
Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Identification of Putative PPRHs Target Sequences in KRAS Promoter and Gene Region
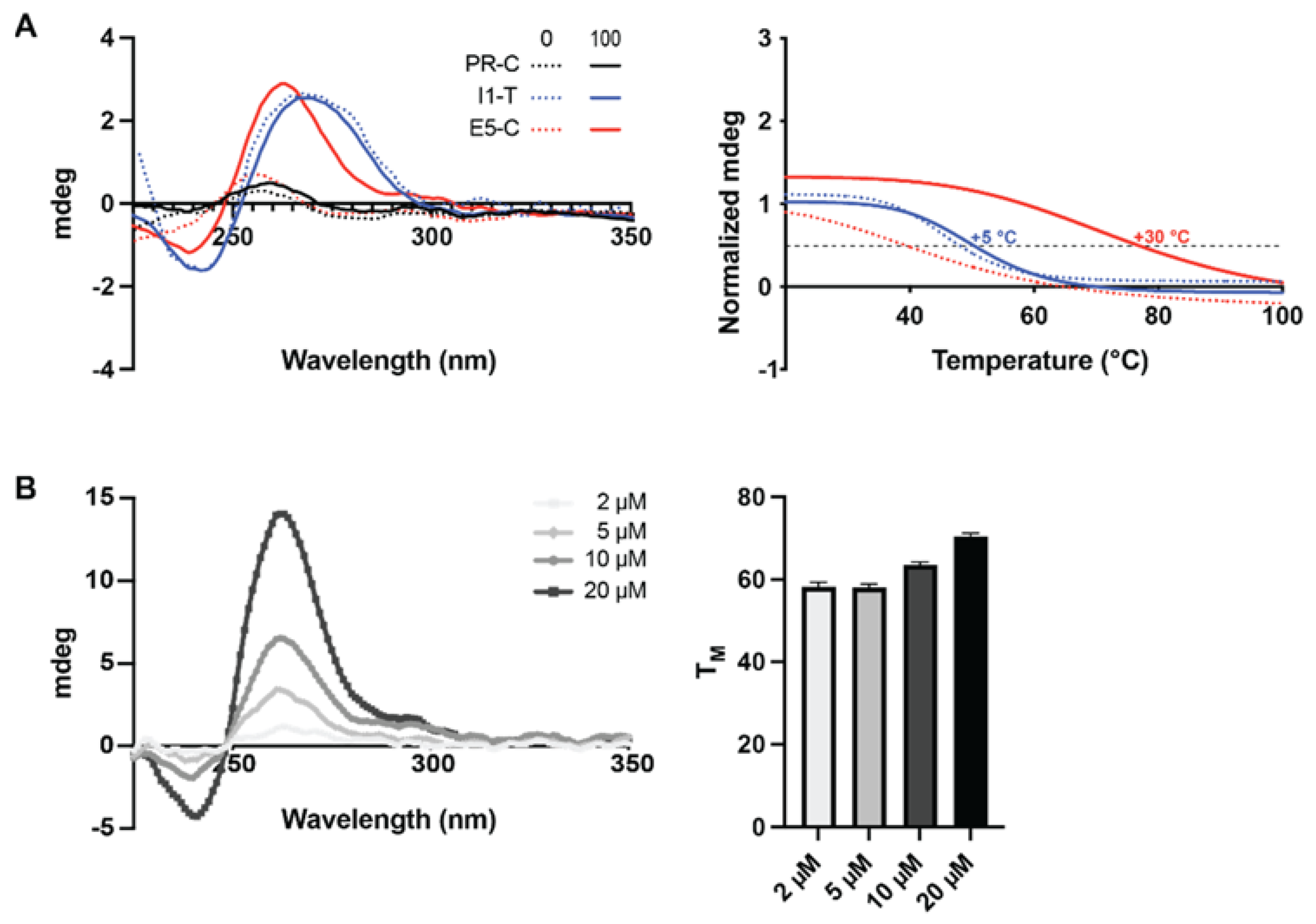
2.2. G4 Formation within Newly Identified G4FS–PR, I1 and E5
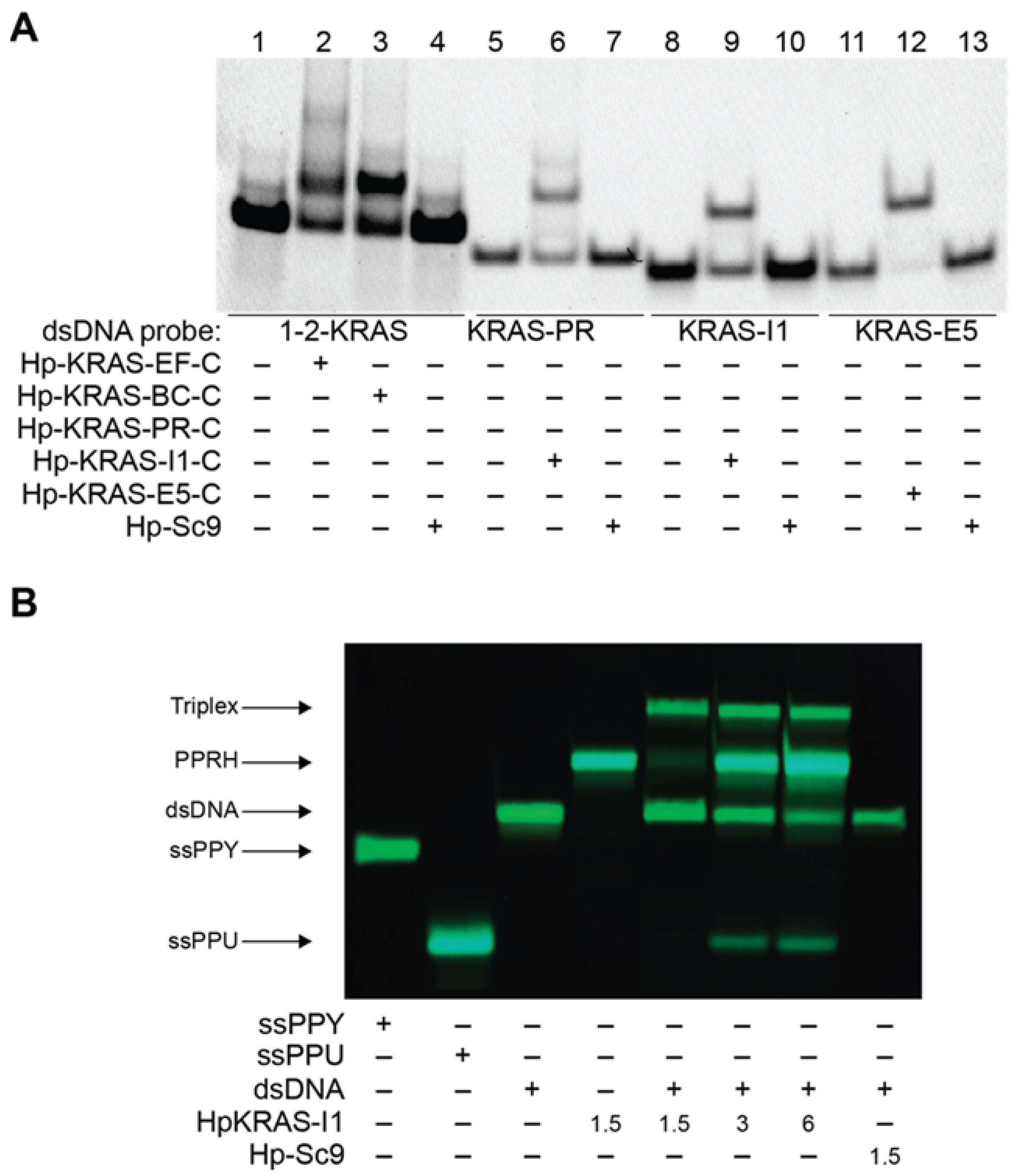
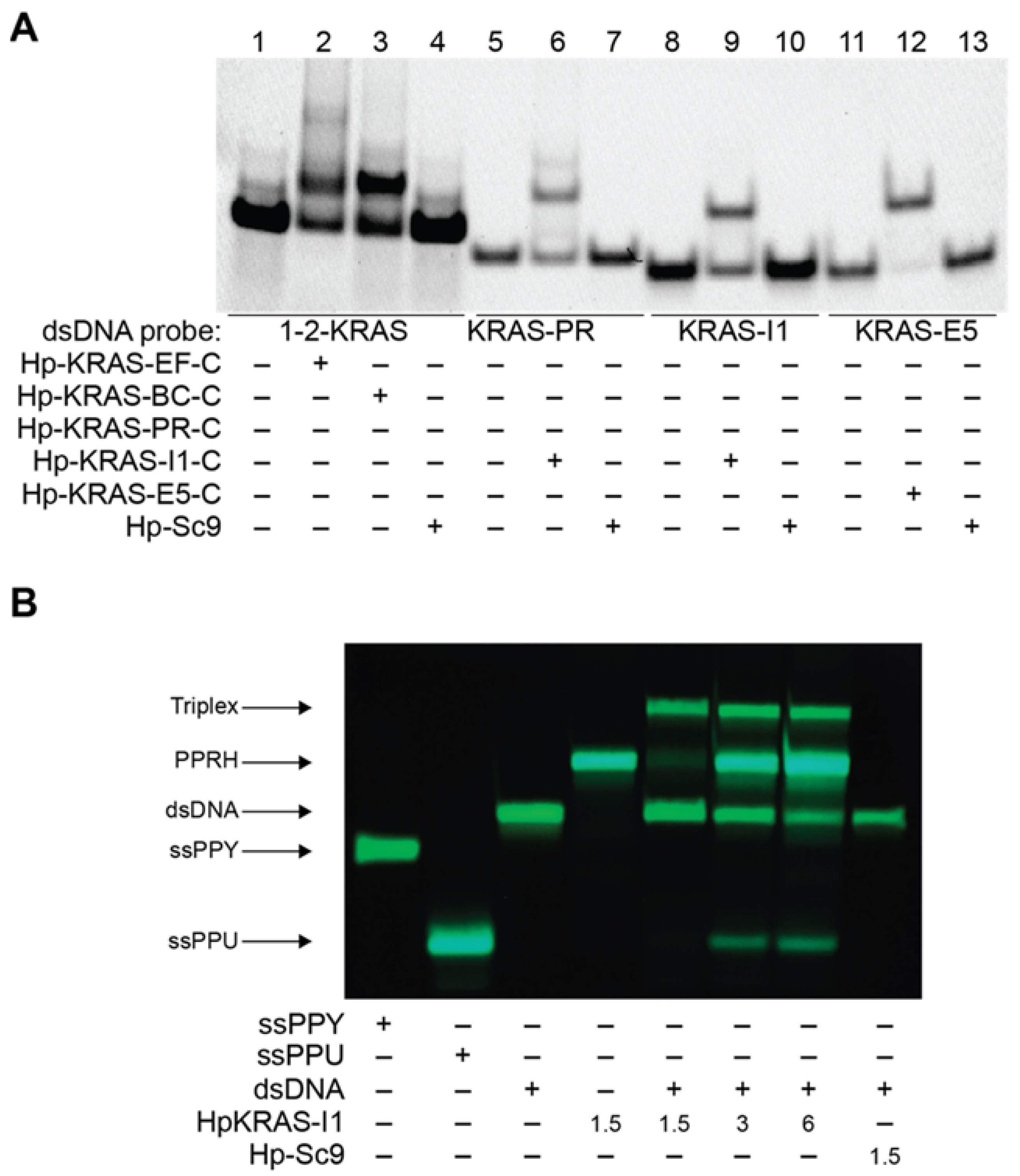
2.3. PPRHs Binding to KRAS Target Sequences and Polupurine Strand Displacement
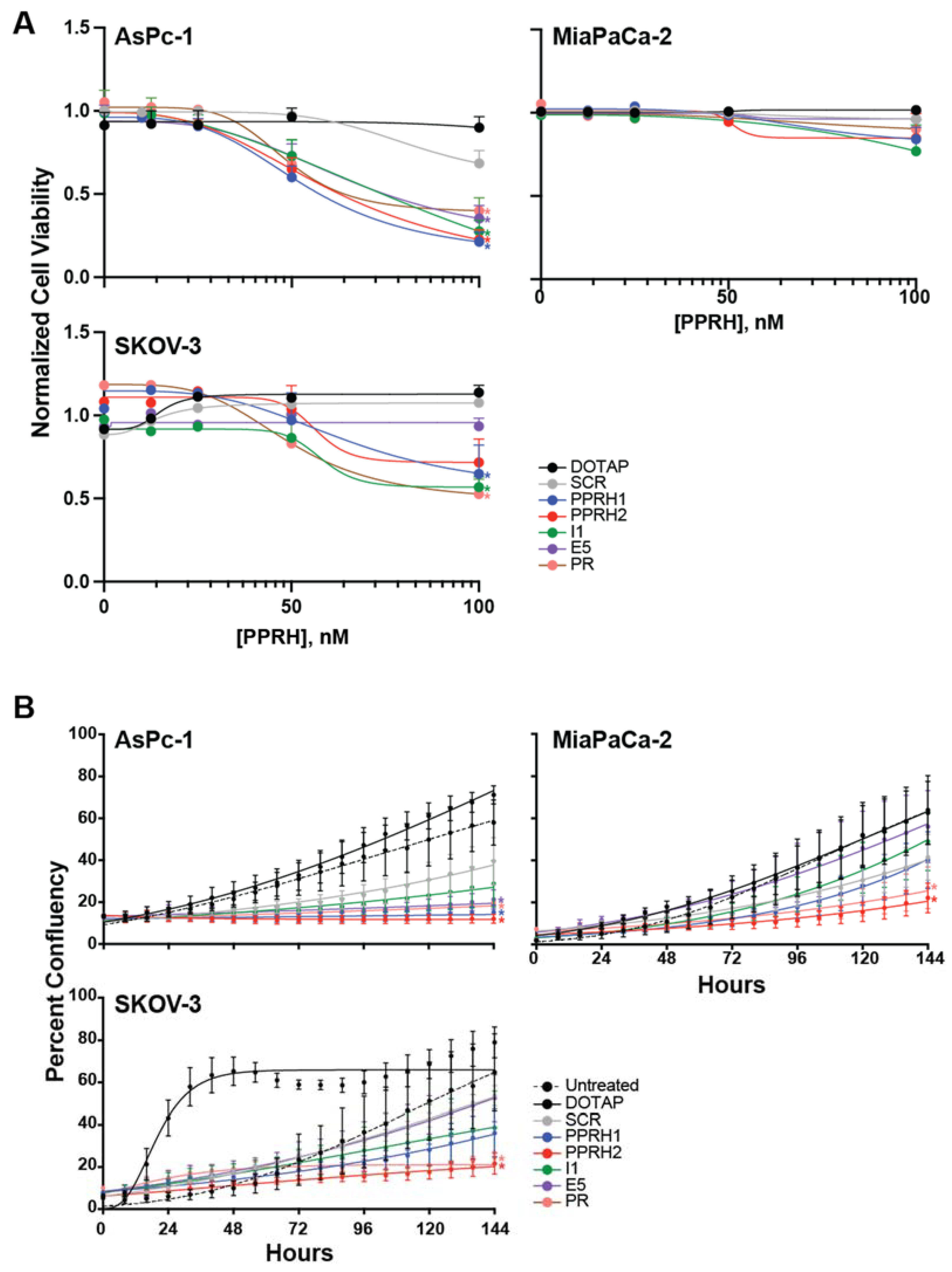
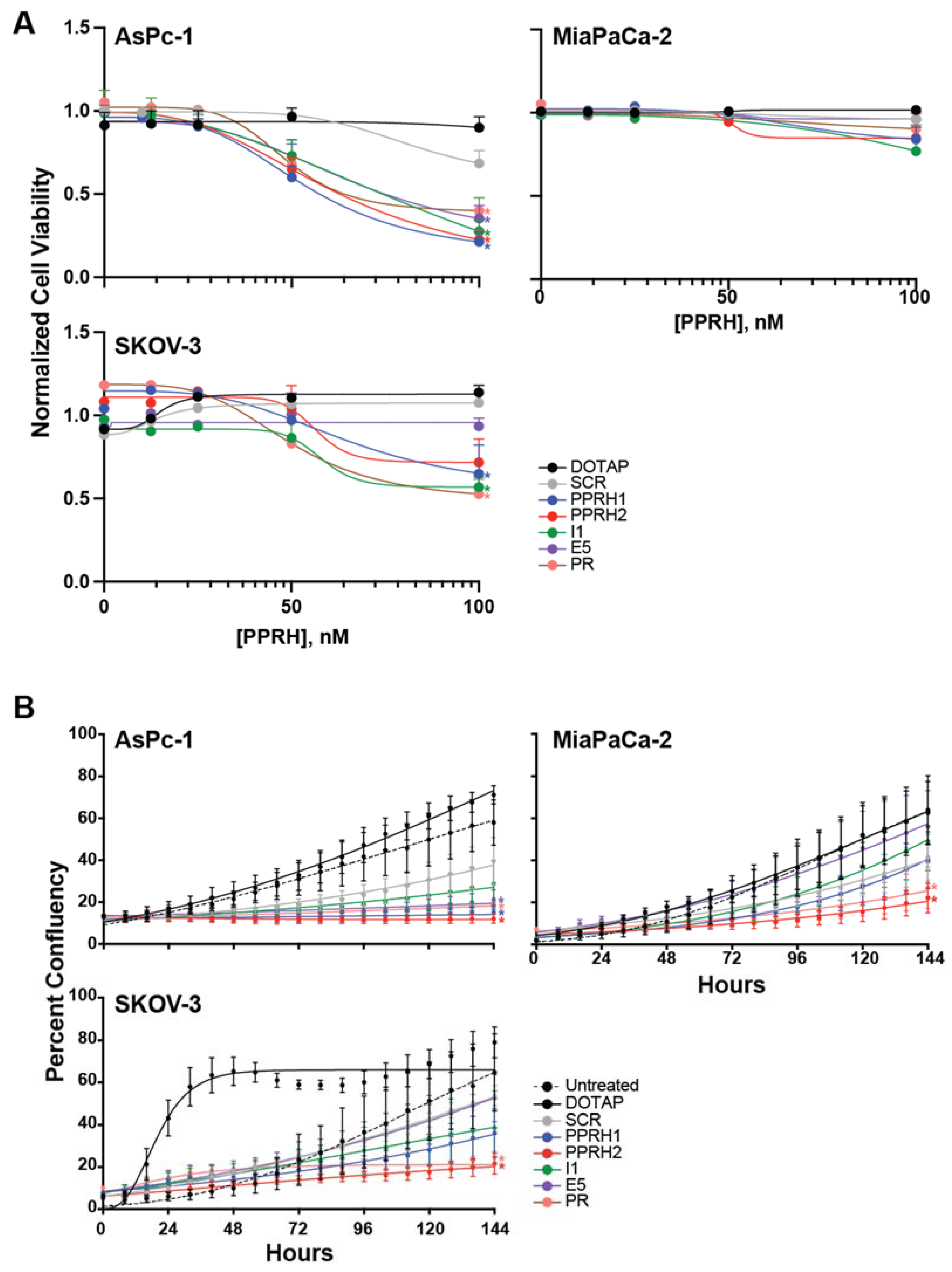
2.4. Effect of KRAS-Targeting PPRHs on Cell Viability and Growth
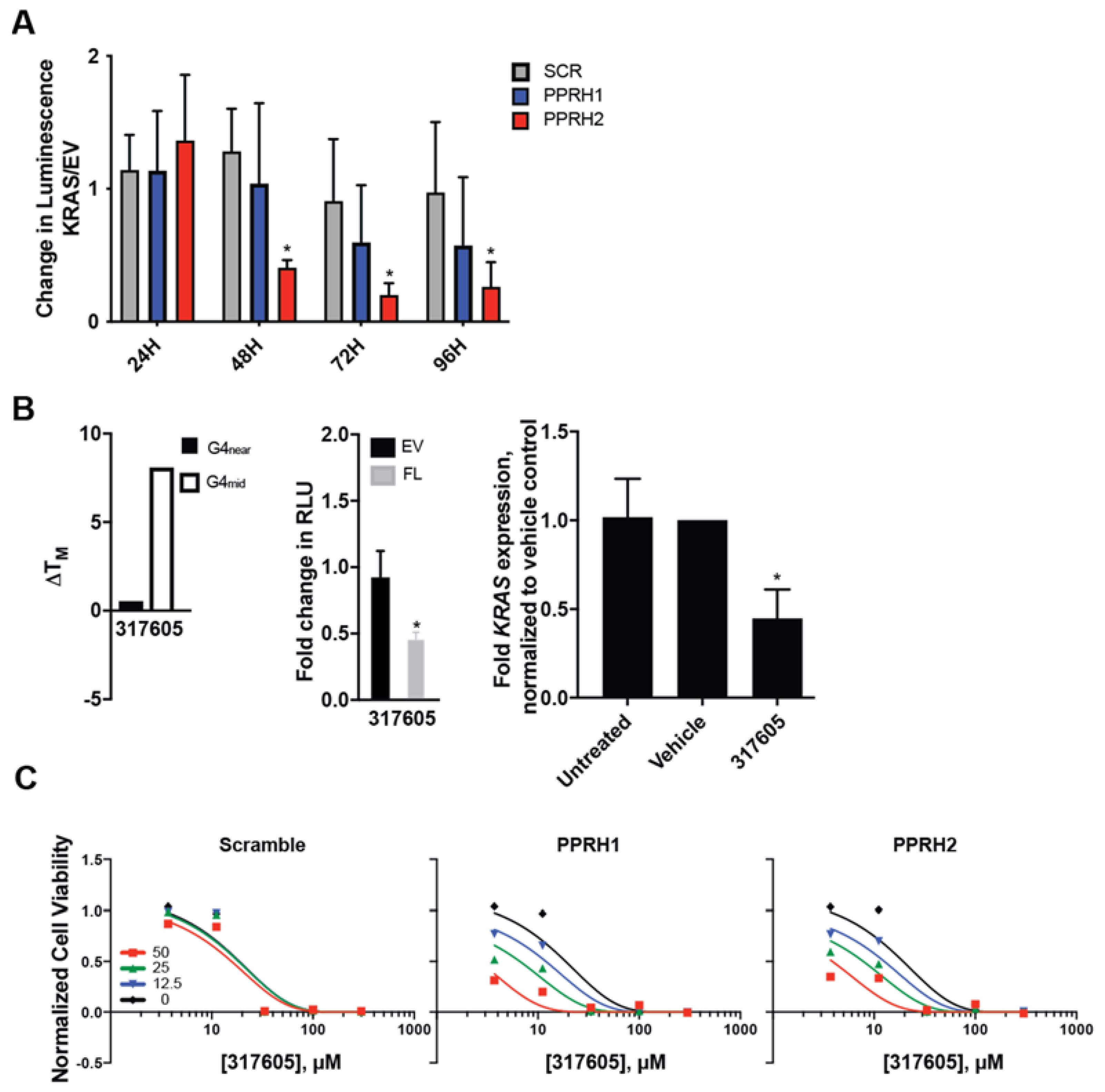
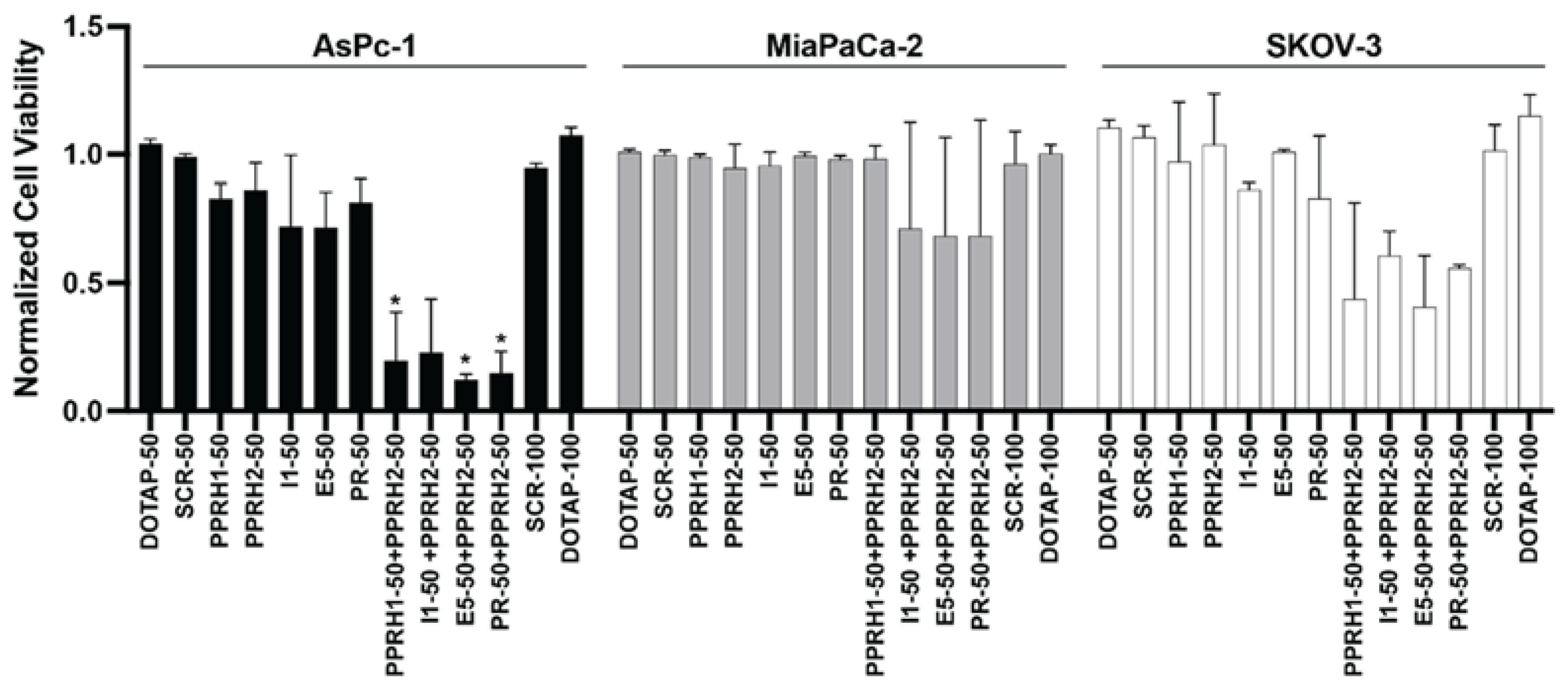
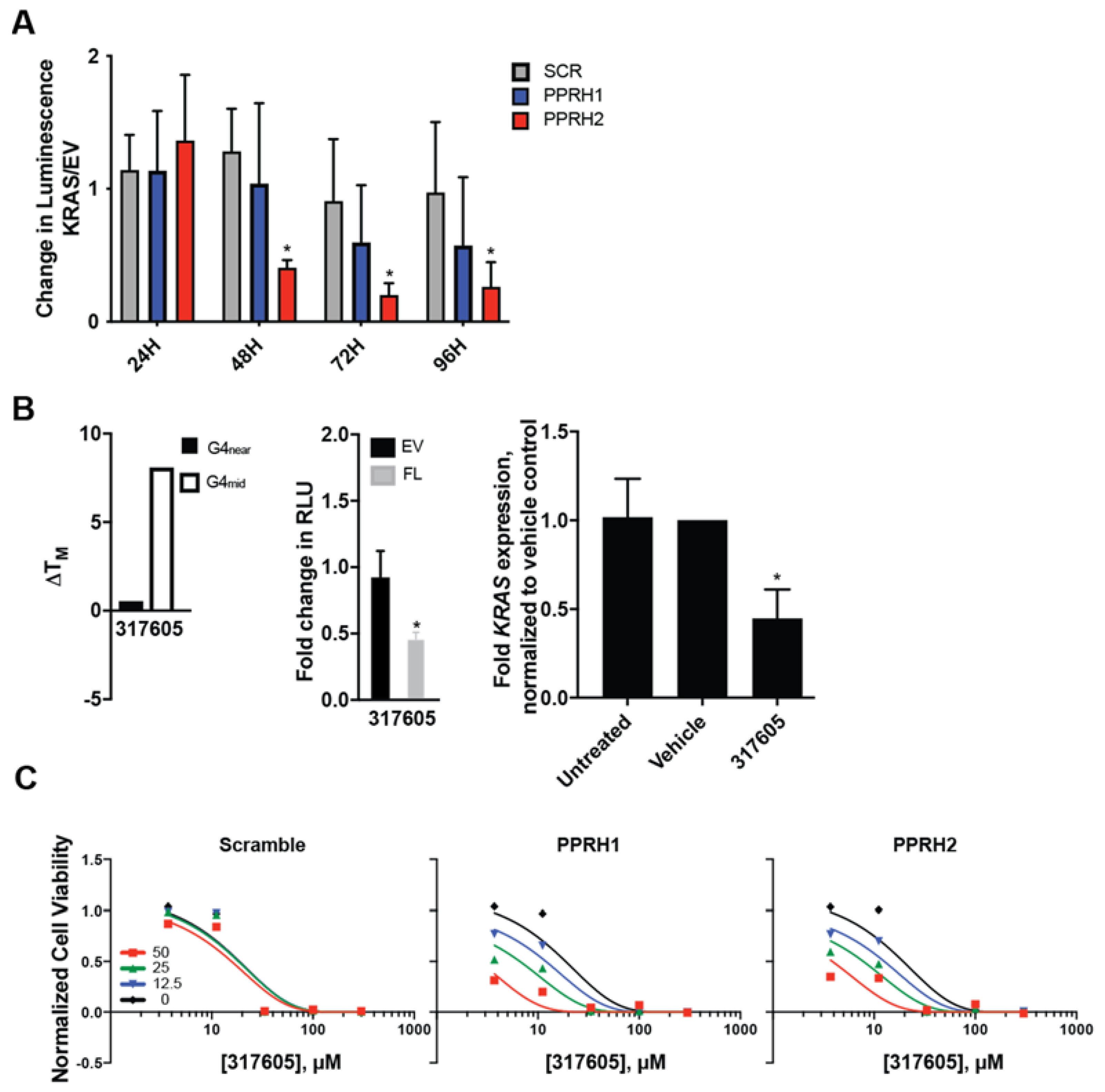
2.5. Combined Effect of Targeting KRAS Promoter G4s with PPRH and Small Molecules
3. Discussion
4. Materials and Methods
4.1. Design of Polypurine Reverse Hoogsteen Hairpins
4.2. Electronic Circular Dichroism (ECD)
4.3. Electrophoretic Mobility Shift Assay (EMSA)
4.4. Strand Displacement Assay upon PPRH Incubation
4.5. Cell Cultures
4.6. Cellular Viability and Cell Growth Studies
4.7. FRET Melt2
4.8. Luciferase Assays
4.9. RT-qPCR
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adjei, A.A. Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst. 2001, 93, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friday, B.B.; Adjei, A.A. K-ras as a target for cancer therapy. Biochim. Biophys. Acta 2005, 1756, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Lyons, J.; Miller, A.L.; Phan, V.T.; Alarcon, I.R.; McCormick, F. Ras signaling and therapies. Adv. Cancer Res. 2009, 102, 1–17. [Google Scholar] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Birkeland, E.; Wik, E.; Mjos, S.; Hoivik, E.A.; Trovik, J.; Werner, H.M.J.; Kusonmano, K.; Petersen, K.; Raeder, M.B.; Holst, F.; et al. KRAS gene amplification and overexpression but not mutation associates with aggressive and metastatic endometrial cancer. Br. J. Cancer 2012, 107, 1997–2004. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Chen, Y.; McGee, J.; Chen, X.; Doman, T.N.; Gong, X.; Zhang, Y.; Hamm, N.; Ma, X.; Higgs, R.E.; Bhagwat, S.V.; et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS ONE 2014, 9, e98293. [Google Scholar] [CrossRef]
- Craig, D.W.; O’Shaughnessy, J.A.; Kiefer, J.A.; Aldrich, J.; Sinari, S.; Moses, T.M.; Wong, S.; Dinh, J.; Christoforides, A.; Blum, J.L.; et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar]
- Goddard, N.C.; McIntyre, A.; Summersgill, B.; Gilbert, D.; Kitazawa, S.; Shipley, J. KIT and RAS signalling pathways in testicular germ cell tumours: New data and a review of the literature. Int. J. Androl. 2007, 30, 337–348, discussion 349; discussion 49. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Chen, G.B.; Matsumura, N.; Lai, H.-C.; Mori, S.; Li, J.; Wong, M.K.; Konishi, I.; Thiery, J.-P.; Goh, L. Histotype-specific copy-number alterations in ovarian cancer. BMC Med. Genom. 2012, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; Garcia, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer 2015, 15, 415. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, A.; Gilbert, D.; Goddard, N.; Looijenga, L.; Shipley, J. Genes, chromosomes and the development of testicular germ cell tumors of adolescents and adults. Genes Chromosomes Cancer 2008, 47, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Podo, F.; Buydens, L.M.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; Huffel, S.V.; van Laarhoven, H.W.M.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Pulciani, S.; Santos, E.; Long, L.K.; Sorrentino, V.; Barbacid, M. ras gene Amplification and malignant transformation. Mol. Cell. Biol. 1985, 5, 2836–2841. [Google Scholar]
- Rahman, M.T.; Nakayama, K.; Rahman, M.; Katagiri, H.; Katagiri, A.; Ishibashi, T.; Ishikawa, M.; Sato, E.; Iida, K.; Nakayama, N.; et al. KRAS and MAPK1 gene amplification in type II ovarian carcinomas. Int. J. Mol. Sci. 2013, 14, 13748–13762. [Google Scholar] [CrossRef] [Green Version]
- Roelofs, H.; Mostert, M.C.; Pompe, K.; Zafarana, G.; van Oorschot, M.; van Gurp, R.J.H.L.M.; Gillis, A.J.M.; Stoop, H.; Beverloo, B.; Oosterhuis, J.W.; et al. Restricted 12p amplification and RAS mutation in human germ cell tumors of the adult testis. Am. J. Pathol. 2000, 157, 1155–1166. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Ali, S.M.; Wang, K.; Palmer, G.; Yelensky, R.; Lipson, D.; Miller, V.A.; Zajchowski, D.; Shawver, L.K.; Stephens, P.J. Comprehensive genomic profiling of epithelial ovarian cancer by next generation sequencing-based diagnostic assay reveals new routes to targeted therapies. Gynecol. Oncol. 2013, 130, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, D.; Cogdell, D.; Hu, L.; Xue, F.; Broaddus, R.; Zhang, W. Genomic characterization of gene copy-number aberrations in endometrial carcinoma cell lines derived from endometrioid-type endometrial adenocarcinoma. Technol. Cancer Res. Treat. 2010, 9, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Yang-Feng, T.L.; Li, S.B.; Leung, W.Y.; Carcangiu, M.L.; Schwartz, P.E. Trisomy 12 and K-ras-2 amplification in human ovarian tumors. Int. J. Cancer 1991, 48, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Sotorasib Edges Closer to Approval. Cancer Discov. 2021, 11, OF2. [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Ahmad, A.; Aboukameel, A.; Bao, B.; Padhye, S.; Philip, P.A.; Sarkar, F.H. Increased Ras GTPase activity is regulated by miRNAs that can be attenuated by CDF treatment in pancreatic cancer cells. Cancer Lett. 2012, 319, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Duursma, A.M.; Agami, R. Ras interference as cancer therapy. Semin. Cancer Biol. 2003, 13, 267–273. [Google Scholar] [CrossRef]
- Hu, Y.; Ou, Y.; Wu, K.; Chen, Y.; Sun, W. miR-143 inhibits the metastasis of pancreatic cancer and an associated signaling pathway. Tumour Biol. 2012, 33, 1863–1870. [Google Scholar] [CrossRef]
- Wickstrom, E. Oligonucleotide treatment of ras-induced tumors in nude mice. Mol. Biotechnol. 2001, 18, 35–55. [Google Scholar] [CrossRef]
- Hansel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L. Four-stranded nucleic acids: Structure, function and targeting of G-quadruplexes. Chem. Soc. Rev. 2008, 37, 1375–1384. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; Miller, J.; Howat, W.J.; Balasubramanian, S. Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues. PLoS ONE 2014, 9, e102711. [Google Scholar] [CrossRef] [PubMed]
- Jordano, J.; Perucho, M. Chromatin structure of the promoter region of the human c-K-ras gene. Nucleic Acids Res. 1986, 14, 7361–7378. [Google Scholar] [CrossRef] [Green Version]
- Jordano, J.; Perucho, M. Initial characterization of a potential transcriptional enhancer for the human c-K-ras gene. Oncogene 1988, 2, 359–366. [Google Scholar]
- Yamamoto, F.; Perucho, M. Characterization of the human c-K-ras gene promoter. Oncogene Res. 1988, 3, 125–130. [Google Scholar]
- Morgan, R.K.; Batra, H.; Gaerig, V.C.; Hockings, J.; Brooks, T.A. Identification and characterization of a new G-quadruplex forming region within the kRAS promoter as a transcriptional regulator. Biochim. Biophys. Acta 2016, 1859, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Coma, S.; Noe, V.; Eritja, R.; Ciudad, C.J. Strand displacement of double-stranded DNA by triplex-forming antiparallel purine-hairpins. Oligonucleotides 2005, 15, 269–283. [Google Scholar] [CrossRef] [Green Version]
- De Almagro, M.C.; Coma, S.; Noe, V.; Ciudad, C.J. Polypurine hairpins directed against the template strand of DNA knock down the expression of mammalian genes. J. Biol. Chem. 2009, 284, 11579–11589. [Google Scholar] [CrossRef] [Green Version]
- De Almagro, M.C.; Mencia, N.; Noe, V.; Ciudad, C.J. Coding polypurine hairpins cause target-induced cell death in breast cancer cells. Hum. Gene Ther. 2011, 22, 451–463. [Google Scholar] [CrossRef]
- Villalobos, X.; Rodriguez, L.; Sole, A.; Lliberós, C.; Mencia, N.; Ciudad, C.J.; Noé, V. Effect of Polypurine Reverse Hoogsteen Hairpins on Relevant Cancer Target Genes in Different Human Cell Lines. Nucleic Acid Ther. 2015, 25, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Bener, G.; A, J.F.; Sanchez de Diego, C.; Pascual Fabregat, I.; Ciudad, C.J.; Noe, V. Silencing of CD47 and SIRPalpha by Polypurine reverse Hoogsteen hairpins to promote MCF-7 breast cancer cells death by PMA-differentiated THP-1 cells. BMC Immunol. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciudad, C.J.; Medina Enriquez, M.M.; Felix, A.J.; Bener, G.; Noe, V. Silencing PD-1 and PD-L1: The potential of PolyPurine Reverse Hoogsteen hairpins for the elimination of tumor cells. Immunotherapy 2019, 11, 369–372. [Google Scholar] [CrossRef]
- Medina Enriquez, M.M.; Felix, A.J.; Ciudad, C.J.; Noe, V. Cancer immunotherapy using PolyPurine Reverse Hoogsteen hairpins targeting the PD-1/PD-L1 pathway in human tumor cells. PLoS ONE 2018, 13, e0206818. [Google Scholar] [CrossRef] [PubMed]
- Aubets, E.; Noe, V.; Ciudad, C.J. Targeting replication stress response using polypurine reverse hoogsteen hairpins directed against WEE1 and CHK1 genes in human cancer cells. Biochem. Pharmacol. 2020, 175, 113911. [Google Scholar] [CrossRef]
- Felix, A.J.; Ciudad, C.J.; Noe, V. Correction of the aprt Gene Using Repair-Polypurine Reverse Hoogsteen Hairpins in Mammalian Cells. Mol. Ther. Nucleic Acids 2020, 19, 683–695. [Google Scholar] [CrossRef]
- Sole, A.; Ciudad, C.J.; Chasin, L.A.; Noe, V. Correction of point mutations at the endogenous locus of the dihydrofolate reductase gene using repair-PolyPurine Reverse Hoogsteen hairpins in mammalian cells. Biochem. Pharmacol. 2016, 110–111, 16–24. [Google Scholar] [CrossRef]
- Huertas, C.S.; Avino, A.; Kurachi, C.; Piqué, A.; Sandoval, J.; Eritja, R.; Esteller, M.; Lechuga, L.M. Label-free DNA-methylation detection by direct ds-DNA fragment screening using poly-purine hairpins. Biosens. Bioelectron. 2018, 120, 47–54. [Google Scholar] [CrossRef]
- Noe, V.; Aubets, E.; Felix, A.J.; Ciudad, C.J. Nucleic acids therapeutics using PolyPurine Reverse Hoogsteen hairpins. Biochem. Pharmacol. 2020, 189, 114371. [Google Scholar] [CrossRef]
- Aubets, E.; Chillon, M.; Ciudad, C.J.; Noe, V. PolyPurine Reverse Hoogsteen Hairpins Work as RNA Species for Gene Silencing. Int. J. Mol. Sci. 2021, 22, 10025. [Google Scholar] [CrossRef]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef]
- Renaud de la Faverie, A.; Guedin, A.; Bedrat, A.; Yatsunyk, L.A.; Mergny, J.L. Thioflavin T as a fluorescence light-up probe for G4 formation. Nucleic Acids Res. 2014, 42, e65. [Google Scholar] [CrossRef] [Green Version]
- Trinh, K.H.; Kadam, U.S.; Rampogu, S.; Cho, Y.; Yang, K.-A.; Kang, C.H.; Lee, K.-W.; Chung, W.S.; Hong, J.C. Development of novel fluorescence-based and label-free noncanonical G4-quadruplex-like DNA biosensor for facile, specific, and ultrasensitive detection of fipronil. J. Hazard. Mater. 2022, 427, 127939. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.K.; Psaras, A.M.; Lassiter, Q.; Raymer, K.; Brooks, T.A. G-quadruplex deconvolution with physiological mimicry enhances primary screening: Optimizing the FRET Melt(2) assay. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194478. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Villalobos, X.; Dakhel, S.; Padilla, L.; Hervas, R.; Hernández, J.L.; Ciudad, C.J.; Noé, V. Polypurine reverse Hoogsteen hairpins as a gene therapy tool against survivin in human prostate cancer PC3 cells in vitro and in vivo. Biochem. Pharmacol. 2013, 86, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, X.; Rodriguez, L.; Prevot, J.; Oleaga, C.; Ciudad, C.J.; Noe, V. Stability and immunogenicity properties of the gene-silencing polypurine reverse Hoogsteen hairpins. Mol. Pharm. 2014, 11, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Cuesta, S.M.; Adhikari, S.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. G-quadruplexes are transcription factor binding hubs in human chromatin. Genome Biol. 2021, 22, 117. [Google Scholar] [CrossRef]
- Krafcikova, M.; Dzatko, S.; Caron, C.; Granzhan, A.; Fiala, R.; Loja, T.; Teulade-Fichou, M.-P.; Fessl, T.; Hänsel-Hertsch, R.; Mergny, J.-L.; et al. Monitoring DNA-Ligand Interactions in Living Human Cells Using NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141, 13281–13285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel-Hertsch, R.; Spiegel, J.; Marsico, G.; Tannahill, D.; Balasubramanian, S. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat Protoc. 2018, 13, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Hansel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Antonio, M.D.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2020, 52, 878–883. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Drug targeting of transcriptional control via G-quadruplexes. Nat. Rev. Drug Discov. 2010. submitted. [Google Scholar]
- Hansel-Hertsch, R.; Simeone, A.; Shea, A.; Hui, W.W.I.; Zyner, K.G.; Marsico, G.; Rueda, O.M.; Bruna, A.; Martin, A.; Zhang, X.; et al. Landscape of G-quadruplex DNA structural regions in breast cancer. Nat. Genet. 2020, 52, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, T.; Rustgi, A.K. Mutant KRAS in the initiation of pancreatic cancer. Biochim. Biophys. Acta 2005, 1756, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Wang, Y.; Kaiser, C.E.; Frett, B.; Li, H.Y. Targeting Mutant KRAS for Anticancer Therapeutics: A Review of Novel Small Molecule Modulators. J. Med. Chem. 2013, 56, 5219–5230. [Google Scholar] [CrossRef] [Green Version]
- Avino, A.; Eritja, R.; Ciudad, C.J.; Noe, V. Parallel Clamps and Polypurine Hairpins (PPRH) for Gene Silencing and Triplex-Affinity Capture: Design, Synthesis, and Use. Curr. Protoc. Nucleic Acid Chem. 2019, 77, e78. [Google Scholar] [CrossRef]
- Ciudad, C.J.; Rodriguez, L.; Villalobos, X.; Felix, A.J.; Noe, V. Polypurine Reverse Hoogsteen Hairpins as a Gene Silencing Tool for Cancer. Curr. Med. Chem. 2017, 24, 2809–2826. [Google Scholar] [CrossRef]
- Noe, V.; Ciudad, C.J. Polypurine Reverse-Hoogsteen Hairpins as a Tool for Exon Skipping at the Genomic Level in Mammalian Cells. International journal of molecular sciences. Int. J. Mol. Sci. 2021, 22, 3784. [Google Scholar] [CrossRef]
- Rodriguez, L.; Villalobos, X.; Sole, A.; Lliberos, C.; Ciudad, C.J.; Noe, V. Improved design of PPRHs for gene silencing. Mol. Pharm. 2015, 12, 867–877. [Google Scholar] [CrossRef]
- Sole, A.; Delagoutte, E.; Ciudad, C.J.; Noe, V.; Alberti, P. Polypurine reverse-Hoogsteen (PPRH) oligonucleotides can form triplexes with their target sequences even under conditions where they fold into G-quadruplexes. Sci. Rep. 2017, 7, 39898. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′-3′) |
|---|---|
| I1 | CGGGAGAGAGGTACGGAGCGGAC |
| E5 | GTGGGGGTGTGGGGGGA |
| PR | CAGGTGGAAGGGGCA |
| Name | Sequence (5′-3′) |
|---|---|
| Probe-1-2-PY-KRAS | 5′-[6FAM]-CCCCCGCTCCTCCCCCGCCGGCCCGGCCCGGCCCCCTCCTTCTCCCC-3′ |
| Probe 1-PPU-KRAS | 5′-GGGGAGAAGGAGGGGGCCGGGCCGG-3’ |
| Probe 2-PPU-KRAS | 5′-GCCGGCGGGGGAGGAGCGGGGG-3′ |
| Probe-PPY-KRAS-PR-UP | 5′-[6FAM]-CTTTTCTCTTCTGCCCCTTCCACC-3′ |
| Probe-PPU-KRAS-PR-UP | 5′-GGTGGAAGGGGCAGAAGAGAAAAG-3′ |
| Probe-PPY-KRAS-I1 | 5′-[6FAM]-TCCGCTCCGTACCTCTCTCCC-3′ |
| Probe-PPU-KRAS-I1 | 5′-GGGAGAGAGGTACGGAGCGGA-3′ |
| Probe-PPY-KRAS-E5 | 5′-[6FAM]-TCTCTCCCCCCACACCCCCAC-3′ |
| Probe-PPU-KRAS-E5 | 5′-GTGGGGGTGTGGGGGGAGAGA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Psaras, A.M.; Valiuska, S.; Noé, V.; Ciudad, C.J.; Brooks, T.A. Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides. Int. J. Mol. Sci. 2022, 23, 2097. https://doi.org/10.3390/ijms23042097
Psaras AM, Valiuska S, Noé V, Ciudad CJ, Brooks TA. Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides. International Journal of Molecular Sciences. 2022; 23(4):2097. https://doi.org/10.3390/ijms23042097
Chicago/Turabian StylePsaras, Alexandra Maria, Simonas Valiuska, Véronique Noé, Carlos J. Ciudad, and Tracy A. Brooks. 2022. "Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides" International Journal of Molecular Sciences 23, no. 4: 2097. https://doi.org/10.3390/ijms23042097
APA StylePsaras, A. M., Valiuska, S., Noé, V., Ciudad, C. J., & Brooks, T. A. (2022). Targeting KRAS Regulation with PolyPurine Reverse Hoogsteen Oligonucleotides. International Journal of Molecular Sciences, 23(4), 2097. https://doi.org/10.3390/ijms23042097