Abstract
Wiedemann–Steiner syndrome (WDSTS) is a Mendelian syndromic intellectual disability (ID) condition associated with hypertrichosis cubiti, short stature, and characteristic facies caused by pathogenic variants in the KMT2A gene. Clinical features can be inconclusive in mild and unusual WDSTS presentations with variable ID (mild to severe), facies (typical or not) and other associated malformations (bone, cerebral, renal, cardiac and ophthalmological anomalies). Interpretation and classification of rare KMT2A variants can be challenging. A genome-wide DNA methylation episignature for KMT2A-related syndrome could allow functional classification of variants and provide insights into the pathophysiology of WDSTS. Therefore, we assessed genome-wide DNA methylation profiles in a cohort of 60 patients with clinical diagnosis for WDSTS or Kabuki and identified a unique highly sensitive and specific DNA methylation episignature as a molecular biomarker of WDSTS. WDSTS episignature enabled classification of variants of uncertain significance in the KMT2A gene as well as confirmation of diagnosis in patients with clinical presentation of WDSTS without known genetic variants. The changes in the methylation profile resulting from KMT2A mutations involve global reduction in methylation in various genes, including homeobox gene promoters. These findings provide novel insights into the molecular etiology of WDSTS and explain the broad phenotypic spectrum of the disease.
1. Introduction
Wiedemann–Steiner syndrome (WDSTS, MIM# 605130) is a rare severe autosomal dominant disorder, characterized by intellectual disability (ID), developmental delay, hypertrichosis cubiti and distinctive facial features [1,2,3,4,5,6,7,8]. The phenotypic spectrum of WDSTS has recently been expanded with clinical features such as ocular abnormalities, recurrent infections of the genitourinary and/or respiratory tract, cardiac or urogenital malformations and skeletal abnormalities, including craniovertebral junction (CVJ) anomalies [6]. The WDSTS phenotype and genotype–phenotype correlation is currently not fully understood. Moreover, the mild/unusual WDSTS presentations may be challenging to be recognized [9].
The introduction of next-generation sequencing (NGS) and the implementation of human phenotype ontology (HPO) have revolutionized diagnostics for rare diseases [10]. Reverse dysmorphology, defined as the delineation of new syndromes primarily by genotype followed by the description of the phenotype, is now preferred [11,12]. While phenotypical evaluation of patients has still remained critical for the process of diagnosis, the clinical diagnosis for chromatin-related disorders is often established only after identification of a causative genetic variant [9].
WDSTS is caused by heterozygous pathogenic variants in the KMT2A (lysine methyltransferase 2A) gene (MIM# 159555), located on chr11q23, previously known as MLL (mixed lineage leukemia). Germline and somatic KMT2A variants are, respectively, associated with WDSTS and multiple neoplastic diseases, along with gene structural rearrangements that are common in acute leukemia [13]. KMT2A encodes a histone H3K4 methyltransferase enzyme that regulates chromatin mediated transcription and is widely expressed in most human tissues. KMT2A is involved in specific complexes mediating the methylation of lysine 4 of histone H3 (H3K4me) and acetylation of lysine 16 of histone H4 (H4K16ac), tags for epigenetic transcriptional activation [14,15,16]. KMT2A is essential for embryonic development, hematopoiesis, and neural development. Known targets include the homeobox (HOX) genes, a family of transcription factors essential for normal embryonic development [17,18].
KMT2A is a 3972 amino acid (aa) multidomain protein (NP_001184033.1), comprising three DNA-binding AT-hooks at the N-terminus, a cysteine-rich CXXC domain, a plant homeodomain (PHD) finger motif, a bromodomain, a transactivation domain (TAD), a FYRN domain, a WDR5 interaction (Win) motif, and a C-terminal SET domain [3]. The SET domain is involved in the histone monomethylation, dimethylation, or trimethylation activity of the protein [19,20,21]. Pathogenic variants in the KMT2A gene lead to defects in chromatin remodeling [15] and are thought to result in global changes in gene expression throughout development leading to abnormalities in multiple body systems. WDSTS or KMT2A-related syndrome is a typical epigenetic machinery disorder, included in chromatin-related disorders, a group of diseases caused by alterations in genes coding for components of the epigenetic apparatus [16]. The proteins associated with chromatin-related disorders act in concert to control the chromatin opening and closing thus regulating gene expression by modification (i.e., methylation, acetylation, etc.) of histones and DNA. Chromatin-related disorders frequently present with overlapping clinical features and inconclusive or ambiguous genetic findings which can confound accurate diagnosis and clinical management [9]. An expanding number of genetic syndromes have been shown to have unique genomic DNA methylation patterns or episignatures. Peripheral blood episignatures can be used for diagnostic testing and for classification of genetic variants. Recently, it has been shown that some diseases influencing DNA methylation have specific methylation signatures, referred to as episignatures [22,23,24,25]. Episignature analysis has recently been implemented as a diagnostic clinical genomic DNA methylation test, in individuals with rare disorders, providing strong evidence of its clinical utility including the ability to provide conclusive diagnostic findings in most subjects tested [26].
A consensus on the clinical classification of genomic variants based on the American College of Medical Genetics and Genomics (ACMG) criteria has recently been attained. WDSTS-associated variants are loss-of-function (LoF) and missense variants. In this study, we identified a unique genome-wide DNA methylation episignature for KMT2A-related syndrome. We compared it with episignatures obtained to a large cohort of patients with various episignature disorders within the EpiSign Knowledge Database (EKD) [22,26], including patients with neurodevelopmental syndromic disorders, especially with regard to Kabuki1, caused by pathogenic variants in KMT2D gene, as some patients with missense and splice site variants in KMT2A have been reported to show phenotype similarities to the ones observed in the Kabuki1 syndrome and because similar to KMT2A, KMT2D also mediates the methylation of lysine 4 of histone H3 [27]. Using in silico studies, aggregated, population and mutations-specific databases, and genome-wide DNA methylation signatures, we were able to definitively classify 56 KMT2A variants.
2. Results
2.1. Demographic and Molecular Characteristics of Patients
The molecular description at diagnosis and demographics of a cohort of 60 patients with clinical diagnosis for WDSTS is shown in Table 1. Fifty-six patients carried KMT2A intragenic variants (missense, nonsense, indel or splice site changes, including variants of uncertain significance (VUS)) and four patients had only a clinical diagnosis of WDSTS or Kabuki syndrome.

Table 1.
Demographic and molecular characteristics of WDSTS cohort.
The molecular description at diagnosis and demographics of a cohort of 74 patients with clinical diagnosis for Kabuki is shown in Table S1. Sixty-six patients carried KMT2D intragenic pathogenic variants (missense, nonsense, indel or splice site variants) and eight patients did not have variant information. All patients had a clinical diagnosis of Kabuki syndrome associated with a Kabuki1 episignature.
2.2. Detection and Verification of an Episignature for WDSTS
Forty-one WDSTS samples with pathogenic KMT2A variants (training set, Pt.1 to Pt.41 in Table 1) and 82 control samples were included for detection of an episignature for WDSTS syndrome. The changes in the methylation status driven by KMT2A pathogenic variants involve an overall (87%) global reduction in methylation (Figure 1).
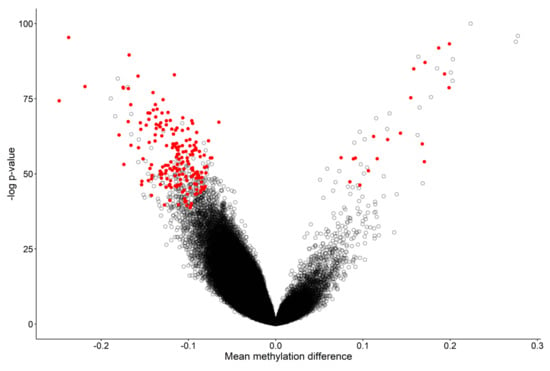
Figure 1.
Volcano plot of methylation difference between 41 WDSTS samples and controls versus statistical significance (-log p-value) of individual probes. Red dots represent selected, significant differentially methylated probes (DMPs). Positive and negative mean methylation difference show hypermethylation and hypomethylation, respectively.
The 207 differentially methylated probes (DMPs) (Figure 1 and Table S2) selected using the three-step process described in the Methods section were used for the purpose of constructing unsupervised and supervised classification models. The methylation levels at these 207 CpG sites were considered as the identifying episignature of the syndrome. In order to assess the robustness of the episignature in differentiating between the case and control samples, hierarchical clustering (Figure 2a) and multidimensional scaling (MDS) analysis (Figure 2b) were performed, resulting in a clear separation between these two groups.
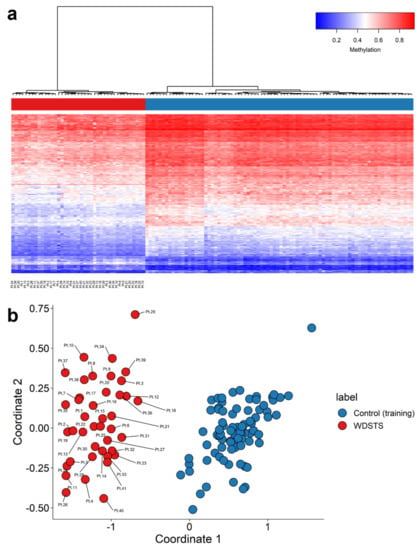
Figure 2.
Identification of a WDSTS episignature. (a) Hierarchical clustering with Ward’s method on Euclidean distance was performed. In the heatmap plot, each row illustrates a selected CpG site, and each column is related to a sample. The heatmap color scale indicates the range of methylation level; from blue (no methylation or 0) to red (full methylation or 1). This plot conveys that the detected episignature clearly differentiates between 41 WDSTS samples and controls; (b) multidimensional scaling (MDS) plot using the selected probes. MDS plot illustrates power of the signature in separating the 41 WDSTS samples and control samples. Blue circles represent control subjects and red circles indicate subjects with pathogenic variants in the KMT2A gene and a confirmed diagnosis of the syndrome.
Forty-one rounds of cross-validation on MDS plot were performed using 40 WDSTS samples as the training set and a single WDSTS sample as the testing set. In all steps, the testing samples were correctly clustered with the training samples, further providing evidence of a robust common DNA methylation episignature (Figure S1).
2.3. Construction of the Binary Prediction Model
Two methylation variant pathogenicity (MVP) plots were generated to confirm specificity of the classification model. In the first MVP plot where the support vector machine classifier (SVM) was trained by comparing the 41 WDSTS samples against controls, the classifier showed a high sensitivity for all WDSTS cases and nine WDSTS (testing) samples (including four samples without known KMT2A variants (Pt.42, Pt.44, Pt.45, Pt.50), one with a canonical -1 splice site variant (Pt.43), one with a nonsense variant (Pt.49), two out of 11 with a missense variant (Pt.46, Pt.47), and one out of two with an in-frame deletion (Pt.48)), with all samples scoring high on the MVP score axis (Figure 3a), further confirming the previous heatmap and MDS results. Some samples from control (testing) as well as other disease cohorts available in EKD (including BAFopathy complex, Börjeson–Forssman–Lehmann syndrome (BFLS), Cornelia de Lange syndrome (CdLS), CHARGE, Down, Kabuki (including Kabuki1 and Kabuki2), Rubinstein–Taybi syndrome (RSTS), Sotos, and Tatton–Brown–Rahman syndrome (TBRS) plus one sample from (Alpha thalassemia/mental retardation X-linked syndrome) ATRX, Blepharophimosis-impaired intellectual development syndrome SMARCA2 Syndrome (BISS), Kleefstra, Mental retardation, X-linked, Snyder–Robinson type (MRXSSR), and SETD1B cohorts) showed an elevated MVP score, suggesting level of similarity in the DNA methylation profiles between these disorders.
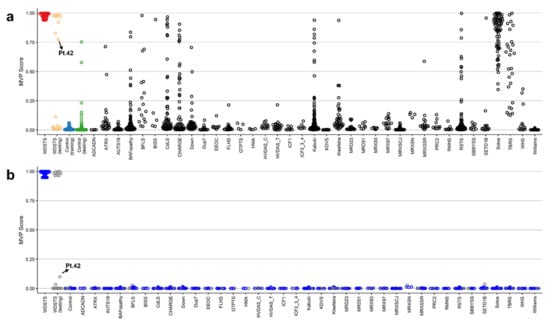
Figure 3.
The methylation variant pathogenicity (MVP) scores plot. (a) The MVP scores were created by the SVM trained by comparing the 41 WDSTS samples against controls; (b) the MVP scores created by the SVM trained by comparing 41 WDSTS samples against controls and 38 neurodevelopmental disorders and congenital anomalies available in the EKD. The blue circles represent the training samples and the grey circles represent the testing samples.
To increase the specificity of the classifier, an SVM was trained by comparing the 41 WDSTS cases against WDSTS (testing) samples, controls, as well as 38 neurodevelopmental disorders (NDDs) and congenital anomalies (CAs) with known episignatures present in the EKD. A high MVP score was seen for 41 WDSTS samples and eight WDSTS (testing) samples (including three out of four with no KMT2A variant information (Pt.44, Pt.45, Pt.50), one with a canonical −1 splice site variant (Pt.43), one with a nonsense variant (Pt.49), two out of 11 with a missense variant (Pt.46, Pt.47), and one out of two with an in-frame deletion (Pt.48)) along with much improved specificity relative to other EpiSign conditions (Figure 3b). Furthermore, one WDSTS (testing) sample with no variant information (Pt.42) had an MVP score of 0.10.
2.4. Validation of WDSTS Signature Using Testing Cohort and Comparison to Kabuki1 Samples
Nineteen WDSTS (testing) were used for validation of WDSTS signature, of which four had WDSTS or Kabuki phenotypes but without known KMT2A variants (Pt.42, Pt.44, Pt.45, Pt.50), one had a canonical -1 splice site variant (Pt.43), one had a nonsense variant (Pt.49), 11 had missense variants (Pt.46, Pt.47, Pt.51, Pt.52, Pt.53, Pt.54, Pt.55, Pt.57, Pt.58, Pt.59, Pt.60), and two had in-frame deletions (Pt.48, Pt.56). Interestingly, all four WDSTS (testing) samples without known KMT2A variants (Pt.42, Pt.44, Pt.45, Pt.50) were grouped with WDSTS samples (Figure 4). However, amongst the remaining 15 samples with known KMT2A variants, 10 (Pt.51 to Pt.60) and five (Pt.43 and Pt.46 to Pt.49) were grouped with control and WDSTS samples, respectively. In addition, using the WDSTS classifier, Kabuki1 samples showed segregation in relation to the control samples. As well, Kabuki1 samples were completely segregated from WDSTS samples, further confirming the results depicted in Figure 3b.
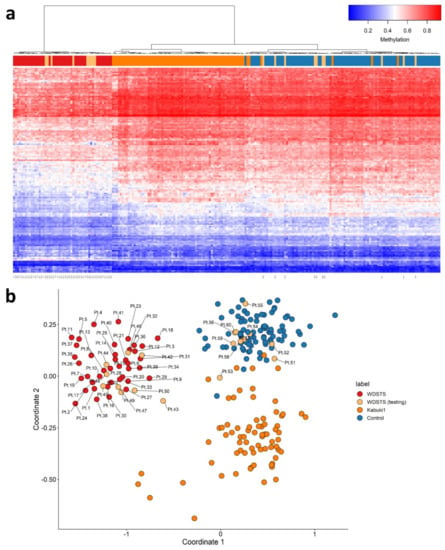
Figure 4.
Adding WDSTS (testing) and Kabuki1 samples to the WDSTS signature. (a) Hierarchical clustering; (b) multidimensional scaling.
2.5. Identification of Differentially Methylated Regions
The identified WDSTS episignature was used to search for differentially methylated regions (DMRs), and resulted in identification of seven DMRs and 207 DMPs (Figure S2 and Table S3). Functional annotation clustering analysis of the DMPs was performed using DAVID [29], and resulted in identification of three significant clusters (Table S4) associated with major terms including (1) homeobox, DNA binding, transcription regulation; (2) regulation of transcription from RNA polymerase II promoter; and (3) T-box transcription factors genes. A relevant proportion of these genes contain hypomethylated regions. This analysis showed enrichment for pathways associated with development and transcription.
2.6. Definitive Classification of KMT2A Variants
The literature described patients already reported with confirmed clinical and molecular diagnoses of WDSTS. Clinical data were collected from literature [1,2,3,4,5,6,7] and accurate HPO terms were selected (Table S5). Using in silico studies, aggregated, population and mutations-specific databases, and genome-wide DNA methylation signatures, we definitively classified 54 KMT2A variants (including 13 missense variants) (Table S6). Variants information was uploaded in MobiDetails [30,31].
3. Discussion
Neurodevelopmental disorders, including the Fragile X and Rett syndromes, disorders of imprinting (such as Angelman and Prader–Willi syndromes), and also the Phelan-McDermid, Sotos, Kleefstra, Coffin Lowry, Kabuki, Charge and ATRX syndromes, are associated with aberrant epigenetic regulation of processes critical for normal brain development [22,26]. The diagnostic utility of genome-wide DNA methylation analysis using peripheral blood has been shown for patients with these NDDs. Over 43 NDDs are currently described with associated distinct DNA methylation episignatures [22,32]. Many of the related genes have a role in the epigenetic machinery such as DNA methylation, histone modification, or chromatin remodeling. Study of the DNA methylation episignatures has been useful to assign a diagnosis to patients with NDDs that remained unresolved by conventional testing or in patients with incorrect initial clinical diagnosis [23,25,26]. EpiSign is the first genome-wide DNA methylation clinical test for patients with NDDs which can be used to assess a clinical diagnostic assessment or to reclassify VUS variants [23].
The intellectual developmental disorder WDSTS was described as a syndromic condition in which ID is associated with hypertrichosis cubiti, short stature, and characteristic facies. Baer et al. described a broad phenotypic spectrum with regard to ID (mild to severe), the facies (typical or not of WDSTS) and associated malformations (bone, cerebral, renal, cardiac and ophthalmological anomalies) [2]. Hypertrichosis cubiti was supposed to be pathognomonic but was found only in 61% of the cases [2]. A majority of patients exhibited suggestive features, but others were less characteristic, only identified by molecular diagnosis [2,9]. The authors suggested that the prevalence of WDSTS is higher than expected in patients with ID, suggesting that KMT2A is a major gene in ID [2].
Following the identification of the causative KMT2A gene in 2012 [33], all types of variants, including missense variants in the KMT2A gene, were reported as the causal variants of the disorder. In this study, DNA methylation data were collected from peripheral blood of a patient cohort including 60 WDSTS patients, of which 55 had known KMT2A variants. The classification model for WDSTS syndrome was built with a training set (41 WDSTS patients) and a control set (82 matched control samples from EKD) using 207 DMPs (Figure 2 and Table S2). In addition, seven DMRs were also identified (Figure S2 and Table S3). The classification model was tested with a testing set (19 patients with uncertain clinical diagnosis) and allowed to reclassify 13 KMT2A missense variants as probably benign (ACMG class 4) or pathogenic (ACMG class 1) (Figure 3 and Figure S3, Table S6). Nine patients from the testing set were finally classified as WDSTS (including four samples without KMT2A variant information (Pt.42, Pt.44, Pt.45, Pt.50), one with a canonical -1 splice site variant (Pt.43), one with a nonsense variant (Pt.49), two out of 11 with a missense variant (Pt.46, Pt.47), and one out of two with an in-frame deletion (Pt.48)). Note that Pt.42 received a low MVP score while it was clustered with KMT2A training samples in both MDS plots and hierarchical clustering heatmap, even though with slightly different level of methylation for some probes in comparison with other KMT2A training samples. There should be something interesting about the Pt.42 but unfortunately no variant information or clinical details were available to make a solid conclusion about this difference. Then, the classification model was tested with a Kabuki1 set (74 patients) (Table S1). Kabuki1 samples were completely segregated from WDSTS samples (Figure 4).
KMT2A is a histone methyltransferase protein deemed as a positive global regulator of gene transcription. This protein belongs to the group of histone-modifying enzymes comprising transactivation domain 9aa TAD [34] and is involved in the epigenetic maintenance of transcriptional memory. Its role as an epigenetic regulator of neuronal function is an ongoing area of research. KMT2A gene encodes a transcriptional coactivator that plays an essential role in regulating gene expression during early development and hematopoiesis. The encoded protein contains multiple conserved functional domains. One of these domains, the SET domain, is responsible for its histone H3 lysine 4 (H3K4) methyltransferase activity which mediates chromatin modifications associated with epigenetic transcriptional activation. Enriched in the nucleus, the KMT2A enzyme mono, di and trimethylates H3K4 [35]. This protein is processed by the enzyme threonine aspartase 1 into two fragments [34,36] and regulates the transcription of specific target genes, including particular HOX genes during development [37,38].
The methylome analysis did highlight a substantial change in the global methylation pattern in WDSTS samples, and resulted in 87% hypomethylated and 13% hypermethylated probes. More detailed information about the 207 CpG probes selected in WDSTS episignature are summarized in Table S2. Full sensitivity and specificity of our model were illustrated in Figure 3b, where all case samples received a high MVP score and all control samples and individuals from the other 38 constitutional disorders and congenital anomalies received a score near zero. Methylation changes involved specific CpGs in regulatory regions, indicating a punctual effect on a relatively small subset of genes and cellular processes. Indeed, only 13% of the DMPs were represented by a hypermethylation change, indicating that the changes in the methylation status driven by KMT2A pathogenic variants concern a global tendency in a reduction in methylation.
Functional annotation clustering analysis of the 207 DMPs using DAVID identified three significant clusters (Table S4) associated with major terms including (1) homeobox, DNA binding, transcription regulation (i.e., HOX genes, PRDM genes, ALX genes, SIX2, WT1); (2) regulation of transcription from RNA polymerase II promoter (i.e., HOX genes, ALX genes, SIX2, WT1); and (3) T-box transcription factors genes (TBX1, TBX2, TBX4). A relevant proportion of these genes contain hypomethylated regions predominantly expressed in brain, but also in bones, kidney, heart, and eye. Among 207 DMPs, a distinctive hypomethylation pattern affecting genes from three of the four HOX clusters (HOXA2, HOXA3, HOXA4, HOXA10, HOXB9, HOXC4, HOXC5, HOXC6), the MIR196A1 gene, two genes from the PRDM protein family (PRDM14, PRDM16), Homeobox protein aristaless-like genes (ALX3 and ALX4), SIX2, T-box transcription factors genes (TBX1, TBX2, TBX4) and WT1 gene were seen. Furthermore, a distinctive hypermethylation pattern affecting genes related to the HOX clusters (HOXA6, HOXA7, HOXA9, HOXA10 and PRDM16) were seen.
In humans, the 39 HOX genes are arranged in four clusters (HOXA, HOXB, HOXC, and HOXD) in chromosomes 7p15, 17q21.2, 12q13, and 2q31, respectively. This highly conserved family belongs to the homeobox class of genes that encode transcription factors required for normal embryonic global development, including brain development [18] and embryology of the bony CVJ [17]. HOX genes function in multiple neuronal classes to shape synaptic specificity during development, suggesting a broader role in circuit assembly. They play key roles in defining the identity, organization, and peripheral connectivity of motor neuron subtypes, and their target effectors are beginning to be defined, the contribution of HOX genes to synaptic specificity in neural circuits within the central nervous system (CNS) remains to be resolved [39]. HOX genes are essentially absent in healthy adult brain, whereas they are detected in malignant brain tumors, namely gliomas [18]. In embryonic stem cells, which do not express HOX genes, whole HOX clusters are fully decorated by H3K27me3, while at their promoter area, this mark co-exists with H3K4me3, constituting the so-called bivalent chromatin. Deposition of H3K4me3 at HOX clusters in mammals relies on the COMPASS-like complexes that contains mammalian Set1 homologs (KMT2A to KMT2G proteins) and the homologues of Drosophila Trx [18,35]. Interestingly, this complex also contains the H3K27me3-demethylase KDM6A that removes H3K27me3 at HOX loci [18]. The dynamics of H3K27me3/H3K4me3 distribution along the different HOX clusters impacts their 3D architecture. The PRDM14, PRDM16 and MIR196A1 genes have been also linked functionally to HOX function. A gene-set enrichment analysis is discussed in additional data file. However, the methylation pattern was analyzed in leukocytes, which might considerably differ in neurons and in other cell lines.
Here, we provide a specific DNA methylation pattern in affected WDSTS patients. Patients with WDSTS which have KMT2A pathogenic variants have a distinct epigenetic signature in peripheral blood from a variety of other NDDs, including syndromes that may clinically overlap with WDSTS, like Kabuki type 1 patients with KMT2D pathogenic variants or Kabuki1 episignature (Figure 4). We demonstrate that WDSTS is characterized by an episignature, which is defined by a particular hypomethylation profile with respect to healthy subjects. WDSTS episignature is robust, enabling a discovery and validation of the highly sensitive and specific signal. Hypermethylation of homeobox gene promoters (including HOX genes), is emerging as a pan-cancer signature with patient-specific DNA methylation patterns [40]. Aberrant DNA methylation is a well-documented signature at HOX loci in glioma [18]. Our result suggests here global hypomethylation of various genes including homeobox gene promoters in WDSTS. KMT2A pathogenic variants may disturb the normal process of H3K4me3 deposition and H3K27me3 removal that are coupled at homeobox gene promoters.
Missense variants may present challenges for assessment of clinical impact on the protein function. In such cases, this WDSTS epigenetic classifier may help solve many clinically ambiguous cases presenting with a neurodevelopmental phenotype. Implementation of a routine genome-wide DNA methylation testing is suggested to be considered in the clinical management of patients with NDDs. The use of DNA obtained from peripheral blood samples makes this assay easily supported by diagnostic laboratories. DNA methylation profiling has the capability to detect episignatures from a variety of clinically related NDDs on the same array. It could be applied as an informative and cost-effective first-tier genetic diagnostic test for patients without prior molecular tests.
While methylation changes in DMRs suggest the possibility of gene expression modifications, further functional genomics analysis would be necessary to better understand the pathophysiology of these epigenetic changes. Investigation of genes affected by the abnormal DNA methylation may lead to the identification of novel targets for more personalized treatment approaches. Our studies reported that the expression of several homeobox containing genes (including HOX or HOX-related genes) is consistently altered in blood of WDSTS patients. Considering the critical functional roles and putative prognostic value of specific HOX genes in cancer, including in malignant glioma, and their complex molecular interactions with upstream regulators and downstream targets, it becomes clear that additional studies are necessary to better understand how HOX genes operate in glioma but also possibly in WDSTS, and whether they may be therapeutically explored in the clinics.
4. Materials and Methods
4.1. Study Cohort
This study included 60 individuals, of which 41 (labeled as WDSTS in Table 1) were used for the purpose of probe selection and construction of the classification model. All the samples and records were de-identified. Informed consent for use of the clinical information was obtained from the patients. This study was approved by the Western University Research Ethics Board (REB 106302) and Ospedale Pediatrico Bambino Gesù Ethical Committee (1702_OPBG_2018). This and all other study procedures complied with the Declaration of Helsinki and French legislation and regulations.
4.2. Methylation Experiment and Selection of Matched Control Subjects
DNA samples extracted from peripheral blood were supplied to Illumina Infinium methylation EPIC (EPIC) bead chip arrays as well as Illumina Infinium HumanMethylation450 (450 K) BeadChip arrays followed by bisulfite conversion, and the methylation analysis was performed at the Western University and Ospedale Pediatrico Bambino Gesù (samples pt. 34–39) in accordance with the manufacturer’s protocol. The 450 K and EPIC arrays cover >450,000 and >850,000 human genomic CpG sites, respectively, which include 99% of RefSeq genes and 96% of CpG islands. The obtained methylated and unmethylated signal intensities were imported into R 4.0.3 for analysis. Normalization was performed according to the Illumina normalization method with background correction using the minfi package [41]. Probes that were located on the X and Y chromosomes, had a detection p-value >0.01, known to contain a SNP at or near CpG interrogation sites, or known to cross-react with other genomic regions were removed, in order to ensure that the difference observed between the two groups is solely based on methylation changes rather than other potentially confounding factors. Where indicated, sex and age of the DNA specimens were predicted using the minfi package (based on the median signal intensities of the probes on the X and Y chromosomes) and the wateRmelon package [42], respectively. Principal component analysis (PCA) was performed in order to observe the overall structure of the batches, as well as to identify outliers. Forty-one WDSTS samples having pathogenic variants in the KMT2A gene with confirmed diagnosis of the syndrome (labelled as WDSTS) were used as the case training set. Eighty-two (case to control ratio of 1:2) age, sex, and array type-matched control samples were selected from the EKD [22] using the MatchIt package [43,44] as the control training set. We performed a PCA subsequent to every matching round to detect outliers and examine the data structure, and removed outlier samples as well as samples with irregular data structure at each trial. This process was iterated until no outlier was observed in the first two components of the PCA.
4.3. DNA Methylation Profiling of WDSTS Syndrome
The procedure of differentially methylated probe selection was performed in accordance with previously published articles [22,45]. Methylation levels, called β values, for each probe were calculated as the ratio of methylated signal intensity to the sum of methylated and unmethylated signal intensities. These values were then converted to M values using logit transformation by the formula log2 (β/(1-β)) in order to obtain homoscedasticity for use in linear regression. Using the limma package [46], we performed linear regression and moderated the obtained p-values with the eBayes function. The DMPs from the comparison between case and control groups were selected in the following three-step process. First, 500 probes with the highest product of methylation difference means between the two groups and the negative of the logarithm of multiple-testing corrected p-values derived from the linear modeling by Benjamini–Hochberg (BH) method were selected. Subsequently, a receiver’s operating characteristic (ROC) curve analysis was performed and 250 probes with the highest area under the ROC curve (AUROC) were retained. Finally, those probes with a pair-wise correlation >0.85, within the case and control samples separately, which was measured using Pearson’s correlation coefficients were removed. This resulted in identification of 207 probes, which were considered as the DNA methylation signature for the WDSTS. In order to examine the robustness of this episignature in differentiating between case and control samples, unsupervised models were applied on the 207 DMPs. They include hierarchical clustering which was performed using Ward’s method on Euclidean distance as well as MDS analysis which was performed by scaling of the pair-wise Euclidean distances between samples. Then, 41 rounds of cross-validation were performed on MDS plot from the 41 WDSTS samples, of which 40 samples were used as the training set and a single sample was used as the testing set at each round.
4.4. Construction of a Classification Model for WDSTS Syndrome
Using the selected DMPs, a binary SVM with linear kernel was constructed by the e1071 package as described previously [22,23,47]. In order to obtain the best hyperparameter (cost) and to assess the accuracy of the classifier, a 10-fold cross-validation was performed during training. In 10-fold cross-validation, at each round, 90% of the samples were used for training and the remaining samples for testing. The model provides an MVP score, ranging from 0 to 1, for each sample. Scores near 1 indicate a high similarity between the methylation profile of that sample and that of the identified episignature, while scores near 0 indicate a low similarity. In order to evaluate the specificity of the classifier, more than 1700 samples with other neurodevelopmental syndromes from the EKD [22] were added to the model.
4.5. Classification of Kabuki1 Samples with Pathogenic KMT2D Variants as well as WDSTS (Testing) Samples
To assess the similarity of the methylation profiles of 74 Kabuki1 samples (Table S1) [22] and perform episignature analysis and variant classification in 19 WDSTS (testing) samples, both hierarchical clustering and MDS analysis were reconstructed using the initial WDSTS and 82 control samples as the training set, plus the 19 WDSTS (testing) and 74 Kabuki1 samples as the testing set.
4.6. Identification of the Differentially Methylated Regions of WDSTS Syndrome
In order to identify the regions that are differentially methylated between the subjects with WDSTS and controls, we used the DMRcate package [48] and selected regions containing a minimum of 3 CpG sites within 1 kb with at least 10% methylation difference between the case and control groups and a Fisher’s multiple comparison p-value < 0.01.
5. Conclusions
In conclusion, the identified WDSTS DNA methylation episignature is added to the list of Mendelian NDDs with known DNA methylation episignatures that can be used for screening and diagnosis of NDD patients. KMT2A pathogenic variants may disturb the normal process of H3K4me3 deposition coupled at gene promoters. The methylome analysis did highlight a substantial change in the global methylation pattern in WDSTS samples, and resulted in 87% hypomethylated and 13% hypermethylated probes. Our studies reported that the expression of several homeobox containing genes (including HOX or HOX-related genes) is consistently altered in the blood of WDSTS patients. If also present in other tissues, dysregulation of normal methylation of homeobox gene expression may explain part of the ID, facies and associated malformations observed in WDSTS patients. These provide novel insights into the molecular etiology of WDSTS and likely explain the broad phenotypic spectrum of the disease.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23031815/s1.
Author Contributions
Conceptualization, A.F., B.S. and A.-S.L.; supervision, B.S. and A.-S.L.; bioinformatics and functional analyses, A.F., S.H. (Sadegheh Haghshenas), M.A.L. and P.B.; DNA methylation microarray experiments, J.K., L.P., E.M. and A.C.; study coordination, sample collection and management, J.K. and H.M.; patient recruitment, collection and interpretation of clinical data, D.G., S.H. (Solveig Heide), M.A., M.N., G.M., G.Z., M.F., E.B., D.B., K.D., P.F., E.S., L.F., A.V., S.M., R.F., C.G., M.P., I.v.d.L. and M.T.; writing—original draft preparation, A.F., S.H. (Sadegheh Haghshenas), B.S. and A.-S.L.; writing—review and editing, A.F., S.H. (Sadegheh Haghshenas), M.T., P.B., A.C., B.S. and A.-S.L. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for this study was provided, in part, by the London Health Sciences Molecular Diagnostics Development Fund and Genome Canada Genomic Applications Partnership Program Grant (Beyond Genomics: Assessing the Improvement in Diagnosis of Rare Diseases using Clinical Epigenomics in Canada, EpiSign-CAN) awarded to B.S, and Italian Ministry of Health (CCR-2017-23669081, RCR-2020-23670068_001 and Ricerca Corrente 2021) and Italian Ministry of Research (FOE 2019) granted to M.T.
Institutional Review Board Statement
This study was approved by the Western University Research Ethics Board (REB 106302). DNA specimens from the subjects included in this study were collected following procedures in accordance with the ethical standards of the declaration of Helsinki protocols and approved by the Review Boards of all involved institutions, with signed informed consents from the participating subjects/families.
Informed Consent Statement
Not applicable.
Data Availability Statement
Availability of data and materials Clinical data of the study cohort, probes defining the methylation episignature associated with KMT2A variants and list of regions differentially methylated in WDSTS are reported in additional files. Additional data are available from the corresponding authors upon request.
Acknowledgments
We thank the patients and their family for their participation in this study.
Conflicts of Interest
The authors declare that they have no competing interests.
Abbreviations
WDSTS: Wiedemann–Steiner syndrome; ID: intellectual disability; SVM: support vector machine classifier; VUS: variant(s) of uncertain significance; HPO: Human Phenotype Ontology; CVJ: craniovertebral junction; NGS: next-generation sequencing; KMT2A: Lysine Methyltransferase 2A; MLL: Mixed Lineage Leukemia; H3K4me: methylation of lysine 4 of histone H3; H4K16ac: acetylation of lysine 16 of histone H4; aa: aminoacids; PHD: plant homeodomain; TAD: transactivation domain; ACMG: American College of Medical Genetics and Genomics; LoF: loss-of-function; EKD: EpiSign Knowledge Database; DMP: differentially methylated probes; MDS: multidimensional scaling; MVP: methylation variant pathogenicity; BAF: BRG1/BRM-associated factor; BFLS: Börjeson–Forssman–Lehmann syndrome; CdLS: Cornelia de Lange syndrome; RSTS: Rubinstein–Taybi syndrome; TBRS: Tatton–Brown–Rahman syndrome; ATRX: Alpha thalassemia/mental retardation X-linked syndrome; BISS: Blepharophimosis-impaired intellectual development syndrome SMARCA2 Syndrome; MRXSSR: Mental retardation, X-linked, Snyder–Robinson type; DMR: differentially methylated region; NDD: Neurodevelopmental disorders; CpG: Cytosine-phosphate-Guanine; CNS: central nervous system; PRDI-BF1: Positive Regulatory Domain I-Binding Factor1; RIZ1: Retinoblastoma protein-Interacting Zinc finger gene 1; HMT: histone methyl-transferases; PCA: principal component analysis; BH: Benjamini–Hochberg; ROC: receiver’s operating characteristic; AUROC: area under the ROC curve.
References
- Aggarwal, A.; Rodriguez-Buritica, D.F.; Northrup, H. Wiedemann-Steiner syndrome: Novel pathogenic variant and review of literature. Eur. J. Med Genet. 2017, 60, 285–288. [Google Scholar] [CrossRef]
- Baer, S.; Afenjar, A.; Smol, T.; Piton, A.; Gérard, B.; Alembik, Y.; Bienvenu, T.; Boursier, G.; Boute, O.; Colson, C.; et al. Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: A study of 33 French cases. Clin. Genet. 2018, 94, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, N.; Giurgea, I.; Goldenberg, A.; Dieux, A.; Afenjar, A.; Ghoumid, J.; Diebold, B.; Mietton, L.; Briand-Suleau, A.; Billuart, P.; et al. Molecular and cellular issues of KMT2A variants involved in Wiedemann-Steiner syndrome. Eur. J. Hum. Genet. 2017, 26, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Yang, Y.; Wang, P.; Huang, H.; Xiong, S.; Sun, L.; Cheng, M.; Song, C.; Cheng, X.; et al. Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients. Orphanet J. Rare Dis. 2018, 13, 178. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.J.S.; Cytrynbaum, C.; Hoang, N.; Ambrozewicz, P.M.; Weksberg, R.; Drmic, I.; Ritzema, A.; Schachar, R.; Walker, S.; Uddin, M.; et al. Expanding the neurodevelopmental phenotypes of individuals with de novo KMT2A variants. npj Genom. Med. 2019, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Giangiobbe, S.; Caraffi, S.G.; Ivanovski, I.; Maini, I.; Pollazzon, M.; Rosato, S.; Trimarchi, G.; Lauriello, A.; Marinelli, M.; Nicoli, D.; et al. Expanding the phenotype of Wiedemann-Steiner syndrome: Craniovertebral junction anomalies. Am. J. Med. Genet. Part A 2020, 182, 2877–2886. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Passaretti, F.F.; Maioli, M.; Cantalupo, G.; Scarano, F.; Lonardo, F. Clinical and molecular spectrum of Wiedemann-Steiner syndrome, an emerging member of the chromatinopathy family. World J. Med. Genet. 2020, 9, 1–11. [Google Scholar] [CrossRef]
- Sheppard, S.E.; Campbell, I.M.; Harr, M.H.; Gold, N.; Li, D.; Bjornsson, H.T.; Cohen, J.S.; Fahrner, J.A.; Fatemi, A.; Harris, J.R.; et al. Expanding the genotypic and phenotypic spectrum in a diverse cohort of 104 individuals with Wiedemann-Steiner syndrome. Am. J. Med Genet. Part A 2021, 185, 1649–1665. [Google Scholar] [CrossRef]
- Squeo, G.M.; Augello, B.; Massa, V.; Milani, D.; Colombo, E.A.; Mazza, T.; Castellana, S.; Piccione, M.; Maitz, S.; Petracca, A.; et al. Customised next-generation sequencing multigene panel to screen a large cohort of individuals with chromatin-related disorder. J. Med. Genet. 2020, 57, 760–768. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2020, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Douzgou, S.; Clayton-Smith, J.; Gardner, S.; Day, R.; Griffiths, P.; Strong, K.; the DYSCERNE Expert Panel. Dysmorphology at a distance: Results of a web-based diagnostic service. Eur. J. Hum. Genet. 2014, 22, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- El Chaer, F.; Keng, M.; Ballen, K.K. MLL-Rearranged Acute Lymphoblastic Leukemia. Curr. Hematol. Malign. Rep. 2020, 15, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical Association and Coordinate Function of the H3 K4 Methyltransferase MLL1 and the H4 K16 Acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Vallianatos, C.N.; Raines, B.; Porter, R.S.; Bonefas, K.M.; Wu, M.C.; Garay, P.M.; Collette, K.M.; Seo, Y.A.; Dou, Y.; Keegan, C.E.; et al. Mutually suppressive roles of KMT2A and KDM5C in behaviour, neuronal structure, and histone H3K4 methylation. Commun. Biol. 2020, 3, 278. [Google Scholar] [CrossRef]
- Pang, D.; Thompson, D.N.P. Embryology and bony malformations of the craniovertebral junction. Child’s Nerv. Syst. 2011, 27, 523–564. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, C.S.; Le Boiteux, E.; Arnaud, P.; Costa, B.M. HOX gene cluster (de)regulation in brain: From neurodevelopment to malignant glial tumours. Cell Mol. Life Sci. 2020, 77, 3797–3821. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, M.S.; Patel, A. Mixed lineage leukemia: A structure-function perspective of the MLL1 protein. FEBS J. 2010, 277, 1832–1842. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Bergamin, E.; Couture, J.-F. The many facets of MLL1 regulation. Biopolymers 2013, 99, 136–145. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.-C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Bend, E.G.; Colaiacovo, S.; Caudle, M.; Chakrabarti, R.; Napier, M.; Brick, L.; Brady, L.; Carere, D.A.; Levy, M.A.; et al. Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions. Am. J. Hum. Genet. 2019, 104, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciolfi, A.; Aref-Eshghi, E.; Pizzi, S.; Pedace, L.; Miele, E.; Kerkhof, J.; Flex, E.; Martinelli, S.; Radio, F.C.; Ruivenkamp, C.A.L.; et al. Frameshift mutations at the C-terminus of HIST1H1E result in a specific DNA hypomethylation signature. Clin. Epigenet. 2020, 12, 7. [Google Scholar] [CrossRef]
- Ciolfi, A.; Foroutan, A.; Capuano, A.; Pedace, L.; Travaglini, L.; Pizzi, S.; Andreani, M.; Miele, E.; Invernizzi, F.; Reale, C.; et al. Childhood-onset dystonia-causing KMT2B variants result in a distinctive genomic hypermethylation profile. Clin. Epigenet. 2021, 13, 157. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical epigenomics: Genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med. 2021, 23, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Sobreira, N.; Brucato, M.; Zhang, L.; Ladd-Acosta, C.; Ongaco, C.; Romm, J.; Doheny, K.F.; Netto, R.C.M.; Bertola, D.; Kim, A.C.; et al. Patients with a Kabuki syndrome phenotype demonstrate DNA methylation abnormalities. Eur. J. Hum. Genet. 2017, 25, 1335–1344. [Google Scholar] [CrossRef]
- Barbosa, M.; Joshi, R.S.; Garg, P.; Martin-Trujillo, A.; Patel, N.; Jadhav, B.; Watson, C.T.; Gibson, W.; Chetnik, K.; Tessereau, C.; et al. Identification of rare de novo epigenetic variations in congenital disorders. Nat. Commun. 2018, 9, 2064. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Baux, D.; Van Goethem, C.; Ardouin, O.; Guignard, T.; Bergougnoux, A.; Koenig, M.; Roux, A.-F. MobiDetails: Online DNA variants interpretation. Eur. J. Hum. Genet. 2020, 29, 356–360. [Google Scholar] [CrossRef]
- Baux, D. Mobidetails_KMT2A. Available online: https://mobidetails.iurc.montp.inserm.fr/MD/auth/variant_list/EpiSignature_KMT2A (accessed on 14 December 2021).
- Courraud, J.; Chater-Diehl, E.; Durand, B.; Vincent, M.; Moreno, M.D.M.M.; Boujelbene, I.; Drouot, N.; Genschik, L.; Schaefer, E.; Nizon, M.; et al. Integrative approach to interpret DYRK1A variants, leading to a frequent neurodevelopmental disorder. Genet. Med. 2021, 23, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.D.; Dafou, D.; McEntagart, M.; Woollard, W.J.; Elmslie, F.V.; Holder-Espinasse, M.; Irving, M.; Saggar, A.K.; Smithson, S.; Trembath, R.; et al. De Novo Mutations in MLL Cause Wiedemann-Steiner Syndrome. Am. J. Hum. Genet. 2012, 91, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Chen, D.Y.; Westergard, T.D.; Fisher, J.K.; Rubens, J.A.; Sasagawa, S.; Kan, J.T.; Korsmeyer, S.J.; Cheng, E.H.-Y.; Hsieh, J.J.-D. Proteolysis of MLL family proteins is essential for Taspase1-orchestrated cell cycle progression. Genes Dev. 2006, 20, 2397–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, B.; Greer, C.B.; Coleman, B.C.; Sweatt, J.D. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin 2019, 12, 7. [Google Scholar] [CrossRef]
- Hsieh, J.J.-D.; Ernst, P.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol. Cell. Biol. 2003, 23, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Terranova, R.; Agherbi, H.; Boned, A.; Meresse, S.; Djabali, M. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc. Natl. Acad. Sci. USA 2006, 103, 6629–6634. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, Y. Structure, function and inhibition of critical protein–protein interactions involving mixed lineage leukemia 1 and its fusion oncoproteins. J. Hematol. Oncol. 2021, 14, 56. [Google Scholar] [CrossRef]
- Philippidou, P.; Dasen, J.S. Hox genes: Choreographers in neural development, architects of circuit organization. Neuron 2013, 80, 12–34. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Liu, Z.; Sun, X.; Xia, X.; Liu, Y.; Liu, L. Pan-cancer genome-wide DNA methylation analyses revealed that hypermethylation influences 3D architecture and gene expression dysregulation in HOXA locus during carcinogenesis of cancers. Front. Cell Dev. Biol. 2021, 9, 649168. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Wong, C.C.Y.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.E.; Imai, K.; King, G.; Stuart, E.A. Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Political Anal. 2007, 15, 199–236. [Google Scholar] [CrossRef] [Green Version]
- Stuart, E.A.; King, G.; Imai, K.; Ho, D. MatchIt: Nonparametric preprocessing for parametric causal inference. J. Stat. Softw. 2011, 42, 1–28. [Google Scholar]
- Sadikovic, B.; Levy, M.A.; Aref-Eshghi, E. Functional annotation of genomic variation: DNA methylation episignatures in neurodevelopmental Mendelian disorders. Hum. Mol. Genet. 2020, 29, R27–R32. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Rodenhiser, D.I.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Hood, R.L.; Bulman, D.E.; Kernohan, K.D.; et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018, 102, 156–174. [Google Scholar] [CrossRef] [Green Version]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).