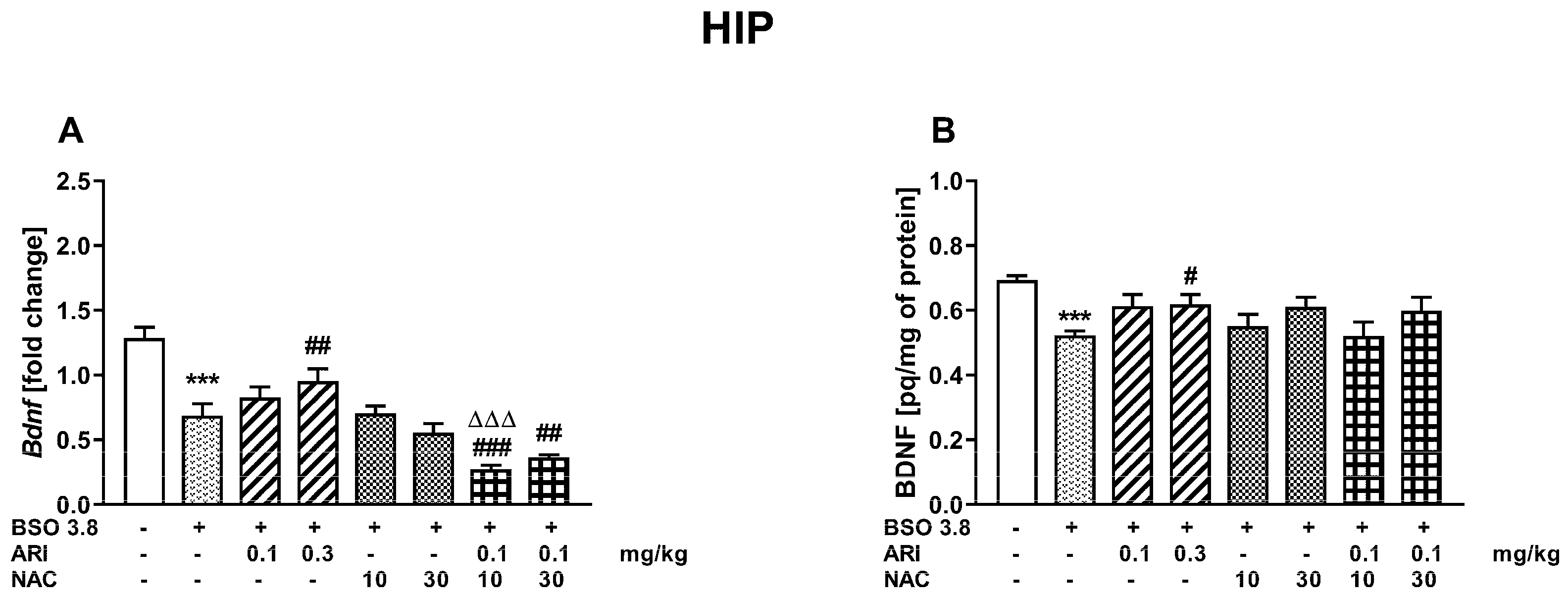
2.3. Chronic Treatment of Adult Sprague-Dawley Rats with ARI and/or NAC Modulates the Changes in BDNF mRNA and Protein Levels in the Prefrontal Cortex and Hippocampus Induced by BSO in the Early Postnatal Life
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Social Interaction Test
4.3. Novel Object Recognition Test
4.4. BDNF Expression Analysis
4.5. ELISA Assay
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rössler, W.; Salize, H.J.; van Os, J.; Riecher-Rössler, A. Size of burden of schizophrenia and psychotic disorders. Eur. Neuropsychopharmacol. 2005, 15, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Tamminga, C.A.; Holcomb, H.H. Phenotype of schizophrenia: A review and formulation. Mol. Psychiatry 2005, 10, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Joyce, E.M.; Roiser, J. Cognitive heterogeneity in schizophrenia. Curr. Opin. Psychiatry 2007, 20, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Nuechterlein, K.H.; Ventura, J.; Subotnik, K.L.; Bartzokis, G. The early longitudinal course of cognitive deficits in schizophrenia. J. Clin. Psychiatry 2014, 75 (Suppl. S2), 25–29. [Google Scholar] [CrossRef] [PubMed]
- Keefe, R.S. The longitudinal course of cognitive impairment in schizophrenia: An examination of data from premorbid through posttreatment phases of illness. J. Clin. Psychiatry 2014, 75 (Suppl. S2), 8–13. [Google Scholar] [CrossRef]
- Keefe, R.S.; Bilder, R.M.; Davis, S.M.; Harvey, P.D.; Palmer, B.W.; Gold, J.M.; Meltzer, H.Y.; Green, M.F.; Capuano, G.; Stroup, T.S.; et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch. Gen. Psychiatry 2007, 64, 633–647. [Google Scholar] [CrossRef]
- Miyamoto, S.; Miyake, N.; Jarskog, L.F.; Fleischhacker, W.W.; Lieberman, J.A. Pharmacological treatment of schizophrenia: A critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol. Psychiatry 2012, 17, 1206–1227. [Google Scholar] [CrossRef]
- van Os, J.; Rutten, B.P.; Poulton, R. Gene-environment interactions in schizophrenia: Review of epidemiological findings and future directions. Schizophr. Bull. 2008, 34, 1066–1082. [Google Scholar] [CrossRef]
- van Os, J.; Kenis, G.; Rutten, B.P. The environment and schizophrenia. Nature 2010, 468, 203–212. [Google Scholar] [CrossRef]
- Moran, P.; Stokes, J.; Marr, J.; Bock, G.; Desbonnet, L.; Waddington, J.; O’Tuathaigh, C. Gene × environment interactions in schizophrenia: Evidence from genetic mouse models. Neural Plast. 2016, 2016, 2173748. [Google Scholar] [CrossRef]
- Lewis, D.A.; Levitt, P. Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 2002, 25, 409–432. [Google Scholar] [CrossRef]
- Rapoport, J.L.; Gogtay, N. Childhood onset schizophrenia: Support for a progressive neurodevelopmental disorder. Int. J. Dev. Neurosci. 2011, 29, 251–258. [Google Scholar] [CrossRef]
- Rapoport, J.L.; Giedd, J.N.; Gogtay, N. Neurodevelopmental model of schizophrenia: Update 2012. Mol. Psychiatry 2012, 17, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Paus, T.T.; Keshavan, M.; Giedd, J.N. Why do many psychiatric disorders emerge during adolescence? Nat. Rev. Neurosci. 2008, 9, 947–957. [Google Scholar] [CrossRef]
- Hoftman, G.D.; Lewis, D.A. Postnatal developmental trajectories of neural circuits in the primate prefrontal cortex: Identifying sensitive periods for vulnerability to schizophrenia. Schizophr. Bull. 2011, 37, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Cuenod, M.; Hensch, T.K. Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophr. Bull. 2015, 41, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Hyman, S.E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 2010, 13, 1161–1169. [Google Scholar] [CrossRef]
- Lee, G.; Zhou, Y. NMDAR hypofunction animal models of schizophrenia. Front. Mol. Neurosci. 2019, 12, 185. [Google Scholar] [CrossRef]
- Lodge, D.J.; Grace, A.A. Gestational methylazoxymethanol acetate administration: A developmental disruption model of schizophrenia. Behav. Brain Res. 2009, 204, 306–312. [Google Scholar] [CrossRef]
- Wischhof, L.; Irrsack, E.; Osorio, C.; Koch, M. Prenatal LPS-exposure--a neurodevelopmental rat model of schizophrenia--differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 17–30. [Google Scholar] [CrossRef]
- Yao, J.K.; Leonard, S.; Reddy, R. Altered glutathione redox state in schizophrenia. Dis. Markers 2006, 22, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Krüger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, D.; Hashimoto, K. Magnetic resonance spectroscopy study of the antioxidant defense system in schizophrenia. Antioxid. Redox Signal. 2011, 15, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Gawryluk, J.W.; Wang, J.F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130. [Google Scholar] [CrossRef]
- Wang, A.M.; Pradhan, S.; Coughlin, J.M.; Trivedi, A.; DuBois, S.L.; Crawford, J.L.; Sedlak, T.W.; Nucifor, F.C., Jr.; Nestadt, G.; Nucifora, L.G.; et al. Assessing brain metabolism with 7-T proton magnetic resonance spectroscopy in patients with first-episode psychosis. JAMA Psychiatry 2019, 76, 314–323. [Google Scholar] [CrossRef]
- Das, T.K.; Javadzadeh, A.; Dey, A.; Sabesan, P.; Théberge, J.; Radua, J.; Palaniyappan, L. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 91, 94–102. [Google Scholar] [CrossRef]
- Palaniyappan, L.; Park, M.T.M.; Jeon, P.; Limongi, R.; Yang, K.; Sawa, A.; Théberge, J. Is there a glutathione centered redox dysregulation subtype of schizophrenia? Antioxidants 2021, 10, 1703. [Google Scholar] [CrossRef]
- Broquist, H.P. Buthionine sulfoximine, an experimental tool to induce glutathione deficiency: Elucidation of glutathione and ascorbate in their role as antioxidants. Nutr. Rev. 1992, 50, 110–111. [Google Scholar] [CrossRef]
- Anderson, M.E. Glutathione: An overview of biosynthesis and modulation. Chem. Biol. Interact. 1998, 111–112, 1–14. [Google Scholar] [CrossRef]
- Cabungcal, J.H.; Preissmann, D.; Delseth, C.; Cuénod, M.; Do, K.Q.; Schenk, F. Transitory glutathione deficit during brain development induces cognitive impairment in juvenile and adult rats: Relevance to schizophrenia. Neurobiol. Dis. 2007, 26, 634–645. [Google Scholar] [CrossRef]
- Górny, M.; Wnuk, A.; Kamińska, A.; Kamińska, K.; Chwatko, G.; Bilska-Wilkosz, A.; Iciek, M.; Kajta, M.; Rogóż, Z.; Lorenc-Koci, E. Glutathione deficiency and alterations in the sulfur amino acids homeostasis during early postnatal development as potential triggering factors for schizophrenia-like behavior in adult rats. Molecules 2019, 24, 4253. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.A.; Leśkiewicz, M.; Kamińska, K.; Rogóż, Z.; Lorenc-Koci, E. Glutathione deficiency during early postnatal development causes schizophrenia-like symptoms and a reduction in BDNF levels in the cortex and hippocampus of adult Sprague-Dawley rats. Int. J. Mol. Sci. 2021, 22, 6171. [Google Scholar] [CrossRef]
- Laruelle, M.; Abi-Dargham, A.; van Dyck, C.H.; Gil, R.; D’Souza, C.D.; Erdos, J.; McCance, E.; Rosenblatt, W.; Fingado, C.; Zoghbi, S.S.; et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl. Acad. Sci. USA 1996, 93, 9235–9240. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Su, T.P.; Saunders, R.; Carson, R.E.; Kolachana, B.S.; de Bartolomeis, A.; Weinberger, D.R.; Weisenfeld, N.; Malhotra, A.K.; Eckelman, W.C.; et al. Schizophrenia is associated with elevated ampheta mine-induced synaptic dopamine concentrations: Evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. USA 1997, 94, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A.; Gil, R.; Krystal, J.; Baldwin, R.; Seibyl, J.; Bowers, M.; van Dyck, C.H.; Charney, D.S.; Innis, R.B.; Laruelle, M. Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am. J. Psychiatry 1998, 155, 761–767. [Google Scholar] [PubMed]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef]
- Day, J.J.; Sweatt, J.D. DNA methylation and memory formation. Nat. Neurosci. 2010, 13, 1319–1323. [Google Scholar] [CrossRef]
- Pidsley, R.; Dempster, E.L.; Mill, J. Brain weight in males is correlated with DNA methylation at IGF2. Mol. Psychiatry 2010, 15, 880–881. [Google Scholar] [CrossRef][Green Version]
- Heyward, F.D.; Sweatt, J.D. DNA methylation in memory formation: Emerging insights. Neuroscientist 2015, 21, 475–489. [Google Scholar] [CrossRef]
- Thome, J.; Foley, P.; Riederer, P. Neurotrophic factors and the maldevelopmental hypothesis of schizophrenic psychoses. J. Neural Transm 1998, 105, 85–100. [Google Scholar] [CrossRef]
- Angelucci, F.; Brenè, S.; Mathé, A.A. BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry 2005, 10, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A. Brain-derived neurotropic factor/TrkB signaling in the pathogenesis and novel pharmacotherapy of schizophrenia. Neurosignals 2008, 16, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef]
- Weickert, C.S.; Hyde, T.M.; Lipska, B.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2003, 8, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Bergen, S.E.; Nguyen, Q.L.; Xu, B.; Monteggia, L.M.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 2005, 25, 372–383. [Google Scholar] [CrossRef]
- Durany, N.; Michel, T.; Zöchling, R.; Boissl, K.W.; Cruz-Sánchez, F.F.; Riederer, P.; Thome, J. Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr. Res. 2001, 52, 79–86. [Google Scholar] [CrossRef]
- Takahashi, M.; Shirakawa, O.; Toyooka, K.; Kitamura, N.; Hashimoto, T.; Maeda, K.; Koizumi, S.; Wakabayashi, K.; Takahashi, H.; Someya, T.; et al. Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol. Psychiatry 2000, 5, 293–300. [Google Scholar] [CrossRef]
- Iritani, S.; Niizato, K.; Nawa, H.; Ikeda, K.; Emson, P.C. Immunohistochemical study of brain-derived neurotrophic factor and its receptor, TrkB, in the hippocampal formation of schizophrenic brains. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 801–807. [Google Scholar] [CrossRef]
- Lipska, B.K.; Jaskiw, G.E.; Weinberger, D.R. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: A potential animal model of schizophrenia. Neuropsychopharmacology 1993, 9, 67–75. [Google Scholar] [CrossRef]
- Tseng, K.Y.; Chambers, R.A.; Lipska, B.K. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav. Brain Res. 2009, 204, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Lipska, B.K.; Khaing, Z.Z.; Weickert, C.S.; Weinberger, D.R. BDNF mRNA expression in rat hippocampus and prefrontal cortex: Effects of neonatal ventral hippocampal damage and antipsychotic drugs. Eur. J. Neurosci. 2001, 14, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Ashe, P.C.; Chlan-Fourney, J.; Juorio, A.V.; Li, X.M. Brain-derived neurotrophic factor (BDNF) mRNA in rats with neonatal ibotenic acid lesions of the ventral hippocampus. Brain Res. 2002, 956, 126–135. [Google Scholar] [CrossRef]
- Fiore, M.; Korf, J.; Antonelli, A.; Talamini, L.; Aloe, L. Long-lasting effects of prenatal MAM exposure on water maze response in aged rats: Correlation with altered brain development and neurotrophins’ expression. Neurotoxicol. Teratol. 2002, 24, 179–191. [Google Scholar] [CrossRef]
- Angelucci, F.; Mathé, A.A.; Aloe, L. Brain-derived neurotrophic factor and tyrosine kinase receptor TrkB in rat brain are significantly altered after haloperidol and risperidone administration. J. Neurosci. Res. 2000, 60, 83–94. [Google Scholar] [CrossRef]
- Bai, O.; Clan-Fourney, J.; Bowen, R.; Keegan, D.; Li, X.M. Expression of brain-derived neurotrophic factor mRNA in rat hippocampus after treatment with antipsychotic drugs. J. Neurosci. Res. 2003, 71, 127–131. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, S.K.; Kim, J.M.; Yoon, J.S.; Kim, Y.H. Effects of quetiapine on the brain-derived neurotrophic factor expression in the hippocampus and neocortex of rats. Neurosci. Lett. 2006, 402, 5–29. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, C.H.; Lee, J.G.; Lee, S.J.; Kim, N.R.; Choi, S.M.; Kim, Y.H. Differential effects of ziprosidone and haloperidol on immobilization stress-induced mRNA expression in the hippocampus and neocortex of rats. J. Psychiatr. Res. 2009, 43, 274–281. [Google Scholar] [CrossRef]
- Dean, O.; Giorlando, F.; Berk, M. N-acetylcysteine in psychiatry: Current therapeutic evidence and potential mechanisms of action. J. Psychiatry Neurosci. 2011, 36, 78–86. [Google Scholar] [CrossRef]
- Yolland, C.O.B.; Phillipou, A.; Castle, D.J.; Neill, E.; Hughes, M.E.; Galletly, C.; Smith, Z.M.; Francis, P.S.; Dean, O.M.; Sarris, J.; et al. Improvement of cognitive function in schizophrenia with N-acetylcysteine: A theoretical review. Nutr. Neurosci. 2020, 23, 139–148. [Google Scholar] [CrossRef]
- Hereta, M.; Kamińska, K.; Rogóż, Z. Co-treatment with antidepressants and aripiprazole reversed the MK-801-induced some negative symptoms of schizophrenia in rats. Pharmacol. Rep. 2019, 71, 768–773. [Google Scholar] [CrossRef]
- Hereta, M.; Kamińska, K.; Białoń, M.; Wąsik, A.; Lorenc-Koci, E.; Rogóż, Z. Effect of combined treatment with aripiprazole and antidepressants on the MK-801-induced deficits in recognition memory in novel recognition test and on the release of monoamines in the rat frontal cortex. Behav. Brain Res. 2020, 393, 112769. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Koprivica, V.; Chen, R.; Tottori, K.; Kikuchi, T.; Altar, A. The antipsychotic aripiprazole is a potent, partial agonist at the human 5-HT1A receptor. Eur. J. Pharmacol. 2002, 441, 137–140. [Google Scholar] [CrossRef]
- Taylor, D.M. Aripiprazole: A review of its pharmacology and clinical use. Int. J. Clin. Pract. 2003, 57, 49–54. [Google Scholar] [PubMed]
- Green, B. Focus on aripiprazole. Curr. Med. Res. Opin. 2004, 20, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Bruins Slot, L.A.; Kleven, M.S.; Newman-Tancredi, A. Effects of novel antipsychotics with mixed D(2) antagonist/5-HT(1A) agonist properties on PCP-induced social interaction deficits in the rat. Neuropharmacology 2005, 49, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine D2 and serotonin 5-HT1A receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef]
- Torrisi, S.A.; Laudani, S.; Contarini, G.; De Luca, A.; Geraci, F.; Managò, F.; Papaleo, F.; Salomone, S.; Drago, F.; Leggio, G.M. Dopamine, cognitive impairments and second-generation antipsychotics: From mechanistic advances to more personalized treatments. Pharmaceuticals 2020, 13, 365. [Google Scholar] [CrossRef]
- DeLeon, A.; Patel, N.C.; Crismon, M.L. Aripiprazole: A comprehensive review of its pharmacology, clinical efficacy, and tolerability. Clin. Ther. 2004, 26, 649–666. [Google Scholar] [CrossRef]
- Ishii, D.; Matsuzawa, D.; Kanahara, N.; Matsuda, S.; Sutoh, C.; Ohtsuka, H.; Nakazawa, K.; Kohno, M.; Hashimoto, K.; Iyo, M.; et al. Effects of aripiprazole on MK-801-induced prepulse inhibition deficits and mitogen-activated protein kinase signal transduction pathway. Neurosci. Lett. 2010, 471, 53–57. [Google Scholar] [CrossRef]
- Carli, M.; Calcagno, E.; Mainolfi, P.; Mainini, E.; Invernizzi, R.W. Effects of aripiprazole, olanzapine, and haloperidol in a model of cognitive deficit of schizophrenia in rats: Relationship with glutamate release in the medial prefrontal cortex. Psychopharmacology 2011, 214, 639–652. [Google Scholar] [CrossRef]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Moghaddam, B. Cognitive dysfunction in schizophrenia: Convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 2006, 63, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Glutamatergic theories of schizophrenia. Isr. J. Psychiatry Relat. Sci. 2010, 47, 4–16. [Google Scholar]
- McCutcheon, R.A.; Krystal, J.H.; Howes, O.D. Dopamine and glutamate in schizophrenia: Biology, symptoms and treatment. World Psychiatry 2020, 19, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Hamberger, A.C.; Chiang, G.H.; Nylén, E.S.; Scheff, S.W.; Cotman, C.W. Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res. 1979, 168, 513–530. [Google Scholar] [CrossRef]
- Erecińska, M.; Silver, I.A. Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol. 1990, 35, 245–296. [Google Scholar] [CrossRef]
- Sanacora, G.; Zarate, C.A.; Krystal, J.H.; Manji, H.K. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat. Rev. Drug Discov. 2008, 7, 426–437. [Google Scholar] [CrossRef]
- Kam, K.; Nicoll, R. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J. Neurosci. 2007, 27, 9192–9200. [Google Scholar] [CrossRef]
- Koga, M.; Serritella, A.V.; Messmer, M.M.; Hayashi-Takagi, A.; Hester, L.D.; Snyder, S.H.; Sawa, A.; Sedlak, T.W. Glutathione is a physiologic reservoir of neuronal glutamate. Biochem. Biophys. Res. Commun. 2011, 409, 596–602. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef] [PubMed]
- Tyzio, R.; Holmes, G.L.; Ben-Ari, Y.; Roustem Khazipov, R. Timing of the developmental switch in GABA(A) mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 2007, 48 (Suppl. 5), 96–105. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T. Glutamate and schizophrenia: Beyond the dopamine hypothesis. Cell Mol. Neurobiol. 2006, 26, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Bymaster, F.P.; Meltzer, H.Y.; Deutch, A.Y.; Duncan, G.E.; Marx, C.E.; Aprille, J.R.; Dwyer, D.S.; Li, X.M.; Mahadik, S.P.; et al. Antipsychotic drugs: Comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol. Rev. 2008, 60, 358–403. [Google Scholar] [CrossRef]
- Berk, M.; Copolov, D.; Dean, O.; Lu, K.; Jeavons, S.; Schapkaitz, I.; Anderson-Hunt, M.; Judd, F.; Katz, F.; Katz, P.; et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol. Psychiatry 2008, 64, 361–368. [Google Scholar] [CrossRef]
- Farokhnia, M.; Azarkolah, A.; Adinehfar, F.; Khodaie-Ardakani, M.R.; Hosseini, S.M.; Yekehtaz, H.; Tabrizi, M.; Rezaei, F.; Salehi, B.; Sadeghi, S.M.; et al. N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: A randomized, double-blind, placebo-controlled study. Clin. Neuropharmacol. 2013, 36, 185–192. [Google Scholar] [CrossRef]
- Breier, A.; Liffick, E.; Hummer, T.A.; Vohs, J.L.; Yang, Z.; Mehdiyoun, N.F.; Visco, A.C.; Metzler, E.; Zhang, Y.; Francis, M.M. Effects of 12-month, double-blind N-acetyl cysteine on symptoms, cognition and brain morphology in early phase schizophrenia spectrum disorders. Schizophr. Res. 2018, 199, 395–402. [Google Scholar] [CrossRef]
- Conus, P.; Seidman, L.J.; Fournier, M.; Xin, L.; Cleusix, M.; Baumann, P.S.; Ferrari, C.; Cousins, A.; Alameda, L.; Gholam-Rezaee, M.; et al. N-acetylcysteine in a double-blind randomized placebo-controlled trial: Toward biomarker-guided treatment in early psychosis. Schizophr. Bull. 2018, 44, 317–327. [Google Scholar] [CrossRef]
- Rapado-Castro, M.; Dodd, S.; Bush, A.I.; Malhi, G.S.; Skvarc, D.R.; On, Z.X.; Berk, M.; Dean, O.M. Cognitive effects of adjunctive N-acetyl cysteine in psychosis. Psychol. Med. 2017, 47, 866–876. [Google Scholar] [CrossRef]
- Skvarc, D.R.; Dean, O.M.; Byrne, L.K.; Gray, L.; Lane, S.; Lewis, M.; Fernandes, B.S.; Berk, M.; Marriott, A. The effect of N-acetylcysteine (NAC) on human cognition—A systematic review. Neurosci. Biobehav. Rev. 2017, 78, 44–56. [Google Scholar] [CrossRef]
- Baker, D.A.; Madayag, A.; Kristiansen, L.V.; Meador-Woodruff, J.H.; Haroutunian, V.; Raju, I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology 2008, 33, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Massie, A.; Boillée, S.; Hewett, S.; Knackstedt, L.; Lewerenz, J. Main path and byways: Non-vesicular glutamate release by system xc(-) as an important modifier of glutamatergic neurotransmission. J. Neurochem. 2015, 135, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.A.; Xi, Z.X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 2002, 22, 9134–9141. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/glutamate antiporter and aripiprazole compensate NMDA antagonist-induced dysfunction of thalamocortical L-glutamatergic transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef]
- Galderisi, S.; Kaiser, S.; Bitter, I.; Nordentoft, M.; Mucci, A.; Sabé, M.; Giordano, G.M.; Nielsen, M.Ø.; Glenthøj, L.B.; Pezzella, P.; et al. EPA guidance on treatment of negative symptoms in schizophrenia. Eur. Psychiatry 2021, 64, e21. [Google Scholar] [CrossRef]
- Lech, M.A.; Kamińska, K.; Leśkiewicz, M.; Lorenc-Koci, E.; Rogóż, Z. Impact of repeated co-treatment with escitalopram and aripiprazole on the schizophrenia-like behaviors and BDNF mRNA expression in the adult Sprague-Dawley rats exposed to glutathione deficit during early postnatal development of the brain. Pharmacol. Rep. 2021, 73, 1712–1723. [Google Scholar] [CrossRef]
- Minarini, A.; Ferrari, S.; Galletti, M.; Giambalvo, N.; Perrone, D.; Rioli, G.; Galeazzi, G.M. N-acetylcysteine in the treatment of psychiatric disorders: Current status and future prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 279–292. [Google Scholar] [CrossRef]
- Nery, F.G.; Tallman, M.J.; Cecil, K.M.; Blom, T.J.; Patino, L.R.; Adler, C.M.; DelBello, M.P. N-acetylcysteine for depression and glutamate changes in the left prefrontal cortex in adolescents and young adults at risk for bipolar disorder: A pilot study. Early Interv. Psychiatry 2022, 16, 195–199. [Google Scholar] [CrossRef]
- Kamińska, K.; Rogóż, Z. The effect of combined treatment with risperidone and antidepressants on the MK-801-induced deficits in the social interaction test in rats. Pharmacol. Rep. 2015, 67, 1183–1187. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogóż, Z.; Kamińska, K.; Lech, M.A.; Lorenc-Koci, E. N-Acetylcysteine and Aripiprazole Improve Social Behavior and Cognition and Modulate Brain BDNF Levels in a Rat Model of Schizophrenia. Int. J. Mol. Sci. 2022, 23, 2125. https://doi.org/10.3390/ijms23042125
Rogóż Z, Kamińska K, Lech MA, Lorenc-Koci E. N-Acetylcysteine and Aripiprazole Improve Social Behavior and Cognition and Modulate Brain BDNF Levels in a Rat Model of Schizophrenia. International Journal of Molecular Sciences. 2022; 23(4):2125. https://doi.org/10.3390/ijms23042125
Chicago/Turabian StyleRogóż, Zofia, Kinga Kamińska, Marta Anna Lech, and Elżbieta Lorenc-Koci. 2022. "N-Acetylcysteine and Aripiprazole Improve Social Behavior and Cognition and Modulate Brain BDNF Levels in a Rat Model of Schizophrenia" International Journal of Molecular Sciences 23, no. 4: 2125. https://doi.org/10.3390/ijms23042125
APA StyleRogóż, Z., Kamińska, K., Lech, M. A., & Lorenc-Koci, E. (2022). N-Acetylcysteine and Aripiprazole Improve Social Behavior and Cognition and Modulate Brain BDNF Levels in a Rat Model of Schizophrenia. International Journal of Molecular Sciences, 23(4), 2125. https://doi.org/10.3390/ijms23042125