
Fractal Analysis of DNA Sequences Using Frequency Chaos Game Representation and Small-Angle Scattering


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theoretical Background
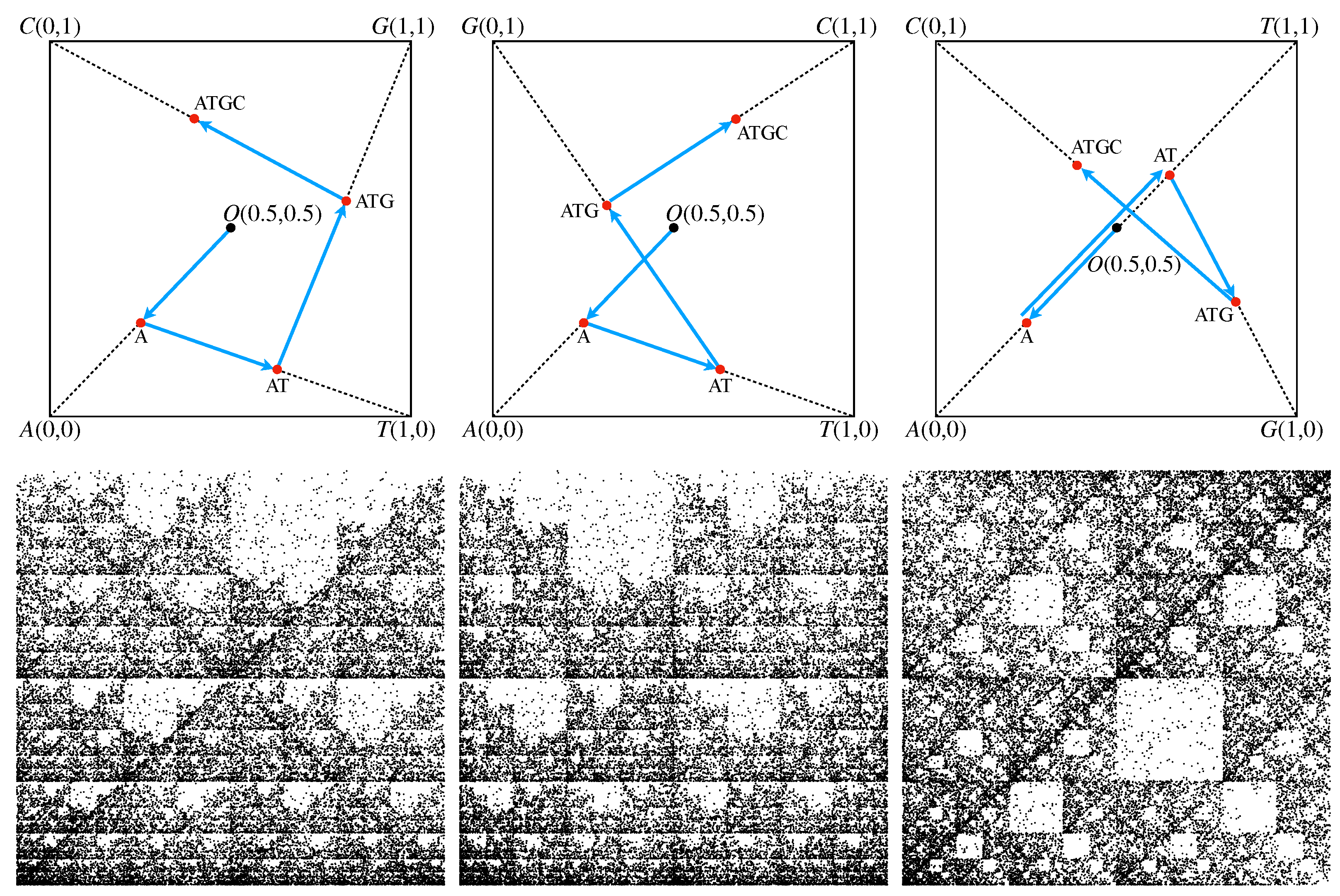
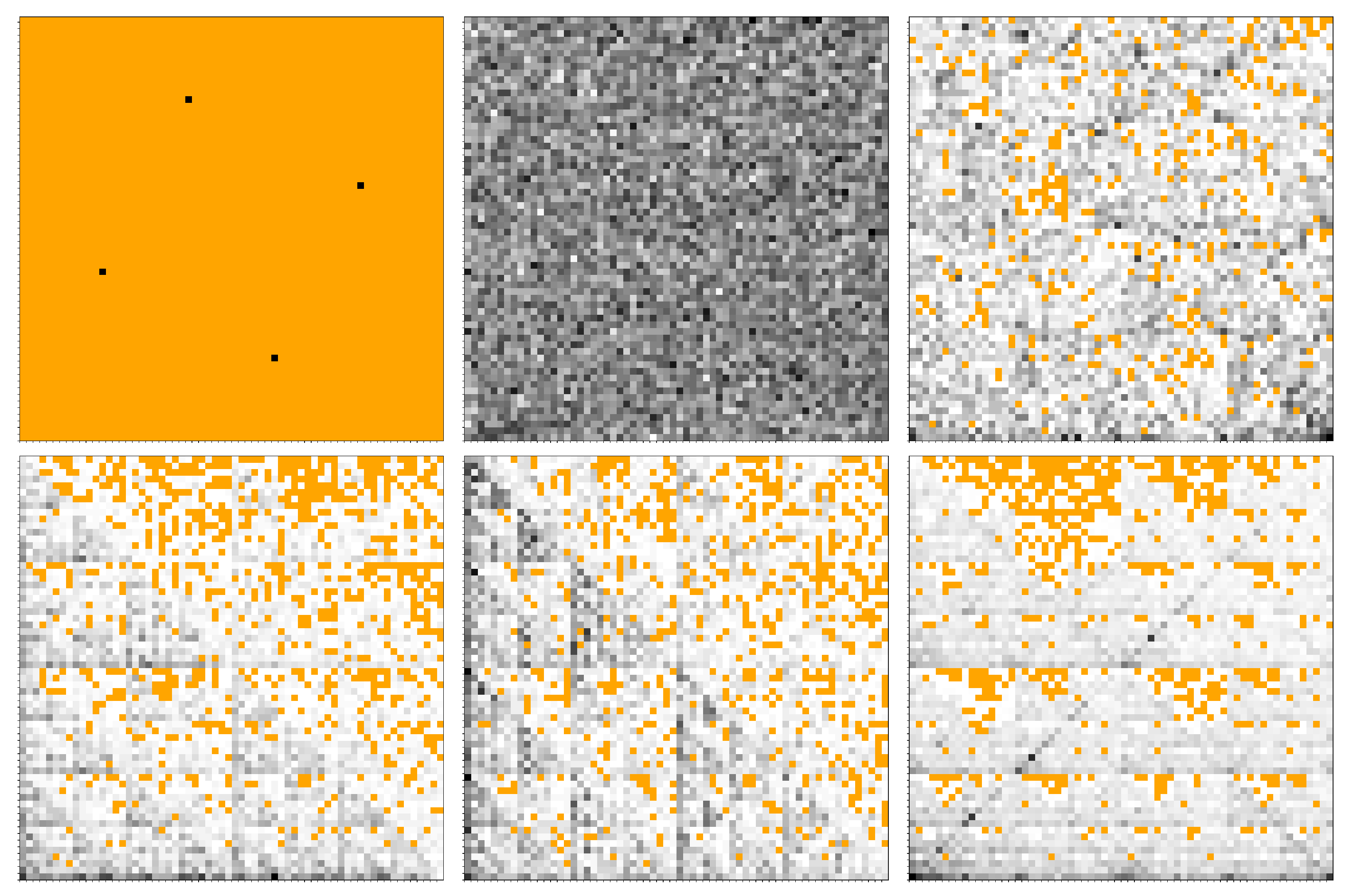
2.1. CGR Representations of Sequences
- 1.
- The four letters “A”, “T”/“U”, “G” and “C” composing the sequence are placed at the vertices of a square centered in the origin.
- 2.
- The first nucleotide in the sequence is placed at the midpoint between the center of the square and the vertex denoted by the same letter as the first nucleotide.
- 3.
- The position of the second nucleotide is obtained by placing it at the midpoint between the position of the first nucleotide and the vertex square denoted by the same letter as the second nucleotide.
- 4.
- The positions of each subsequent nucleotide are obtained as the midpoint between the position of the previous nucleotide and the vertex square corresponding to the current nucleotide.
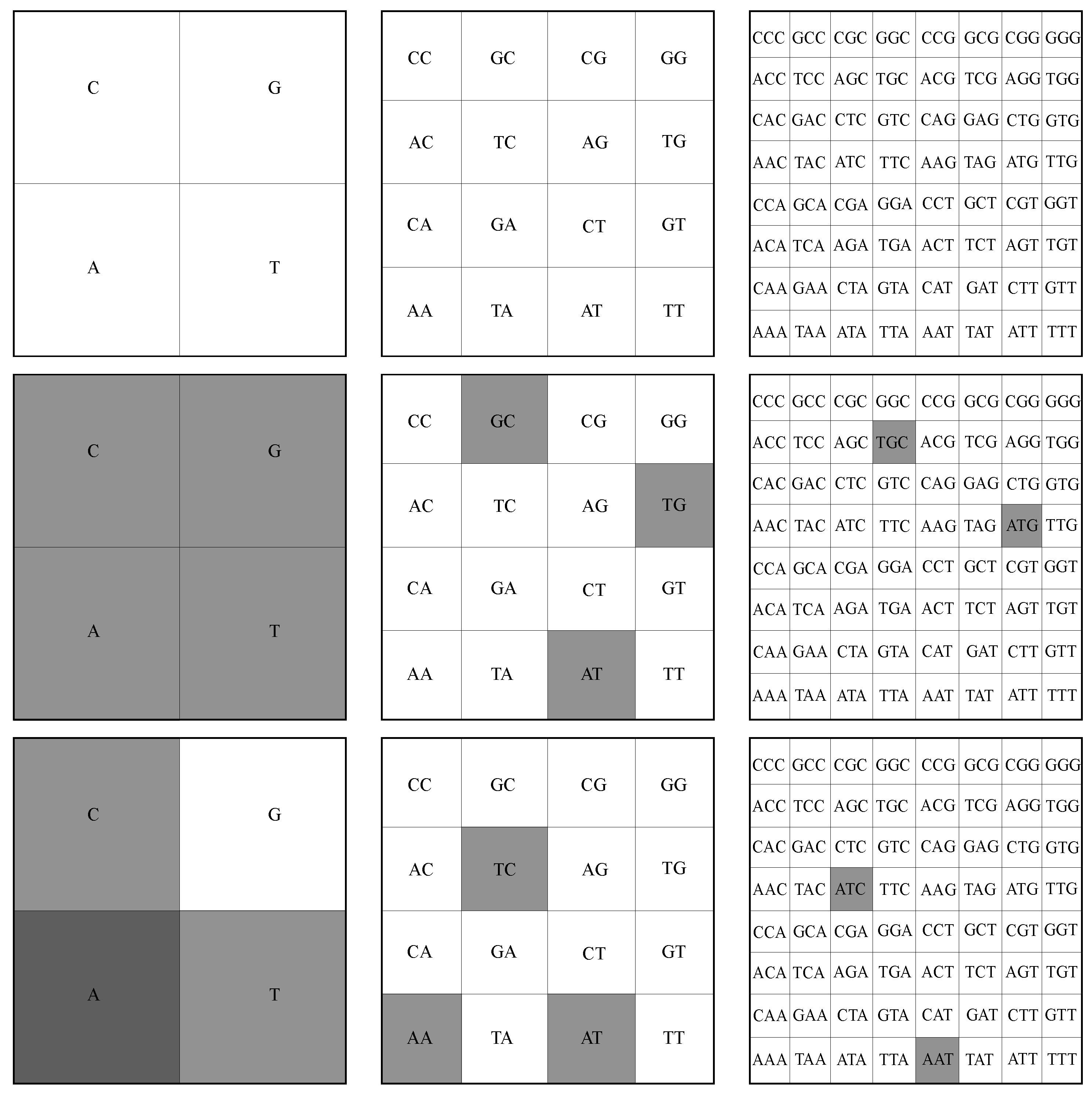
2.2. FCGR Representation of Sequences
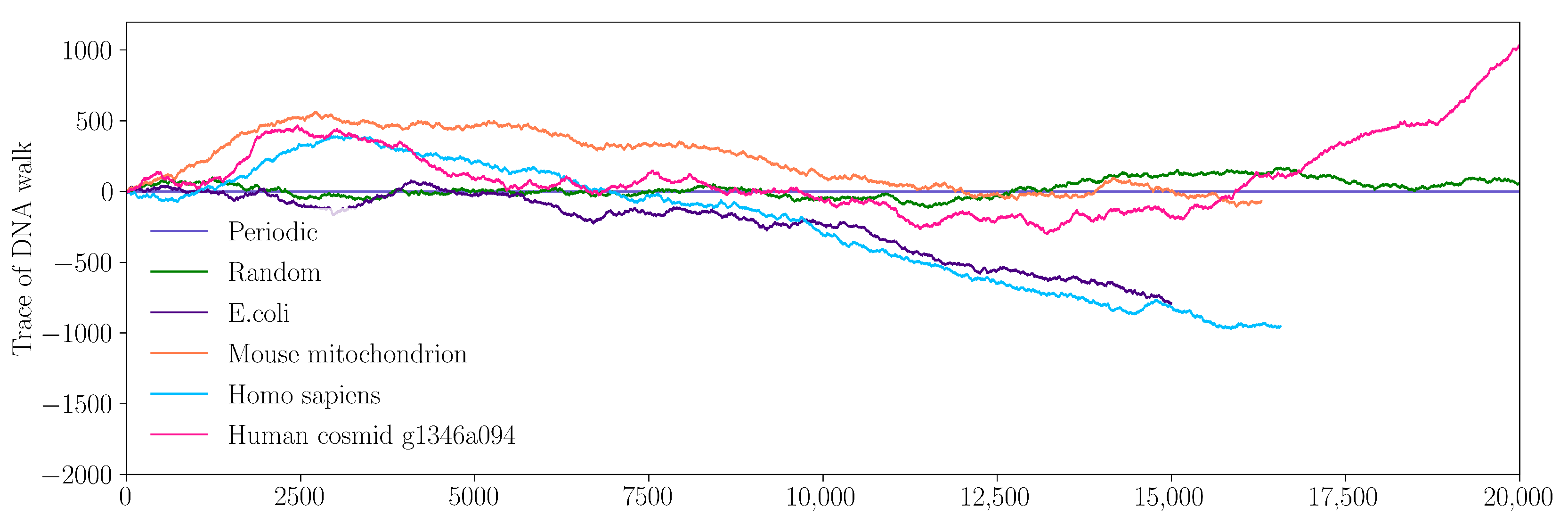
2.3. Time Series and Fractal Theory
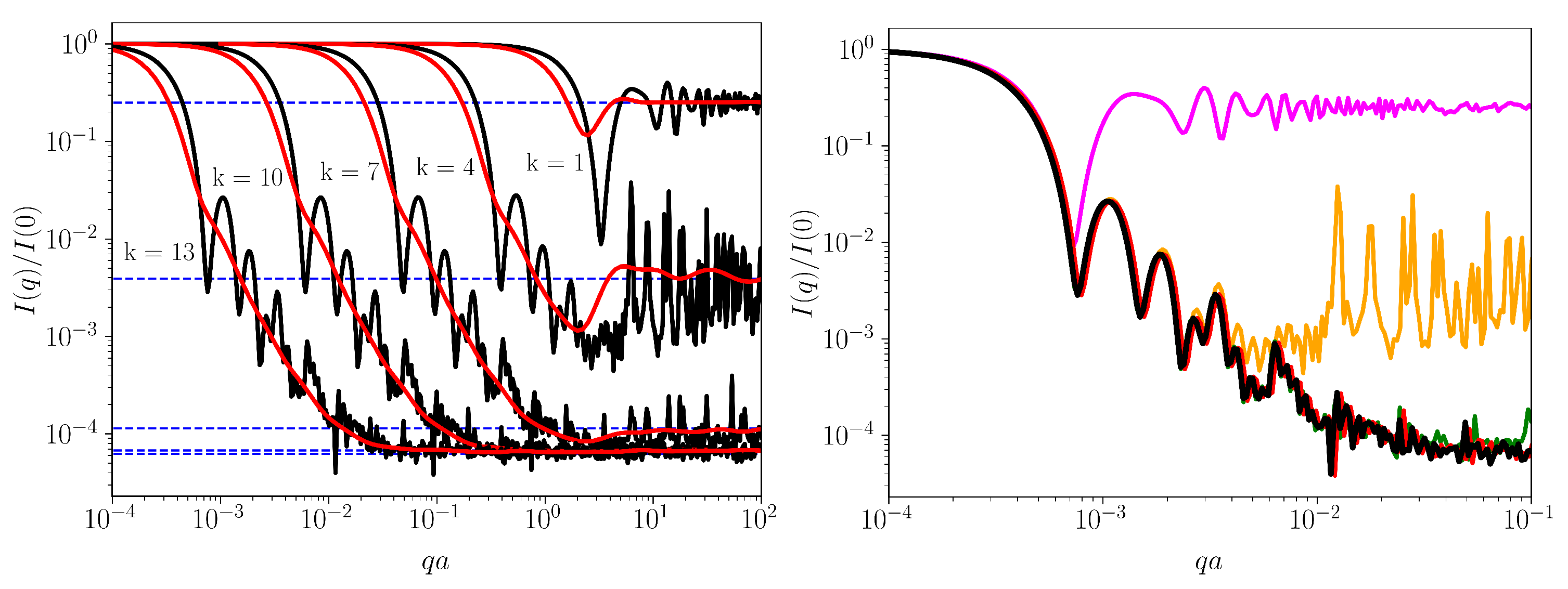
2.4. Small-Angle Scattering Technique
3. Results and Discussion
3.1. Time Series Analysis
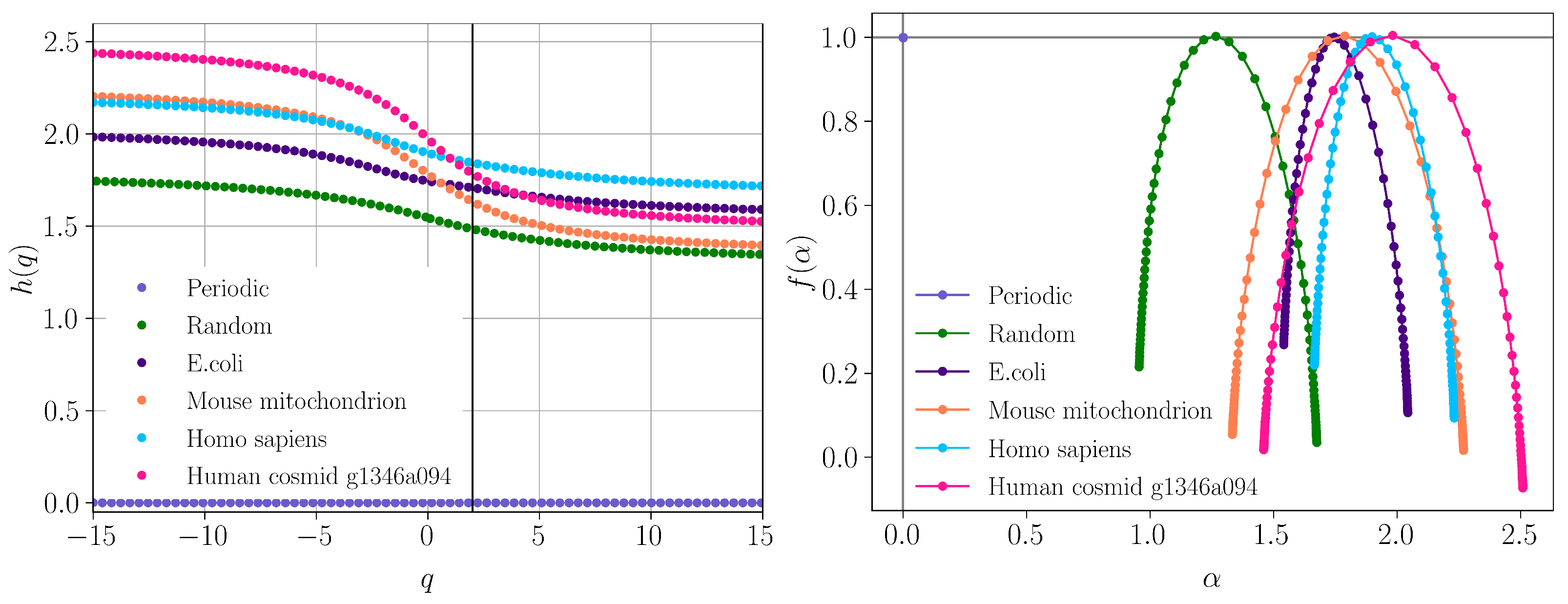
3.2. Multifractal Detrended Fluctuation Analysis
3.3. FCGR Analysis
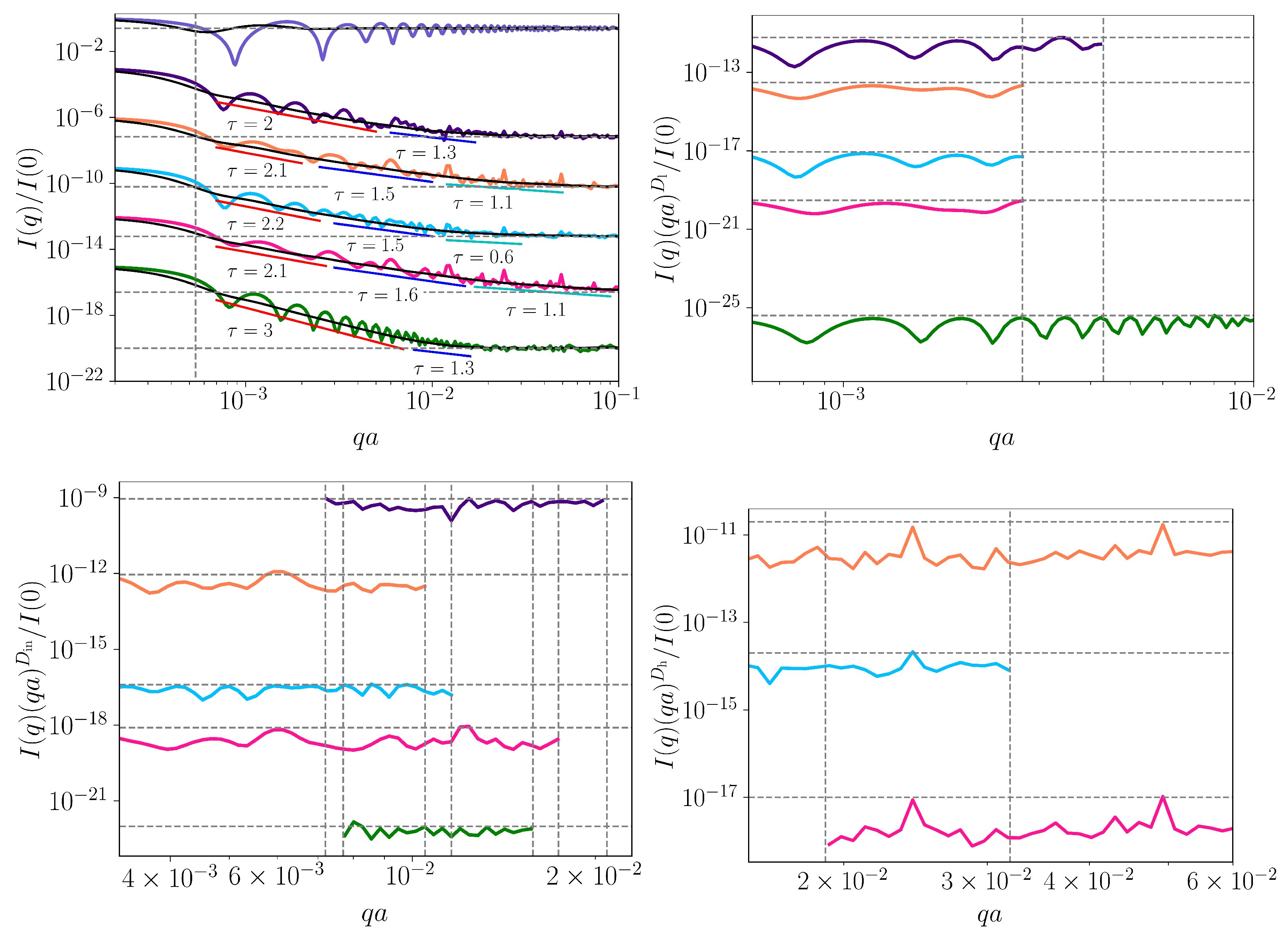
3.4. Small-Angle Scattering Analysis
4. Conclusions
- Differentiate between sequences with power-law correlations and without them. This can be derived from the value of the scattering exponent in the power-law decay of SAS intensity, i.e., for power-law correlated sequences, and for uncorrelated sequences with uniform distribution of nucleotides.
- Differentiate between simple power-law correlations (i.e., mass fractals) and a superposition of power-law correlations over different ranges (i.e., surface fractals), for fractal sequences. This can also be derived from the value of the exponent in the power-law decay of SAS intensity: for mass fractals and for surface fractals. In the former case, the corresponding fractal dimension resulting from the FCGR is , while, in the latter case, .
- Reveal the presence of a succession of power-law correlations at different scales.
- Reveal the scaling factors at each scale (in addition to the scattering exponents ), i.e., how groups of sequences of certain length combine to form repeating patterns, for exact self-similar fractal sequences.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Hsieh, P.; Mao, Y.; Liskovykh, M.A.; Koren, S.; Nurk, S.; Mercuri, L.; Dishuck, P.C.; Rhie, A.; et al. The structure, function and evolution of a complete human chromosome 8. Nature 2021, 593, 101–107. [Google Scholar] [CrossRef]
- Nowoshilow, S.; Schloissnig, S.; Fei, J.F.; Dahl, A.; Pang, A.W.C.; Pippel, M.; Winkler, S.; Hastie, A.R.; Young, G.; Roscito, J.G.; et al. The axolotl genome and the evolution of key tissue formation regulators. Nature 2018, 554, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Schloissnig, S.; Franchini, P.; Du, K.; Woltering, J.M.; Irisarri, I.; Wong, W.Y.; Nowoshilow, S.; Kneitz, S.; Kawaguchi, A.; et al. Giant lungfish genome elucidates the conquest of land by vertebrates. Nature 2021, 590, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lee, W.; Jia, L.; Ji, F.; Tsai, L. Statistical correlation of nucleotides in a DNA sequence. Phys. Rev. E 1998, 58, 861–871. [Google Scholar] [CrossRef]
- Thummadi, N.; Charutha, S.; Pal, M.; Manimaran, P. Multifractal and cross-correlation analysis on mitochondrial genome sequences using chaos game representation. Mitochondrion 2021, 60, 121–128. [Google Scholar] [CrossRef]
- Arneodo, A.; Bacry, E.; Graves, P.V.; Muzy, J.F. Characterizing Long-Range Correlations in DNA Sequences from Wavelet Analysis. Phys. Rev. Lett. 1995, 74, 3293–3296. [Google Scholar] [CrossRef] [Green Version]
- Buldyrev, S.; Dokholyan, N.; Goldberger, A.; Havlin, S.; Peng, C.K.; Stanley, H.; Viswanathan, G. Analysis of DNA sequences using methods of statistical physics. Phys. A Stat. Mech. Appl. 1998, 249, 430–438. [Google Scholar] [CrossRef]
- Peng, C.K.; Buldyrev, S.V.; Goldberger, A.L.; Havlin, S.; Sciortino, F.; Simons, M.; Stanley, H.E. Long-range correlations in nucleotide sequences. Nature 1992, 356, 168–170. [Google Scholar] [CrossRef]
- Audit, B.; Thermes, C.; Vaillant, C.; dÁubenton Carafa, Y.; Muzy, J.F.; Arneodo, A. Long-Range Correlations in Genomic DNA: A Signature of the Nucleosomal Structure. Phys. Rev. Lett. 2001, 86, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.; Silva, J.; Anselmo, D.; Alcaniz, J.; da Silva, W.; Costa, M. An alternative description of power law correlations in DNA sequences. Phys. A Stat. Mech. Appl. 2020, 545, 123735. [Google Scholar] [CrossRef]
- Arneodo, A.; dÁubenton Carafa, Y.; Bacry, E.; Graves, P.; Muzy, J.; Thermes, C. Wavelet based fractal analysis of DNA sequences. Phys. D Nonlinear Phenom. 1996, 96, 291–320. [Google Scholar] [CrossRef]
- Leong, P.; Morgenthaler, S. Random walk and gap plots of DNA sequences. Bioinformatics 1995, 11, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Kantelhardt, J.W.; Zschiegner, S.A.; Koscielny-Bunde, E.; Havlin, S.; Bunde, A.; Stanley, H. Multifractal detrended fluctuation analysis of nonstationary time series. Phys. A Stat. Mech. Appl. 2002, 316, 87–114. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, H. Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, J.S.; Carriço, J.A.; Maretzek, A.; Noble, P.A.; Fletcher, M. Analysis of genomic sequences by Chaos Game Representation. Bioinformatics 2001, 17, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, M.; Satish, B.; Srinivas, K.; Rao, P.M.; Manimaran, P. Multifractal detrended cross-correlation analysis of coding and non-coding DNA sequences through chaos-game representation. Phys. A Stat. Mech. Appl. 2015, 436, 596–603. [Google Scholar] [CrossRef]
- Anitas, E.M. Small-Angle Scattering and Multifractal Analysis of DNA Sequences. Int. J. Mol. Sci. 2020, 21, 4651. [Google Scholar] [CrossRef]
- Tavassoly, I.; Tavassoly, O.; Rad, M.S.R.; Dastjerdi, N.M. Multifractal Analysis of Chaos Game Representation Images of Mitochondrial DNA. In Proceedings of the 2007 Frontiers in the Convergence of Bioscience and Information Technologies, Jeju, Korea, 11–13 October 2007; pp. 224–229. [Google Scholar] [CrossRef]
- Yu, Z.G.; Wang, B. A time series model of CDS sequences in complete genome. Chaos Solitons Fractals 2001, 12, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Buldyrev, S.; Goldberger, A.; Havlin, S.; Simons, M.; Stanley, H. Finite-size effects on long-range correlations: Implications for analyzing DNA sequences. Phys. Rev. E 1993, 47, 3730. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.W. Small-angle scattering studies of disordered, porous and fractal systems. J. Appl. Crystallogr. 1991, 24, 414–435. [Google Scholar] [CrossRef]
- Movahed, M.S.; Jafari, G.R.; Ghasemi, F.; Rahvar, S.; Tabar, M.R.R. Multifractal detrended fluctuation analysis of sunspot time series. J. Stat. Mech. Theory Exp. 2006, 2006, P02003. [Google Scholar] [CrossRef] [Green Version]
- Mandelbrot, B.B. Self-Affine Fractals and Fractal Dimension. Phys. Scr. 1985, 32, 257–260. [Google Scholar] [CrossRef]
- Cherny, A.Y.; Anitas, E.M.; Osipov, V.A.; Kuklin, A.I. Deterministic fractals: Extracting additional information from small-angle scattering data. Phys. Rev. E 2011, 84, 036203. [Google Scholar] [CrossRef] [Green Version]
- Cherny, A.Y.; Anitas, E.M.; Osipov, V.A.; Kuklin, A.I. Scattering from surface fractals in terms of composing mass fractals. J. Appl. Crystallogr. 2017, 50, 919–931. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anitas, E.M. Fractal Analysis of DNA Sequences Using Frequency Chaos Game Representation and Small-Angle Scattering. Int. J. Mol. Sci. 2022, 23, 1847. https://doi.org/10.3390/ijms23031847
Anitas EM. Fractal Analysis of DNA Sequences Using Frequency Chaos Game Representation and Small-Angle Scattering. International Journal of Molecular Sciences. 2022; 23(3):1847. https://doi.org/10.3390/ijms23031847
Chicago/Turabian StyleAnitas, Eugen Mircea. 2022. "Fractal Analysis of DNA Sequences Using Frequency Chaos Game Representation and Small-Angle Scattering" International Journal of Molecular Sciences 23, no. 3: 1847. https://doi.org/10.3390/ijms23031847
APA StyleAnitas, E. M. (2022). Fractal Analysis of DNA Sequences Using Frequency Chaos Game Representation and Small-Angle Scattering. International Journal of Molecular Sciences, 23(3), 1847. https://doi.org/10.3390/ijms23031847