
The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders


Abstract
:1. Introduction
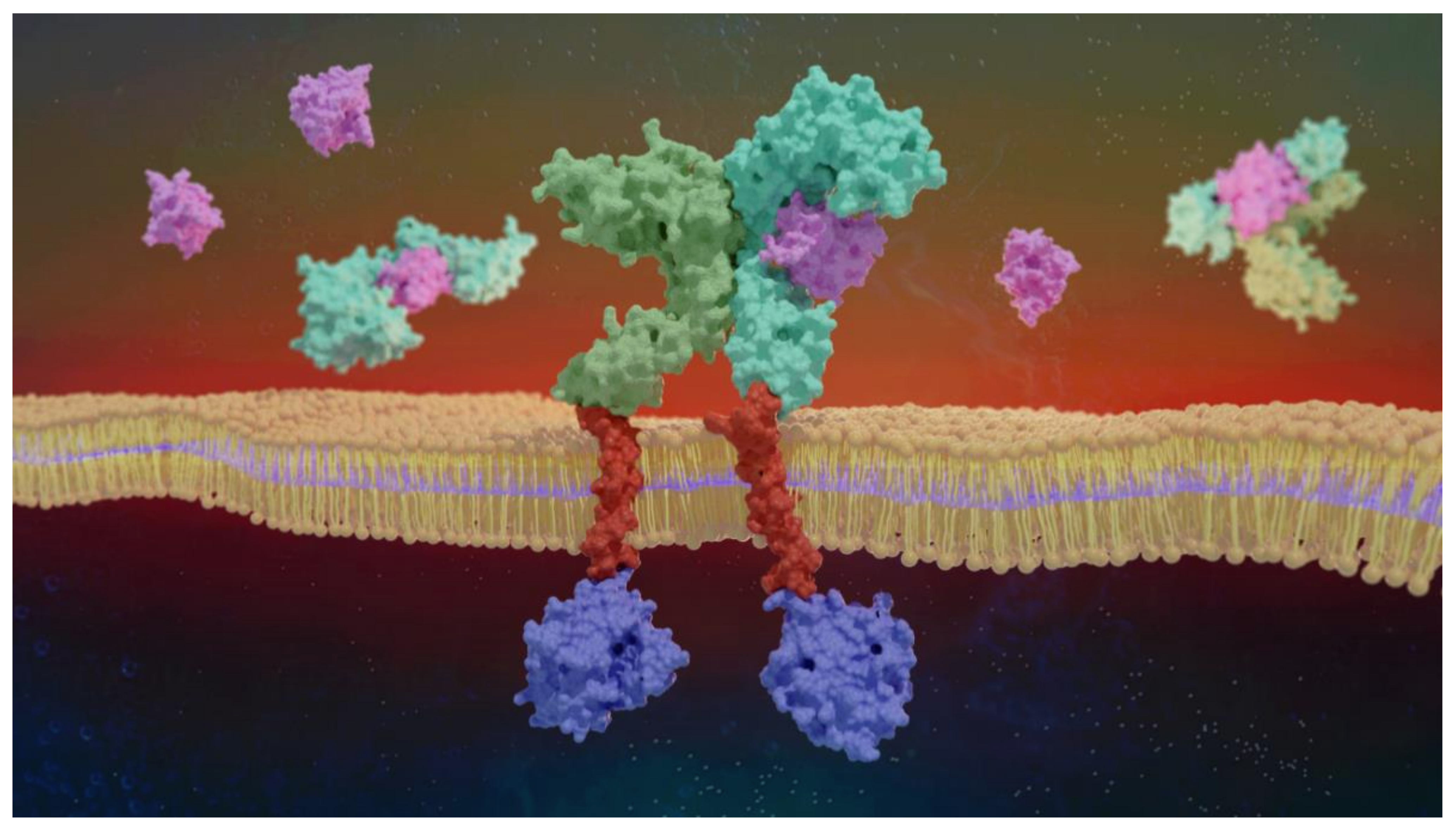
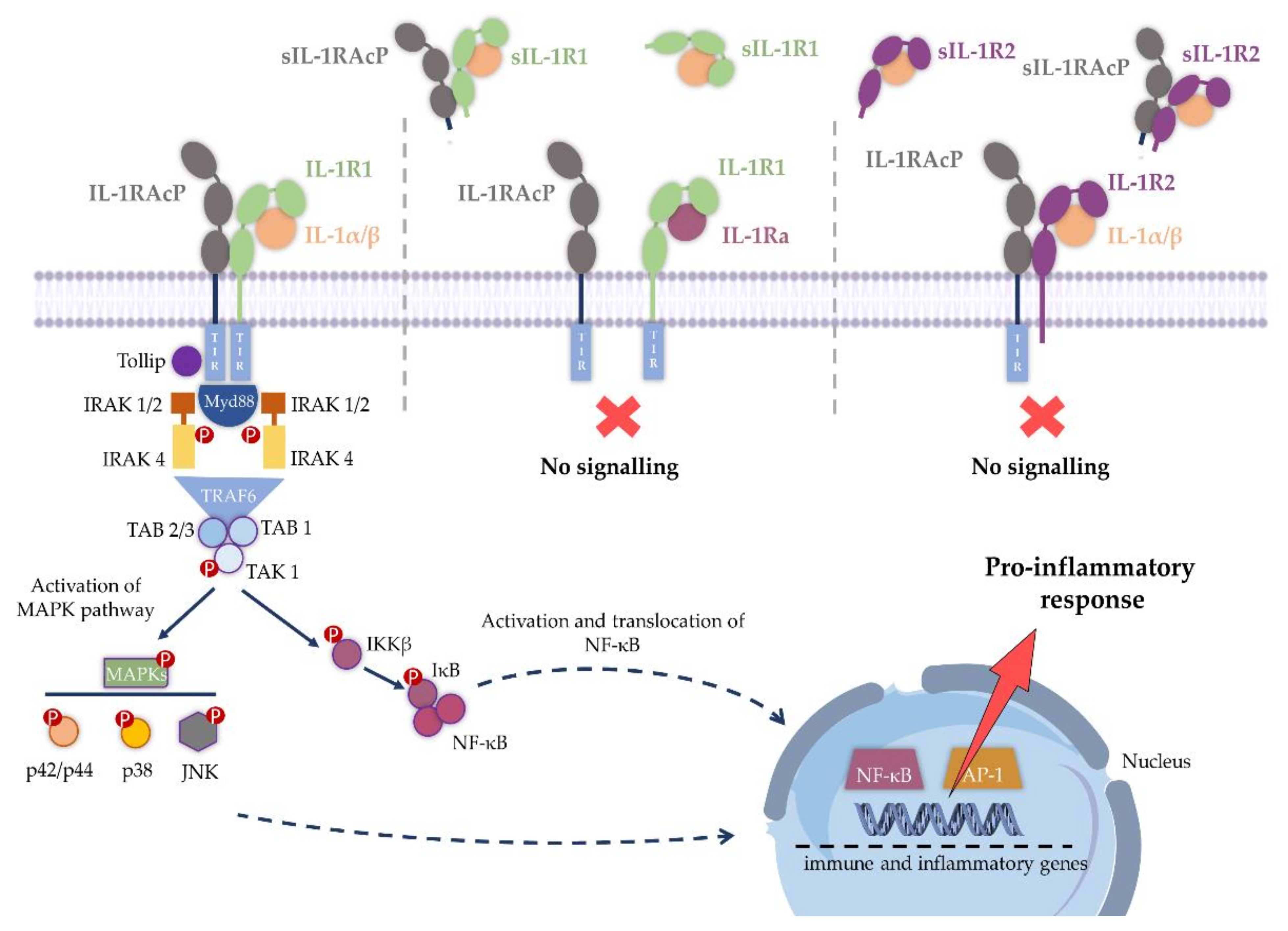
2. IL-1R1 Signaling
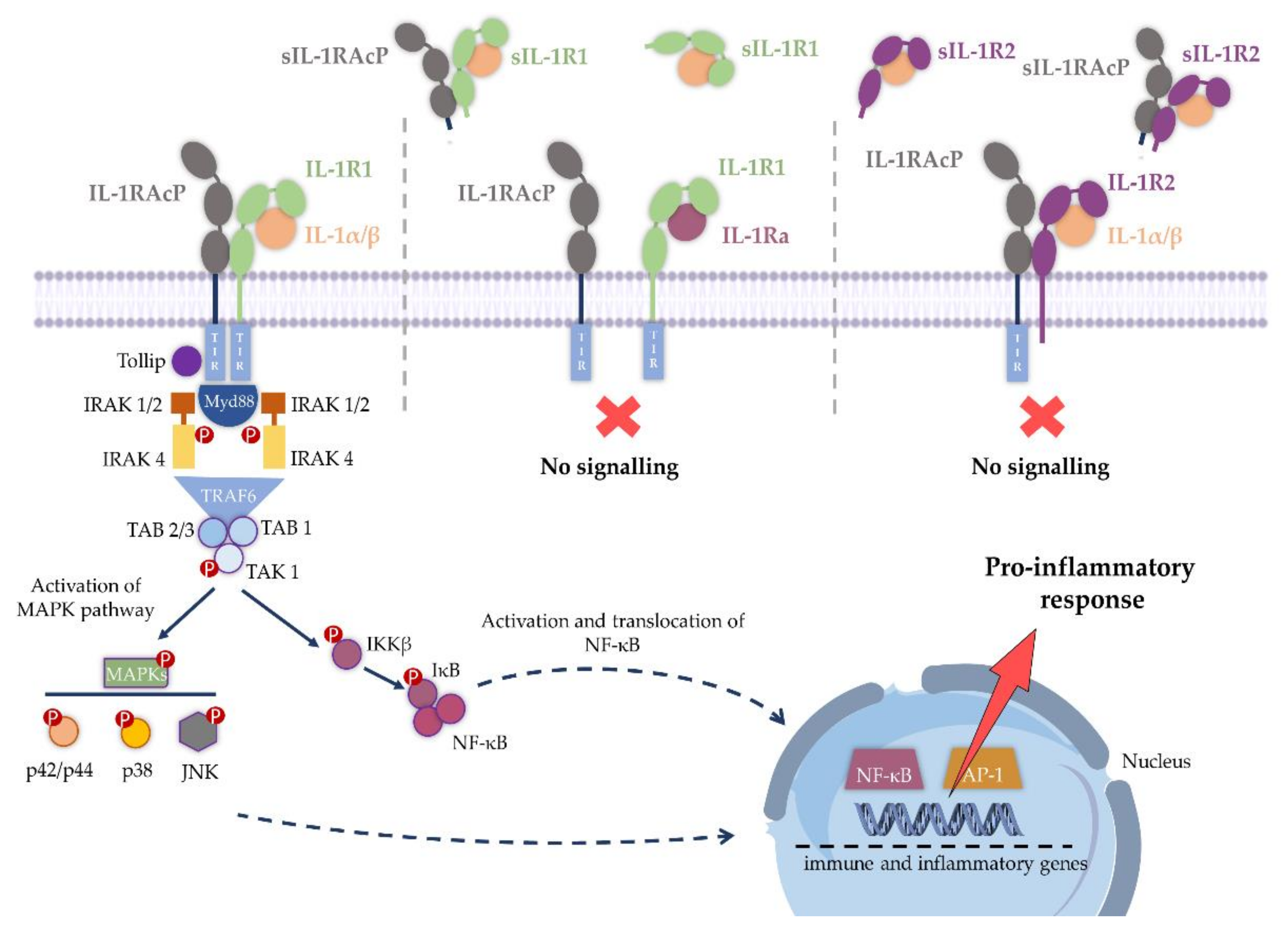
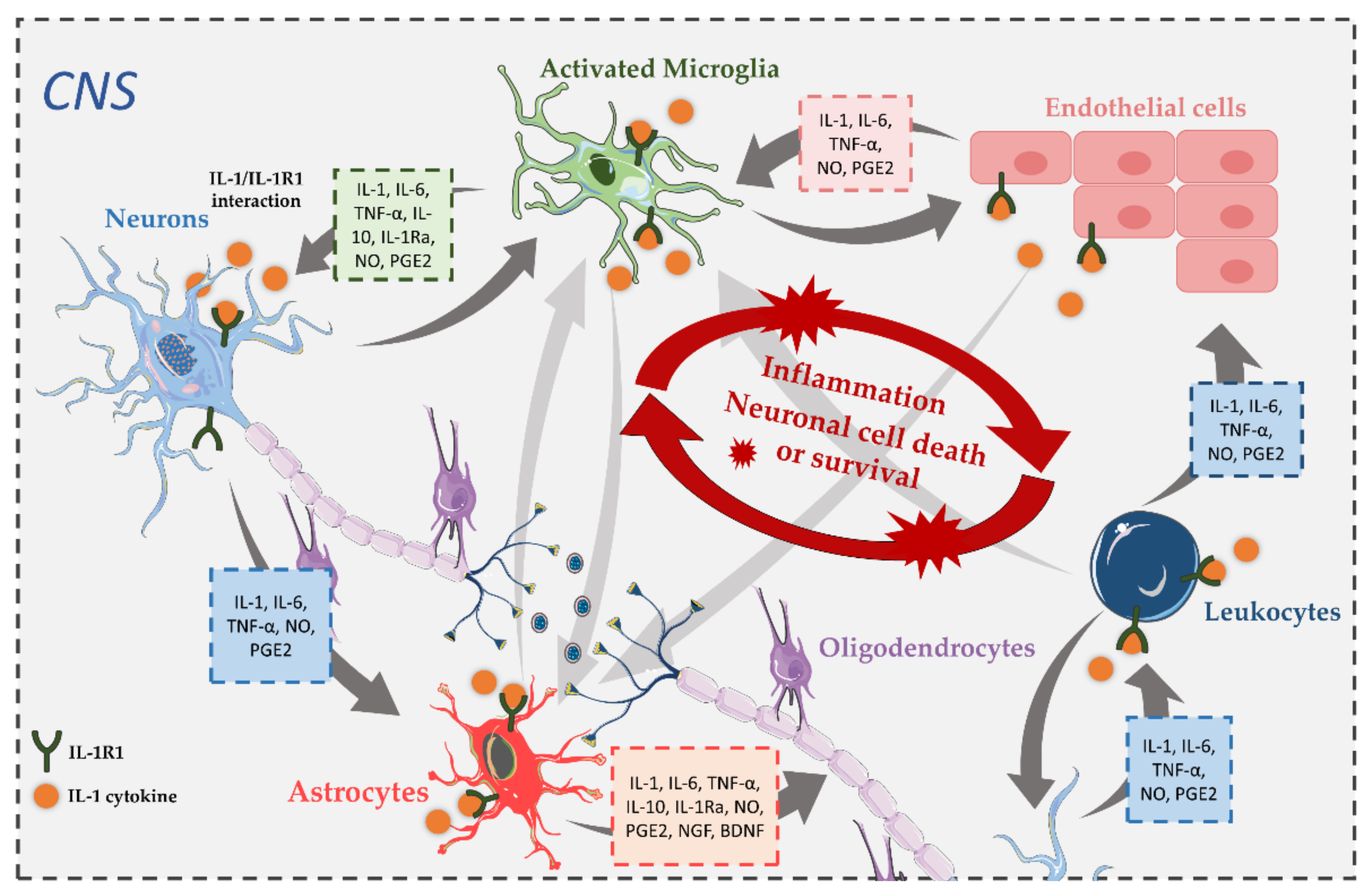
3. Expression of IL-1R1 in the CNS
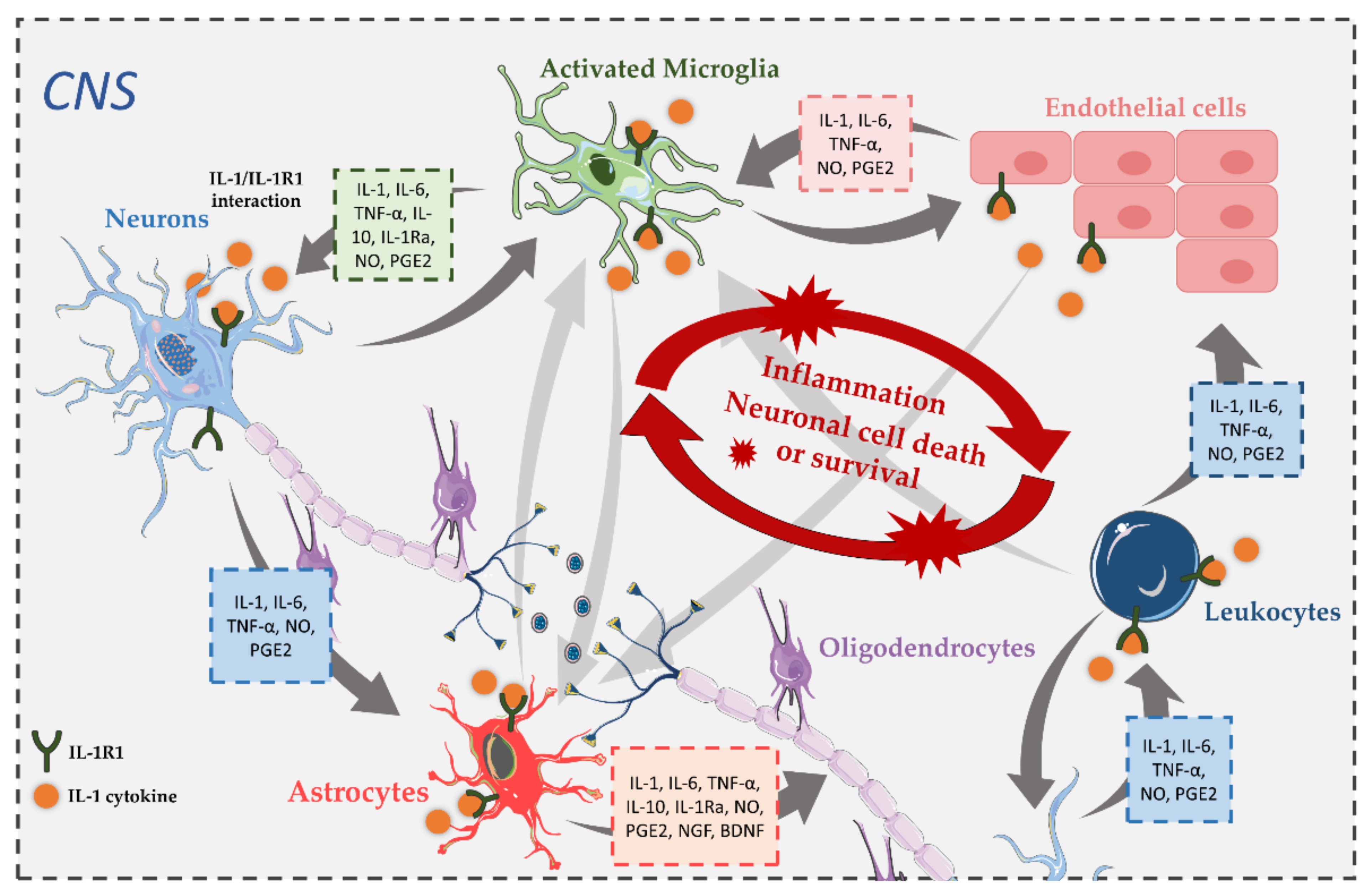
4. The Role and Involvement of IL-1 Pathways in CNS Disorders
4.1. IL-1R1 in Alzheimer’s Disease
4.2. IL-1R1 in Parkinson’s Disease
4.3. IL-1R1 in Amyotrophic Lateral Sclerosis
4.4. IL-1R1 in Multiple Sclerosis
4.5. IL-1R1 in Schizophrenia
4.6. IL-1R1 in Epilepsy
4.7. IL-1R1 in Traumatic Brain Injury and Stroke
4.8. IL-1R1 in Prion Diseases
4.9. IL-1R1 in Other CNS Diseases
5. Tractability of IL-1R1 as a Pharmacological Target
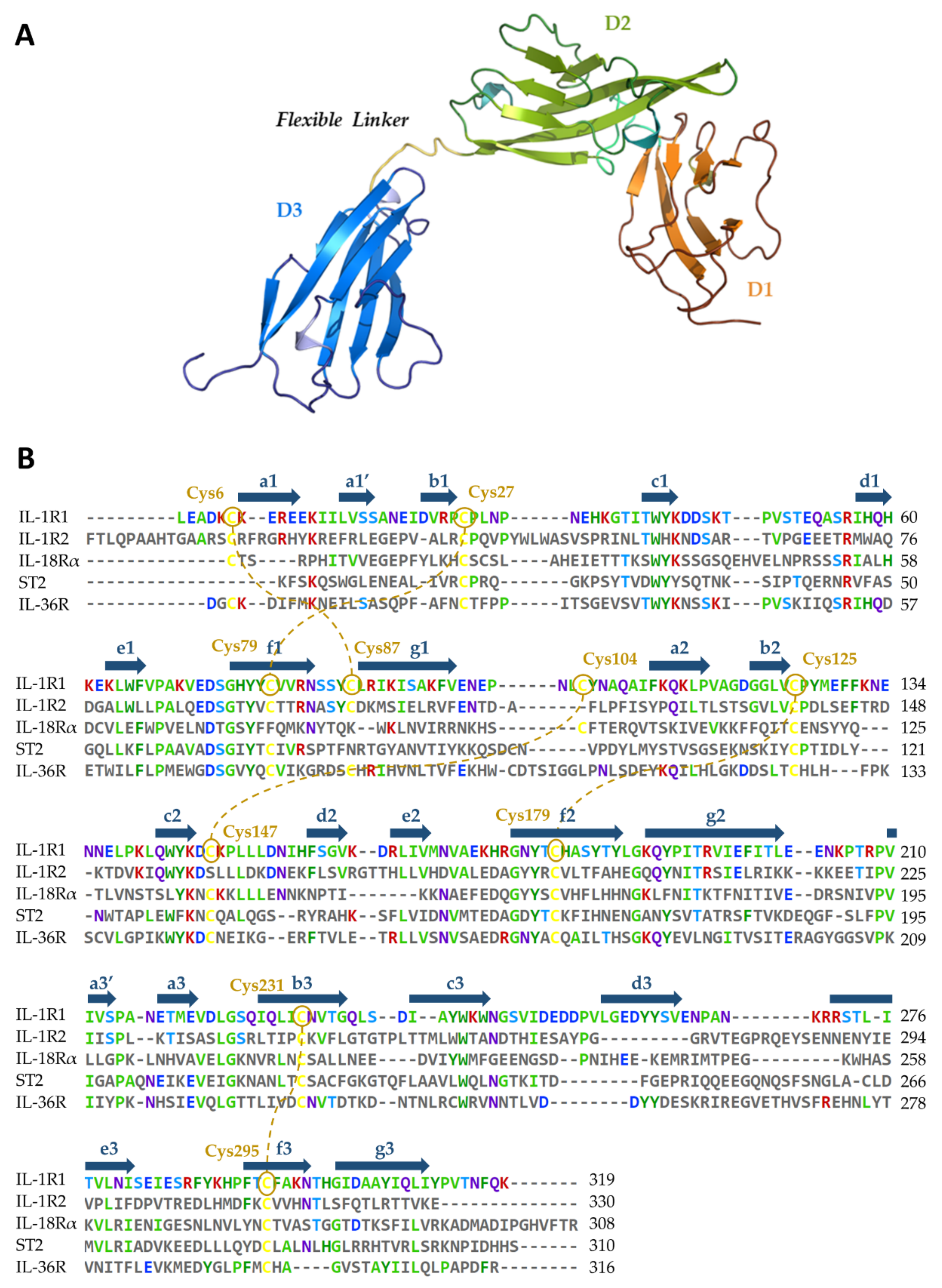
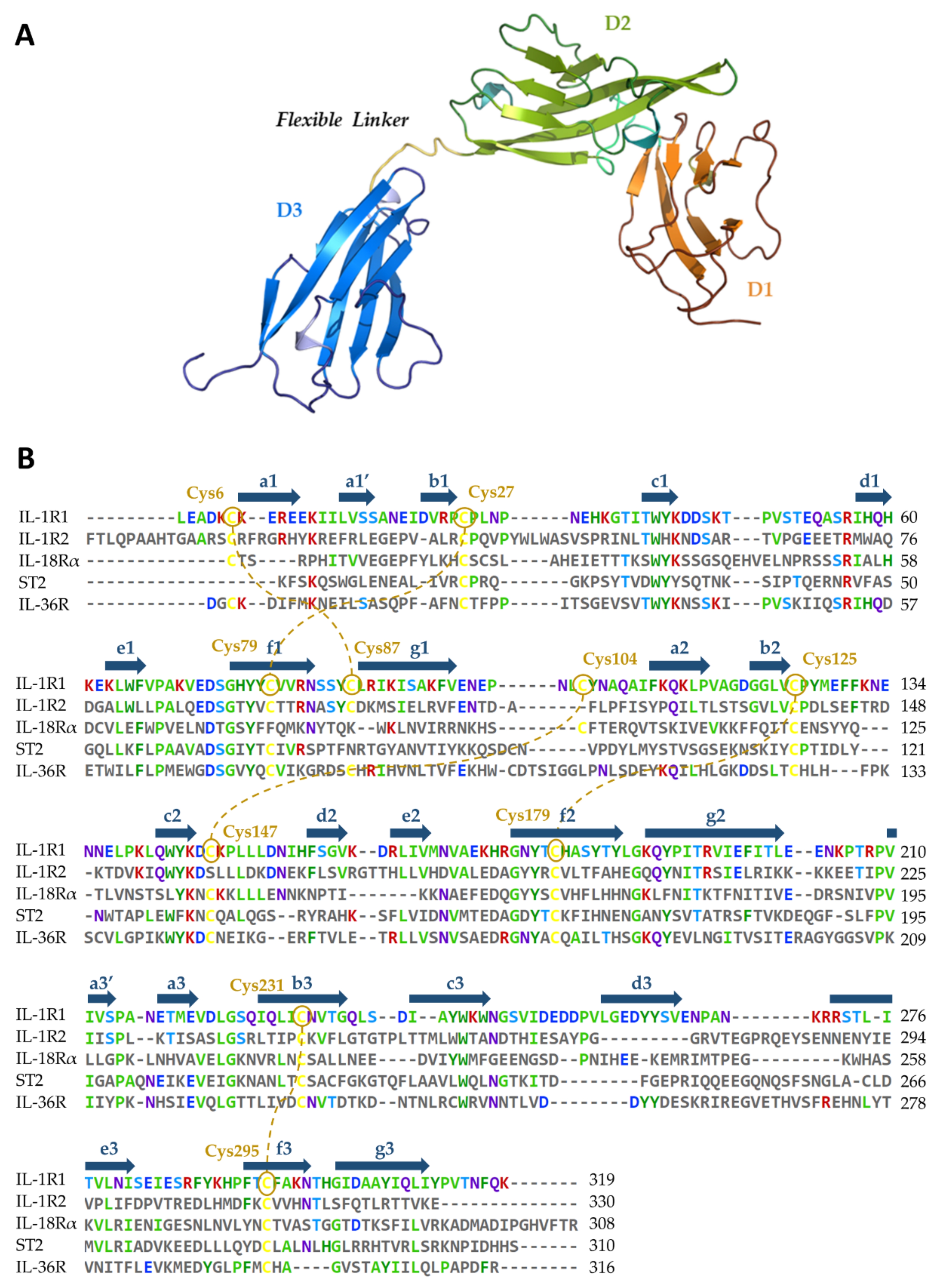
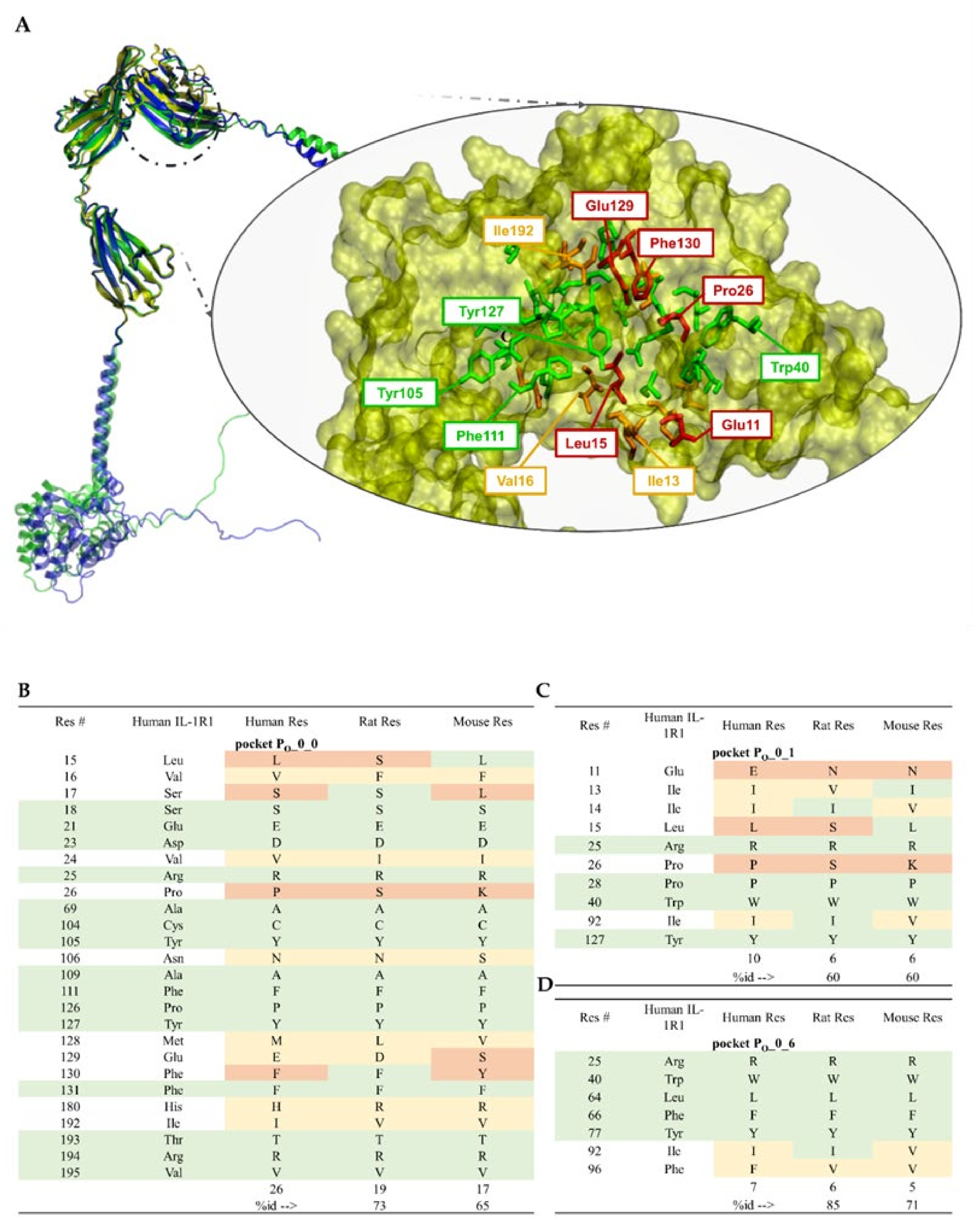
5.1. Availability of Protein Structures
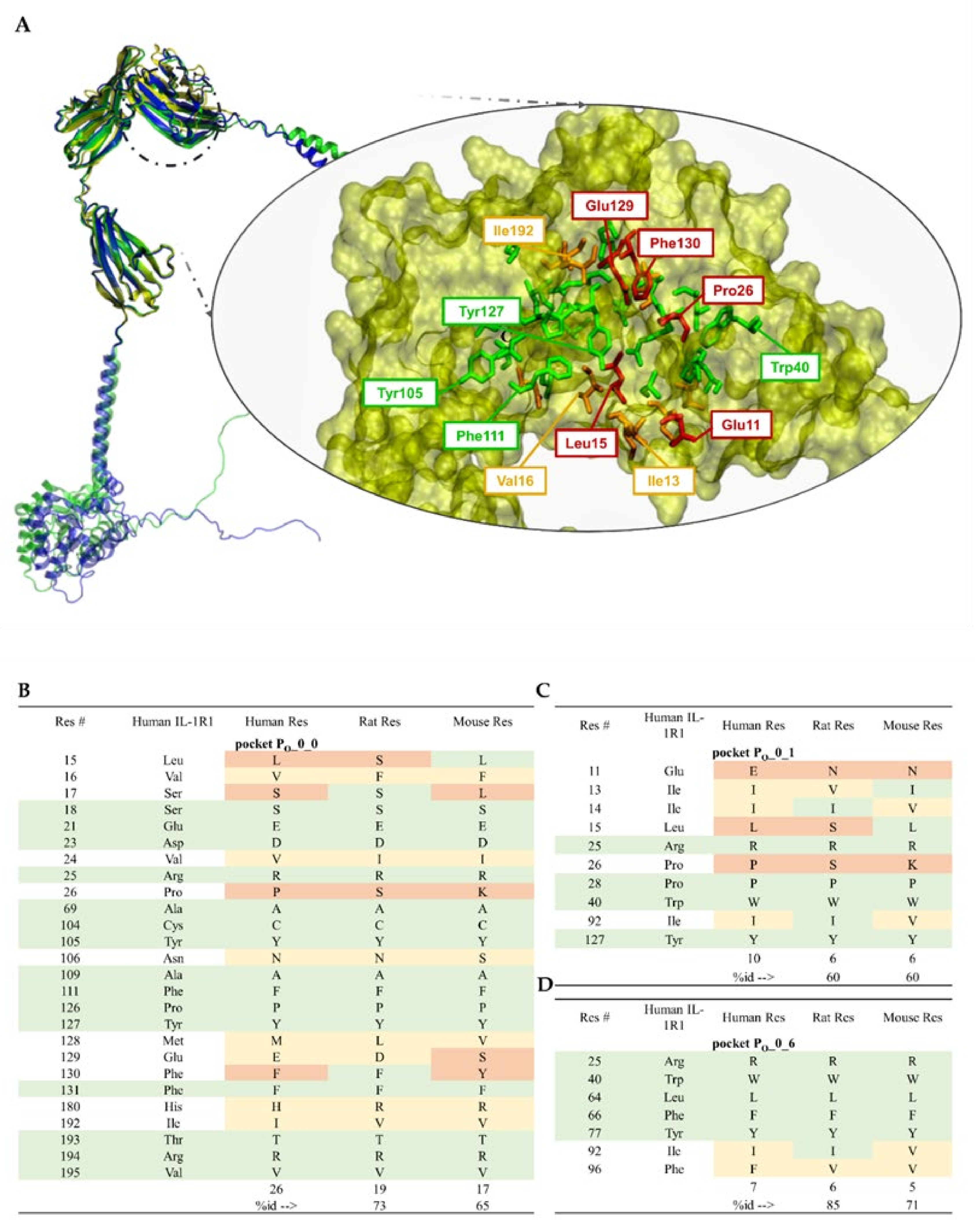
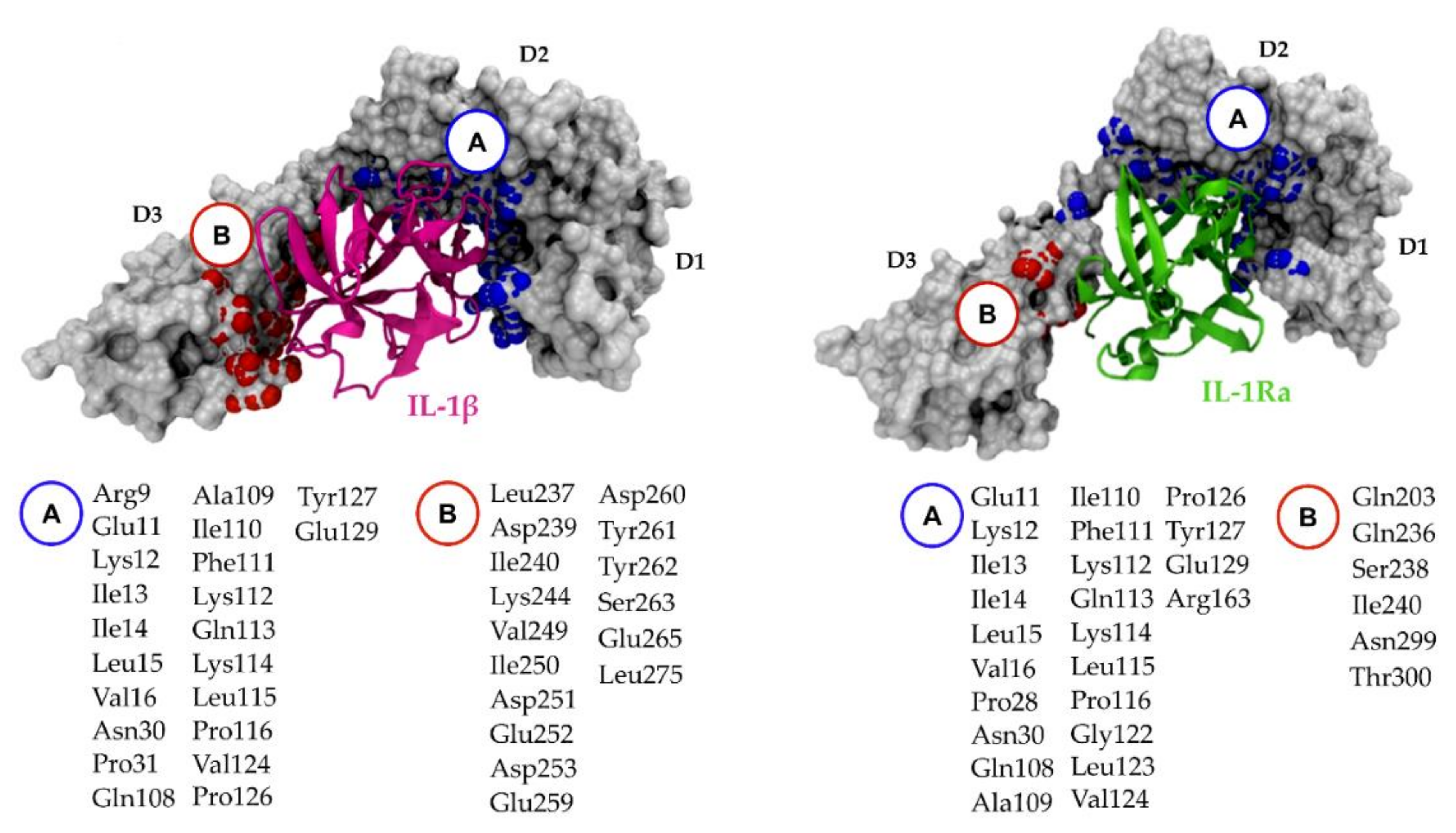
5.2. Relevant IL-1R1–Protein Interactions
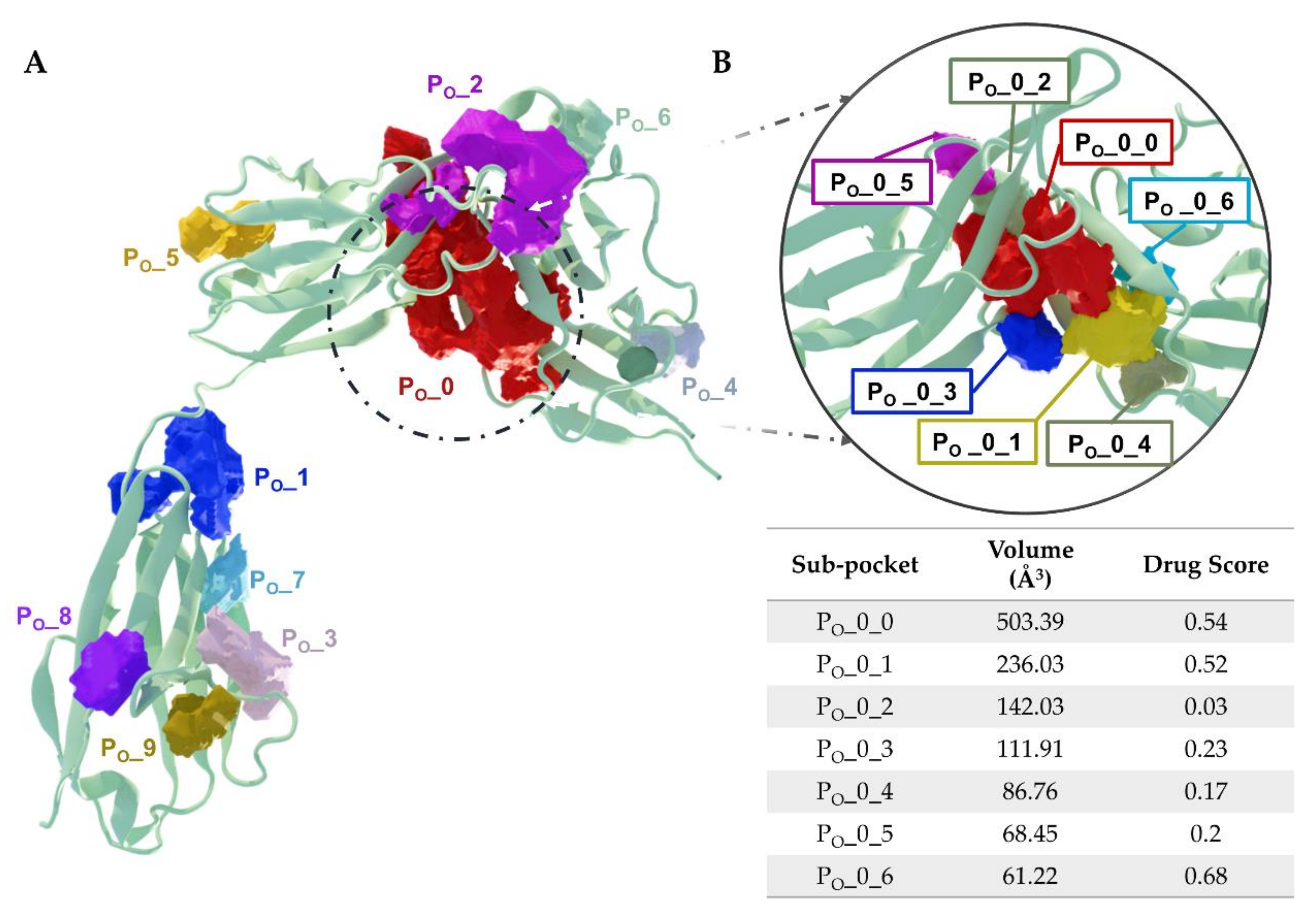
5.3. Putative IL-1R1 Druggable Binding Sites
- PO_0_0: Leu15, Val16, Ser17, Ser18, Glu21, Asp23, Val24, Arg25, Pro26, Ala69, Cys104, Tyr105, Asn106, Ala109, Phe111, Pro126, Tyr127, Met128, Glu129, Phe130, Phe131, His180, Ile192, Thr193, Arg194 and Val195;
- PO_0_1: Glu11, Ile13, Ile14, Leu15, Arg25, Pro26, Pro28, Trp40, Ile92 and Tyr127;
- PO_0_6: Arg25, Trp40, Leu64, Phe66, Tyr77, Ile92, Ser93, Ala94 and Phe96.
5.4. Ligand-Binding Site Differences across IL-1R1 Orthologs
6. Animal Disease Models Relating to IL-1 Pathways
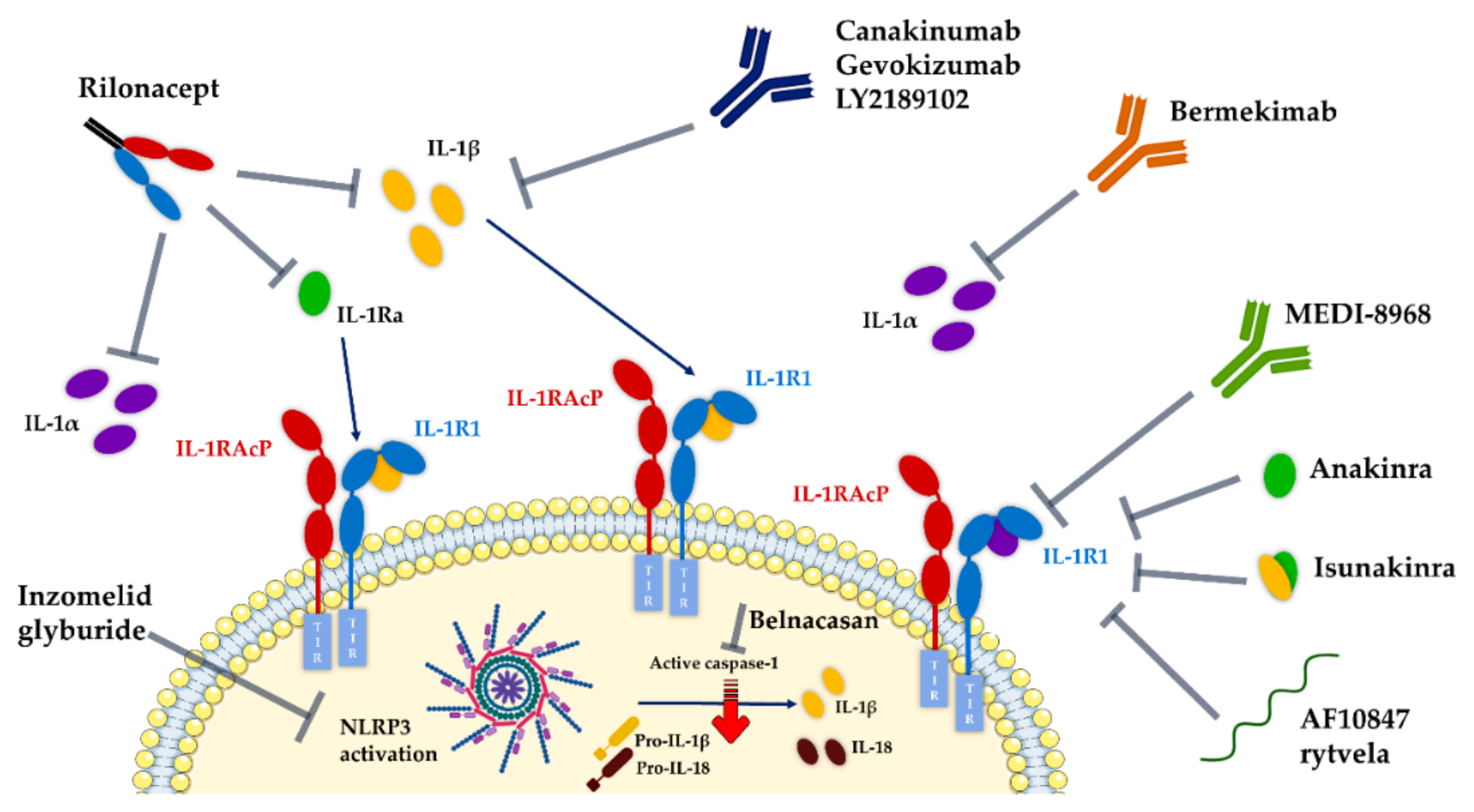
7. Pipeline of IL-1 Therapeutic Modulators
7.1. Development Status of Therapeutics Targeting IL-1R1
- Anakinra (Kineret®, Amgen), a recombinant non-glycosylated form of the natural human IL-1Ra, distinguishable by the addition of an N-terminal methionine residue, reached the US and Europe drug markets in 2001 and 2002, respectively, to treat rheumatoid arthritis. Fundamentally, this molecule targets the IL-1R1-ECD, competitively inhibiting IL-1α and IL-1β binding and blocking intracellular signal transduction. Anakinra is rapidly removed from the body by renal filtration due to its small size (17.3 kD). However, it requires daily self-administration via subcutaneous injection, which may result in adverse side effects such as injection site reactions, missed doses and ultimately decreased patient treatment compliance [217,240]. Interestingly, a recent study reported that anakinra crossed a human in vitro model of the BBB derived from human umbilical cord blood stem cells at a 4–7-fold higher rate than the monoclonal antibodies bermekimab (IL-1α antagonist) and canakinumab (IL-1β antagonist) [241];
- EBI-005 (isunakinra), a human IL-1β and IL-1Ra chimeric protein, developed in 2013 by Eleven Biotherapeutics, has been shown to bind IL-1R1 at a higher affinity than IL-1β (KD = 0.014 nM for EBI-005; KD = 2.0 nM for IL-1β). This biologic was optimized for topical ocular administration in patients with dry eye disease and allergic conjunctivitis [189]. However, phase III clinical trials were halted after EBI-005 failed to achieve primary endpoints;
- The monoclonal antibody (mAb) AMG108 (licensed to AstraZeneca and Medlmmune and now termed MEDI-78998) binds IL-1R1-ECD, blocking IL-1β-mediated signaling pathways. In preclinical studies, this human mAb has shown efficacy in models of osteoarthritis; however, no significant clinical benefits were observed in phase II trials [242].
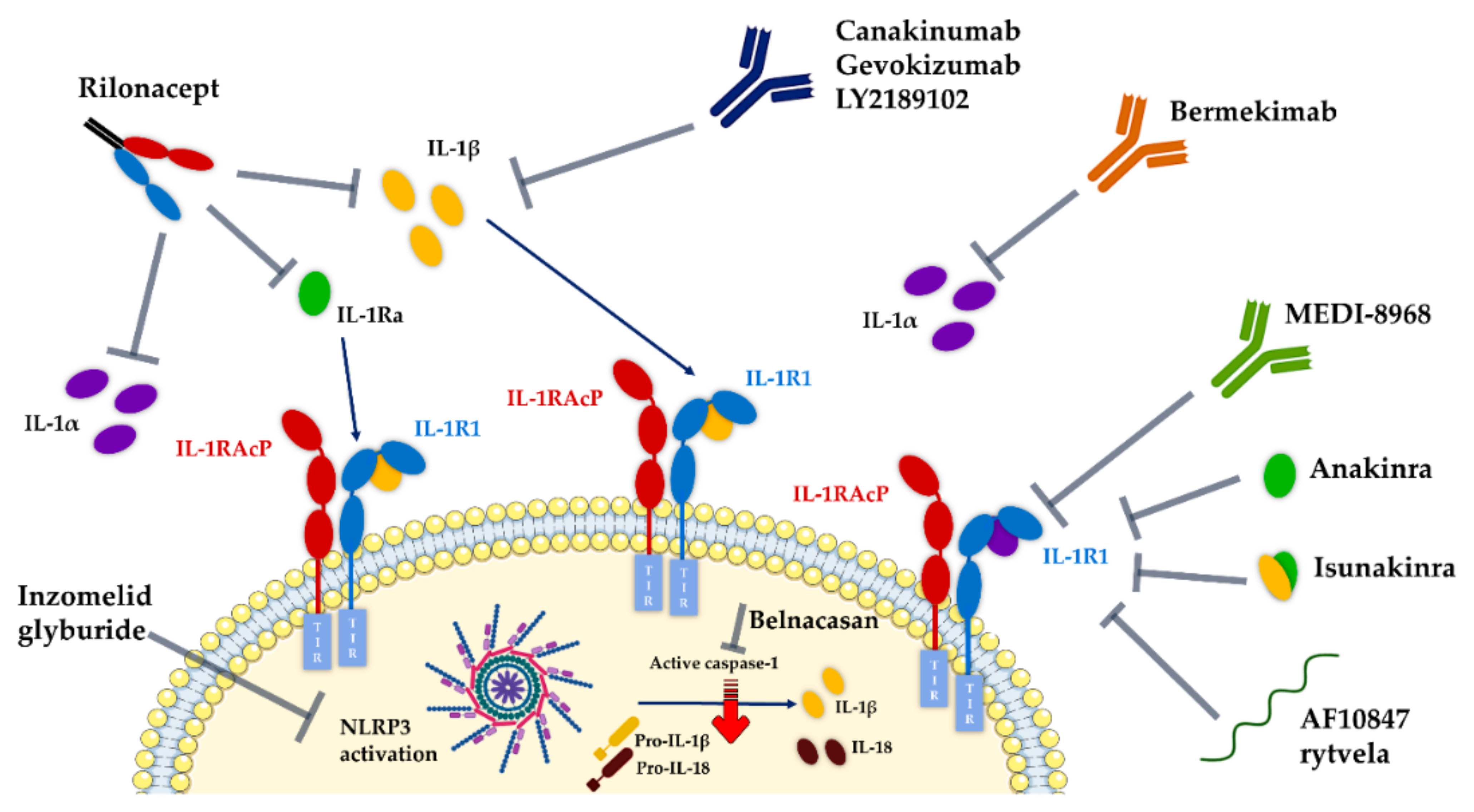
7.2. New and Alternative Approaches Targeting IL-1/IL-1R1 Pathways
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fantuzzi, G. 3. Evolution of message discovery: The Interleukin-1 family. In Body Messages; Harvard University Press: Cambridge, MA, USA, 2016; pp. 29–73. [Google Scholar]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Elzinga, B.M.; Twomey, C.; Powell, J.C.; Harte, F.; McCarthy, J.V. Interleukin-1 receptor type 1 is a substrate for γ-secretase- dependent regulated intramembrane proteolysis. J. Biol. Chem. 2009, 284, 1394–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural basis of IL-1 family cytokine signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Quan, N. Microglia and CNS Interleukin-1: Beyond immunological concepts. Front. Neurol. 2018, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [Green Version]
- Gottschlich, A.; Endres, S.; Kobold, S. Therapeutic strategies for targeting IL-1 in cancer. Cancers 2021, 13, 477. [Google Scholar] [CrossRef]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Neuroinflammation: A potential therapeutic target. Expert Opin. Ther. Targets 2005, 9, 887–900. [Google Scholar] [CrossRef]
- Brambilla, R. Neuroinflammation, the thread connecting neurological disease. Acta Neuropathol. 2019, 137, 689–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the role of neuroinflammation in the progression of Parkinson’s disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelobaba, I.; Savic, D.; Lavrnja, I. Multiple sclerosis and neuroinflammation: The overview of current and prospective therapies. Curr. Pharm. Des. 2017, 23, 693–730. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Baylr, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, D.; Fernandes, A. Neuroinflammation and depression: Microglia activation, extracellular microvesicles and microRNA dysregulation. Front. Cell Neurosci. 2015, 9, 476. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Weidinger, E.; Leitner, B.; Schwarz, M.J. The role of inflammation in schizophrenia. Front. Neurosci. 2015, 9, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.A.; Joesting, J.J.; Freund, G.G. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav. Immun. 2013, 32, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlüter, T.; Schelmbauer, C.; Karram, K.; Mufazalov, I.A. Regulation of IL-1 signaling by the decoy receptor IL-1R2. J. Mol. Med. 2018, 96, 983–992. [Google Scholar] [CrossRef]
- Dale, M.; Nicklin, M.J.H. Interleukin-1 receptor cluster: Gene organization ofIL1R2, IL1R1, IL1RL2(IL-1Rrp2),IL1RL1(T1/ST2), andIL18R1(IL-1Rrp) on human chromosome 2q. Genomics 1999, 57, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Bazan, J.F.; Garcia, K.C. Structure of the activating IL-1 receptor signaling complex. Nat. Struct. Mol. Biol. 2012, 19, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Deredge, D.; Bowers, A.L.; Luchini, A.; Bonsor, D.A.; Beadenkopf, R.; Liotta, L.; Wintrode, P.L.; Sundberg, E.J. IL-1 family cytokines use distinct molecular mechanisms to signal through their shared co-receptor. Immunity 2017, 47, 510–523.e4. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The TLR and IL-1 signalling network at a glance. J. Cell Sci. 2014, 127, 2383–2390. [Google Scholar] [CrossRef] [Green Version]
- Auron, P.E. The interleukin 1 receptor: Ligand interactions and signal transduction. Cytokine Growth Factor Rev. 1998, 9, 221–237. [Google Scholar] [CrossRef]
- Arend, W.P.; Malyak, M.; Guthridge, C.J.; Gabay, C. Interleukin-1 receptor antagonist: Role in biology. Annu. Rev. Immunol. 1998, 16, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Dripps, D.J.; Brandhuber, B.J.; Thompson, R.C.; Eisenberg, S.P. Interleukin-1 (IL-1) receptor antagonist binds to the 80-kDa IL-1 receptor but does not initiate IL-1 signal transduction. J. Biol. Chem. 1991, 266, 10331–10336. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kent, B.R. 3D Scientific Visualization with Blender; Morgan & Claypool Publishers: San Rafael, CA, USA, 2015; ISBN 978-1-6270-5612-0. [Google Scholar]
- Symons, J.A.; Eastgate, J.A.; Duff, G.W. Purification and characterization of a novel soluble receptor for interleukin 1. J. Exp. Med. 1991, 174, 1251–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, K.; Bartlett, A.H.; Chen, Y.; Park, P.W. Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2010, 293, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.E.; Hanna, R.; Friend, D.; Moore, H.; Chen, H.; Farese, A.M.; MacVittie, T.J.; Virca, G.D.; Sims, J.E. The soluble form of IL-1 receptor accessory protein enhances the ability of soluble type II IL-1 receptor to inhibit IL-1 action. Immunity 2003, 18, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Goldstein, J.D.; Mermoud, L.; Diaz-Barreiro, A.; Palmer, G. IL-1 family antagonists in mouse and human skin inflammation. Front. Immunol. 2021, 12, 652846. [Google Scholar] [CrossRef]
- Cunningham, E.; Wada, E.; Carter, D.; Tracey, D.; Battey, J.; De Souza, E. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J. Neurosci. 1992, 12, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- French, R.A.; VanHoy, R.W.; Chizzonite, R.; Zachary, J.F.; Dantzer, R.; Parnet, P.; Bluthé, R.-M.; Kelley, K.W. Expression and localization of p80 and p68 interleukin-1 receptor proteins in the brain of adult mice. J. Neuroimmunol. 1999, 93, 194–202. [Google Scholar] [CrossRef]
- Rothwell, N.; Luheshi, G.N. Interleukin 1 in the brain: Biology, pathology and therapeutic target. Trends Neurosci. 2000, 23, 618–625. [Google Scholar] [CrossRef]
- Todd, L.; Palazzo, I.; Suarez, L.; Liu, X.; Volkov, L.; Hoang, T.V.; Campbell, W.A.; Blackshaw, S.; Quan, N.; Fischer, A.J. Reactive microglia and IL1β/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J. Neuroinflamm. 2019, 16, 118. [Google Scholar] [CrossRef] [Green Version]
- Gajtkó, A.; Bakk, E.; Hegedűs, K.; Ducza, L.; Holló, K. IL-1β induced cytokine expression by spinal astrocytes can play a role in the maintenance of chronic inflammatory pain. Front. Physiol. 2020, 11, 543331. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nemeth, D.P.; McKim, D.B.; Zhu, L.; DiSabato, D.J.; Berdysz, O.; Gorantla, G.; Oliver, B.; Witcher, K.G.; Wang, Y.; et al. Cell-type-specific Interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity 2019, 50, 317–333.e6. [Google Scholar] [CrossRef] [Green Version]
- Pinteaux, E.; Parker, L.C.; Rothwell, N.J.; Luheshi, G.N. Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J. Neurochem. 2002, 83, 754–763. [Google Scholar] [CrossRef]
- Lin, H.-W.; Basu, A.; Druckman, C.; Cicchese, M.; Krady, J.K.; Levison, S.W. Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J. Neuroinflamm. 2006, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Ohtaki, H.; Tsumuraya, T.; Song, D.; Ohara, K.; Asano, M.; Iwakura, Y.; Atsumi, T.; Shioda, S. Interleukin-1 participates in the classical and alternative activation of microglia/macrophages after spinal cord injury. J. Neuroinflamm. 2012, 9, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnow, S.M.; Knoll, J.G.; Verghese, S.C.; Levasseur, P.R.; Marks, D.L. Amplification and propagation of interleukin-1β signaling by murine brain endothelial and glial cells. J. Neuroinflamm. 2017, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Friedman, W.J. Cytokines regulate expression of the type 1 Interleukin-1 receptor in rat hippocampal neurons and glia. Exp. Neurol. 2001, 168, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-J.; Jiang, M.; Zhou, H.; Liu, W.; Wang, C.; Kang, Z.; Han, B.; Zhang, Q.; Chen, X.; Xiao, J.; et al. TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and promotes neuroinflammation. J. Clin. Investig. 2018, 128, 5399–5412. [Google Scholar] [CrossRef]
- Bruttger, J.; Karram, K.; Wörtge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 2015, 43, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.-H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Investig. 2020, 130, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-F.; Huang, L.-D.; Yu, P.-P.; Hu, J.-G.; Yin, L.; Wang, L.; Xu, X.-M.; Lu, P.-H. Upregulation of type I interleukin−1 receptor after traumatic spinal cord injury in adult rats. Acta Neuropathol. 2006, 111, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krady, J.K.; O’Malley, M.; Styren, S.D.; DeKosky, S.T.; Levison, S.W. The type 1 Interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J. Neurosci. 2002, 22, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hori, T.; Mori, T.; Kuriyama, K.; Mizuno, K. Recombinant human interleukin-1 β alters the activity of preoptic thermosensitive neurons in vitro. Brain Res. Bull. 1989, 23, 209–213. [Google Scholar] [CrossRef]
- Krueger, J.M.; Fang, J.; Taishi, P.; Chen, Z.; Kushikata, T.; Gardi, J. Sleep: A physiologic role for IL-1β and TNF-α a. Ann. N. Y. Acad. Sci. 1998, 856, 148–159. [Google Scholar] [CrossRef]
- Koo, J.W.; Duman, R.S. IL-1 is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. USA 2008, 105, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Del Rey, A.; Balschun, D.; Wetzel, W.; Randolf, A.; Besedovsky, H.O. A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6, and TNFα operates during long-term potentiation and learning. Brain Behav. Immun. 2013, 33, 15–23. [Google Scholar] [CrossRef]
- Depino, A.M.; Alonso, M.; Ferrari, C.; del Rey, A.; Anthony, D.; Besedovsky, H.; Medina, J.H.; Pitossi, F. Learning modulation by endogenous hippocampal IL-1: Blockade of endogenous IL-1 facilitates memory formation. Hippocampus 2004, 14, 526–535. [Google Scholar] [CrossRef]
- Gui, W.-S.; Wei, X.; Mai, C.-L.; Murugan, M.; Wu, L.-J.; Xin, W.-J.; Zhou, L.-J.; Liu, X.-G. Interleukin-1β overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol. Pain 2016, 12, 174480691664678. [Google Scholar] [CrossRef] [Green Version]
- Hewett, S.J.; Jackman, N.A.; Claycomb, R.J. Interleukin-1β in central nervous system injury and repair. Eur. J. Neurodegener. Dis. 2012, 1, 195–211. [Google Scholar] [PubMed]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L.; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, C.; El-Okl, M.; Williams, A.L.; Cunningham, C.; Wilcockson, D.; Perry, V.H. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 788–789. [Google Scholar] [CrossRef]
- Italiani, P.; Puxeddu, I.; Napoletano, S.; Scala, E.; Melillo, D.; Manocchio, S.; Angiolillo, A.; Migliorini, P.; Boraschi, D.; Vitale, E.; et al. Circulating levels of IL-1 family cytokines and receptors in Alzheimer’s disease: New markers of disease progression? J. Neuroinflamm. 2018, 15, 342. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Tanaka, S.; Ishii, A.; Ohtaki, H.; Shioda, S.; Yoshida, T.; Numazawa, S. Activation of microglia induces symptoms of Parkinson’s disease in wild-type, but not in IL-1 knockout mice. J. Neuroinflamm. 2013, 10, 907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meer, J.W.M.; Simon, A. Blocking IL-1 to slow down progression of ALS? Proc. Natl. Acad. Sci. USA 2010, 107, 12741–12742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1 accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuinness, M.; Powers, J.; Bias, W.; Schmeckpeper, B.; Segal, A.; Gowda, V.; Wesselingh, S.; Berger, J.; Griffin, D.; Smith, K. Human leukocyte antigens and cytokine expression in cerebral inflammatory demyelinative lesions of X-linked adrenoleukodystrophy and multiple sclerosis. J. Neuroimmunol. 1997, 75, 174–182. [Google Scholar] [CrossRef]
- Hauser, S.L.; Doolittle, T.H.; Lincoln, R.; Brown, R.H.; Dinarello, C.A. Cytokine accumulations in CSF of multiple sclerosis patients. Neurology 1990, 40, 1735. [Google Scholar] [CrossRef] [PubMed]
- Seppi, D.; Puthenparampil, M.; Federle, L.; Ruggero, S.; Toffanin, E.; Rinaldi, F.; Perini, P.; Gallo, P. Cerebrospinal fluid IL-1β correlates with cortical pathology load in multiple sclerosis at clinical onset. J. Neuroimmunol. 2014, 270, 56–60. [Google Scholar] [CrossRef]
- Chung, J.Y.; Krapp, N.; Wu, L.; Lule, S.; McAllister, L.M.; Edmiston, W.J.; Martin, S.; Levy, E.; Songtachalert, T.; Sherwood, J.S.; et al. Interleukin-1 receptor 1 deletion in focal and diffuse experimental traumatic brain injury in mice. J. Neurotrauma 2019, 36, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Bertani, I.; Iori, V.; Trusel, M.; Maroso, M.; Foray, C.; Mantovani, S.; Tonini, R.; Vezzani, A.; Chiesa, R. Inhibition of IL-1β signaling normalizes NMDA-dependent neurotransmission and reduces seizure susceptibility in a mouse model of Creutzfeldt–Jakob disease. J. Neurosci. 2017, 37, 10278–10289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.-L.; Kim, M.-O.; Morgello, S.; Lee, S.C. Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J. Neuroimmunol. 2001, 115, 182–191. [Google Scholar] [CrossRef]
- Hu, S.J.; Calippe, B.; Lavalette, S.; Roubeix, C.; Montassar, F.; Housset, M.; Levy, O.; Delarasse, C.; Paques, M.; Sahel, J.-A.; et al. Upregulation of P2RX7 in Cx3cr1-deficient mononuclear phagocytes leads to increased Interleukin-1 secretion and photoreceptor neurodegeneration. J. Neurosci. 2015, 35, 6987–6996. [Google Scholar] [CrossRef]
- Patel, H.C.; Boutin, H.; Allan, S.M. Interleukin-1 in the brain: Mechanisms of action in acute neurodegeneration. Ann. N. Y. Acad. Sci. 2003, 992, 39–47. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Sciacca, F.; Ferri, C.; Licastro, F.; Veglia, F.; Biunno, I.; Gavazzi, A.; Calabrese, E.; Martinelli Boneschi, F.; Sorbi, S.; Mariani, C.; et al. Interleukin-1B polymorphism is associated with age at onset of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 927–931. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates Tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Licastro, F.; Pedrini, S.; Caputo, L.; Annoni, G.; Davis, L.J.; Ferri, C.; Casadei, V.; Grimaldi, L.M.E. Increased plasma levels of interleukin-1, interleukin-6 and α-1-antichymotrypsin in patients with Alzheimer’s disease: Peripheral inflammation or signals from the brain? J. Neuroimmunol. 2000, 103, 97–102. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Brissoni, B.; Agostini, L.; Kropf, M.; Martinon, F.; Swoboda, V.; Lippens, S.; Everett, H.; Aebi, N.; Janssens, S.; Meylan, E.; et al. Intracellular trafficking of Interleukin-1 receptor I requires tollip. Curr. Biol. 2006, 16, 2265–2270. [Google Scholar] [CrossRef]
- Cadete Martini, A.; Gomez-Arboledas, A.; Forner, S.; Rodriguez-Ortiz, C.J.; McQuade, A.; Danhash, E.; Phan, J.; Javonillo, D.; Ha, J.-V.; Tram, M.; et al. Amyloid-beta impairs TOM1-mediated IL-1R1 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 21198–21206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Hunot, S.; Dugas, N.; Faucheux, B.; Hartmann, A.; Tardieu, M.; Debré, P.; Agid, Y.; Dugas, B.; Hirsch, E.C. FcεRII/CD23 Is Expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. J. Neurosci. 1999, 19, 3440–3447. [Google Scholar] [CrossRef] [Green Version]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Pott Godoy, M.C.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008, 131, 1880–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pott Godoy, M.C.; Ferrari, C.C.; Pitossi, F.J. Nigral neurodegeneration triggered by striatal AdIL-1 administration can be exacerbated by systemic IL-1 expression. J. Neuroimmunol. 2010, 222, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Stojakovic, A.; Paz-Filho, G.; Arcos-Burgos, M.; Licinio, J.; Wong, M.-L.; Mastronardi, C.A. Role of the IL-1 pathway in dopaminergic neurodegeneration and decreased voluntary movement. Mol. Neurobiol. 2017, 54, 4486–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, H.; Wang, Q.; Zhao, W.; Liu, J.; Wang, D.; Muhammad, B.; Liu, X.; Quan, N.; Zhang, H.; Zhang, F.; et al. IL-1β/IL-1R1 signaling induced by intranasal lipopolysaccharide infusion regulates alpha-Synuclein pathology in the olfactory bulb, substantia nigra and striatum. Brain Pathol. 2020, 30, 1102–1118. [Google Scholar] [CrossRef]
- Lobsiger, C.S.; Cleveland, D.W. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 2007, 10, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Vaz, A.R.; Pinto, S.; Ezequiel, C.; Cunha, C.; Carvalho, L.A.; Moreira, R.; Brites, D. Phenotypic Effects of wild-type and mutant SOD1 expression in N9 murine microglia at steady state, inflammatory and immunomodulatory conditions. Front. Cell. Neurosci. 2019, 13, 109. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hao, W.; Dawson, A.; Liu, S.; Fassbender, K. Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J. Biol. Chem. 2009, 284, 3691–3699. [Google Scholar] [CrossRef] [Green Version]
- Hensley, K.; Floyd, R.A.; Gordon, B.; Mou, S.; Pye, Q.N.; Stewart, C.; West, M.; Williamson, K. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2002, 82, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Esch, E.; Hartmann, P.; Goswami, A.; Nikolin, S.; Weis, J.; Beyer, C.; Johann, S. Expression profile of pattern recognition receptors in skeletal muscle of SOD1 (G93A) amyotrophic lateral sclerosis (ALS) mice and sporadic ALS patients. Neuropathol. Appl. Neurobiol. 2018, 44, 606–627. [Google Scholar] [CrossRef]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Carlesi, C.; Giungato, P.; Puxeddu, I.; Borroni, B.; Bossù, P.; Migliorini, P.; Siciliano, G.; Boraschi, D. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J. Neuroinflamm. 2014, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Maier, A.; Deigendesch, N.; Müller, K.; Weishaupt, J.H.; Krannich, A.; Röhle, R.; Meissner, F.; Molawi, K.; Münch, C.; Holm, T.; et al. Interleukin-1 antagonist Anakinra in amyotrophic lateral sclerosis—A pilot study. PLoS ONE 2015, 10, e0139684. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Günther, R.; Akgün, K.; Hermann, A.; Ziemssen, T. Peripheral proinflammatory Th1/Th17 immune cell shift is linked to disease severity in amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 5941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Roos, P.M.; Larsson, S.C. Interleukin-1 receptor antagonist, interleukin-2 receptor alpha subunit and amyotrophic lateral sclerosis. Eur. J. Neurol. 2020, 27, 1913–1917. [Google Scholar] [CrossRef]
- Lin, C.-C.; Edelson, B.T. New insights into the role of IL-1β in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Immunol. 2017, 198, 4553–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellergård, J.; Edström, M.; Vrethem, M.; Ernerudh, J.; Dahle, C. Natalizumab treatment in multiple sclerosis: Marked decline of chemokines and cytokines in cerebrospinal fluid. Mult. Scler. J. 2010, 16, 208–217. [Google Scholar] [CrossRef]
- Dujmovic, I.; Mangano, K.; Pekmezovic, T.; Quattrocchi, C.; Mesaros, S.; Stojsavljevic, N.; Nicoletti, F.; Drulovic, J. The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: The elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J. Neuroimmunol. 2009, 207, 101–106. [Google Scholar] [CrossRef]
- Symons, J.A.; Bundick, R.V.; Suckling, A.J.; Rumsby, M.G. Cerebrospinal fluid interleukin 1 like activity during chronic relapsing experimental allergic encephalomyelitis. Clin. Exp. Immunol. 1987, 68, 648–654. [Google Scholar] [PubMed]
- Bauer, J.; Berkenbosch, F.; Van Dam, A.-M.; Dijkstra, C.D. Demonstration of interleukin-1β in Lewis rat brain during experimental allergic encephalomyelitis by immunocytochemistry at the light and ultrastructural level. J. Neuroimmunol. 1993, 48, 13–21. [Google Scholar] [CrossRef]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by Interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.G.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17–producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17–producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, Y.; Markovic-Plese, S. Activated IL-1RI signaling pathway induces Th17 cell differentiation via interferon regulatory factor 4 signaling in patients with relapsing-remitting multiple sclerosis. Front. Immunol. 2016, 7, 543. [Google Scholar] [CrossRef] [Green Version]
- Capone, A.; Bianco, M.; Ruocco, G.; De Bardi, M.; Battistini, L.; Ruggieri, S.; Gasperini, C.; Centonze, D.; Sette, C.; Volpe, E. Distinct expression of inflammatory features in T helper 17 cells from multiple sclerosis patients. Cells 2019, 8, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.-X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Schiffenbauer, J.; Streit, W.J.; Butfiloski, E.; LaBow, M.; Edwards, C.; Moldawer, L.L. The induction of EAE Is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clin. Immunol. 2000, 95, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Mufazalov, I.A.; Schelmbauer, C.; Regen, T.; Kuschmann, J.; Wanke, F.; Gabriel, L.A.; Hauptmann, J.; Müller, W.; Pinteaux, E.; Kurschus, F.C.; et al. IL-1 signaling is critical for expansion but not generation of autoreactive GM-CSF+ Th17 cells. EMBO J. 2017, 36, 102–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, C.A.; Baker, P.E.; Roux, E.R.; Picha, K.S.; Toivola, B.; Waugh, S.; Kennedy, M.K. Experimental autoimmune encephalomyelitis is exacerbated by IL-1 alpha and suppressed by soluble IL-1 receptor. J. Immunol. 1991, 146, 2983–2989. [Google Scholar] [PubMed]
- Martin, D. Protective effect of the interleukin-1 receptor antagonist (IL-1ra) on experimental allergic encephalomyelitis in rats. J. Neuroimmunol. 1995, 61, 241–245. [Google Scholar] [CrossRef]
- Badovinac, V.; Mostarica-Stojković, M.; Dinarello, C.A.; Stošić-Grujičić, S. Interleukin-1 receptor antagonist suppresses experimental autoimmune encephalomyelitis (EAE) in rats by influencing the activation and proliferation of encephalitogenic cells. J. Neuroimmunol. 1998, 85, 87–95. [Google Scholar] [CrossRef]
- Aubé, B.; Lévesque, S.A.; Paré, A.; Chamma, É.; Kébir, H.; Gorina, R.; Lécuyer, M.-A.; Alvarez, J.I.; De Koninck, Y.; Engelhardt, B.; et al. Neutrophils mediate blood–spinal cord barrier disruption in demyelinating neuroinflammatory diseases. J. Immunol. 2014, 193, 2438–2454. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, S.A.; Paré, A.; Mailhot, B.; Bellver-Landete, V.; Kébir, H.; Lécuyer, M.-A.; Alvarez, J.I.; Prat, A.; Vaccari, J.P.R.; Keane, R.W.; et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1β–driven neuroinflammation during autoimmune encephalomyelitis in mice. J. Exp. Med. 2016, 213, 929–949. [Google Scholar] [CrossRef]
- Ching, S.; He, L.; Lai, W.; Quan, N. IL-1 type I receptor plays a key role in mediating the recruitment of leukocytes into the central nervous system. Brain Behav. Immun. 2005, 19, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Paré, A.; Mailhot, B.; Lévesque, S.A.; Juzwik, C.; Ignatius Arokia Doss, P.M.; Lécuyer, M.-A.; Prat, A.; Rangachari, M.; Fournier, A.; Lacroix, S. IL-1β enables CNS access to CCR2 hi monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1194–E1203. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Powell, N.; Zhang, H.; Belevych, N.; Ching, S.; Chen, Q.; Sheridan, J.; Whitacre, C.; Quan, N. Endothelial IL-1R1 is a critical mediator of EAE pathogenesis. Brain Behav. Immun. 2011, 25, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Hauptmann, J.; Johann, L.; Marini, F.; Kitic, M.; Colombo, E.; Mufazalov, I.A.; Krueger, M.; Karram, K.; Moos, S.; Wanke, F.; et al. Interleukin-1 promotes autoimmune neuroinflammation by suppressing endothelial heme oxygenase-1 at the blood–brain barrier. Acta Neuropathol. 2020, 140, 549–567. [Google Scholar] [CrossRef]
- Comer, A.L.; Carrier, M.; Tremblay, M.-È.; Cruz-Martín, A. The Inflamed brain in schizophrenia: The convergence of genetic and environmental risk factors that lead to uncontrolled neuroinflammation. Front. Cell. Neurosci. 2020, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.E.; Walker, A.K.; Weickert, C.S. Neuroinflammation in schizophrenia: The role of nuclear factor kappa B. Transl. Psychiatry 2021, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Lesh, T.A.; Careaga, M.; Rose, D.R.; McAllister, A.K.; Van de Water, J.; Carter, C.S.; Ashwood, P. Cytokine alterations in first-episode schizophrenia and bipolar disorder: Relationships to brain structure and symptoms. J. Neuroinflamm. 2018, 15, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Asmari, A.; Khan, M.W. Inflammation and schizophrenia: Alterations in cytokine levels and perturbation in antioxidative defense systems. Hum. Exp. Toxicol. 2014, 33, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Katila, H.; Appelberg, B.; Hurme, M.; Rimón, R. Plasma levels of interleukin-1β and interleukin-6 in schizophrenia, other psychoses, and affective disorders. Schizophr. Res. 1994, 12, 29–34. [Google Scholar] [CrossRef]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Peng, W.; Wang, J.; Zhou, W.; Zhou, Y.; Ying, B. Plasma levels of IL-1Ra are associated with schizophrenia. Psychiatry Clin. Neurosci. 2018, 73, pcn.12794. [Google Scholar] [CrossRef]
- Potvin, S.; Stip, E.; Sepehry, A.A.; Gendron, A.; Bah, R.; Kouassi, E. Inflammatory cytokine alterations in schizophrenia: A systematic quantitative review. Biol. Psychiatry 2008, 63, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Izumi, R.; Hino, M.; Wada, A.; Nagaoka, A.; Kawamura, T.; Mori, T.; Sainouchi, M.; Kakita, A.; Kasai, K.; Kunii, Y.; et al. Detailed postmortem profiling of inflammatory mediators expression revealed post-inflammatory alternation in the superior temporal gyrus of schizophrenia. Front. Psychiatry 2021, 12, 653821. [Google Scholar] [CrossRef]
- Pandey, G.N.; Ren, X.; Rizavi, H.S.; Zhang, H. Proinflammatory cytokines and their membrane-bound receptors are altered in the lymphocytes of schizophrenia patients. Schizophr. Res. 2015, 164, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Söderlund, J.; Schröder, J.; Nordin, C.; Samuelsson, M.; Walther-Jallow, L.; Karlsson, H.; Erhardt, S.; Engberg, G. Activation of brain interleukin-1β in schizophrenia. Mol. Psychiatry 2009, 14, 1069–1071. [Google Scholar] [CrossRef] [Green Version]
- Frydecka, D.; Krzystek-Korpacka, M.; Lubeiro, A.; Stramecki, F.; Stańczykiewicz, B.; Beszłej, J.A.; Piotrowski, P.; Kotowicz, K.; Szewczuk-Bogusławska, M.; Pawlak-Adamska, E.; et al. Profiling inflammatory signatures of schizophrenia: A cross-sectional and meta-analysis study. Brain Behav. Immun. 2018, 71, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Barak, V.; Barak, Y.; Levine, J.; Nisman, B.; Roisman, I. Changes in Interleukin-1β and soluble Interleukin-2 receptor levels in CSF and serum of schizophrenic patients. J. Basic Clin. Physiol. Pharmacol. 1995, 6, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Costantini, E.; Greig, N.H. Cytokine imbalance in schizophrenia. From research to clinic: Potential implications for treatment. Front. Psychiatry 2021, 12, 5362574. [Google Scholar] [CrossRef]
- Vezzani, A.; Conti, M.; De Luigi, A.; Ravizza, T.; Moneta, D.; Marchesi, F.; De Simoni, M.G. Interleukin-1β immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: Functional evidence for enhancement of electrographic seizures. J. Neurosci. 1999, 19, 5054–5065. [Google Scholar] [CrossRef] [Green Version]
- Dubé, C.; Vezzani, A.; Behrens, M.; Bartfai, T.; Baram, T.Z. Interleukin-1β contributes to the generation of experimental febrile seizures. Ann. Neurol. 2005, 57, 152–155. [Google Scholar] [CrossRef] [Green Version]
- Kostic, D.; Carlson, R.; Henke, D.; Rohn, K.; Tipold, A. Evaluation of IL-1β levels in epilepsy and traumatic brain injury in dogs. BMC Neurosci. 2019, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- De Simoni, M.G.; Perego, C.; Ravizza, T.; Moneta, D.; Conti, M.; Marchesi, F.; De Luigi, A.; Garattini, S.; Vezzani, A. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur. J. Neurosci. 2000, 12, 2623–2633. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Ravizza, T.; Vezzani, A. Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience 2006, 137, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iori, V.; Iyer, A.M.; Ravizza, T.; Beltrame, L.; Paracchini, L.; Marchini, S.; Cerovic, M.; Hill, C.; Ferrari, M.; Zucchetti, M.; et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol. Dis. 2017, 99, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Moneta, D.; Conti, M.; Richichi, C.; Ravizza, T.; De Luigi, A.; De Simoni, M.G.; Sperk, G.; Andell-Jonsson, S.; Lundkvist, J.; et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11534–11539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auvin, S.; Shin, D.; Mazarati, A.; Sankar, R. Inflammation induced by LPS enhances epileptogenesis in immature rat and may be partially reversed by IL1RA. Epilepsia 2010, 51, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilena, R.; Mauri, E.; Aronica, E.; Bernasconi, P.; Bana, C.; Cappelletti, C.; Carrabba, G.; Ferrero, S.; Giorda, R.; Guez, S.; et al. Therapeutic effect of Anakinra in the relapsing chronic phase of febrile infection–related epilepsy syndrome. Epilepsia Open 2019, 4, 344–350. [Google Scholar] [CrossRef]
- Lai, Y.; Muscal, E.; Wells, E.; Shukla, N.; Eschbach, K.; Hyeong Lee, K.; Kaliakatsos, M.; Desai, N.; Wickström, R.; Viri, M.; et al. Anakinra usage in febrile infection related epilepsy syndrome: An international cohort. Ann. Clin. Transl. Neurol. 2020, 7, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Kuraishi, Y.; Yabuuchi, K.; Yamazaki, A.; Satoh, M. Induction of Interleukin-1β mRNA in rat brain after transient forebrain ischemia. J. Neurochem. 1992, 58, 390–392. [Google Scholar] [CrossRef]
- Pinteaux, E.; Trotter, P.; Simi, A. Cell-specific and concentration-dependent actions of interleukin-1 in acute brain inflammation. Cytokine 2009, 45, 1–7. [Google Scholar] [CrossRef]
- Stroemer, R.P.; Rothwell, N.J. Exacerbation of ischemic brain damage by localized striatal injection of Interleukin-1β in the rat. J. Cereb. Blood Flow Metab. 1998, 18, 833–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColl, B.W.; Rothwell, N.J.; Allan, S.M. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via Interleukin-1- and neutrophil-dependent mechanisms. J. Neurosci. 2007, 27, 4403–4412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradillo, J.M.; Murray, K.N.; Coutts, G.A.; Moraga, A.; Oroz-Gonjar, F.; Boutin, H.; Moro, M.A.; Lizasoain, I.; Rothwell, N.J.; Allan, S.M. Reparative effects of interleukin-1 receptor antagonist in young and aged/co-morbid rodents after cerebral ischemia. Brain Behav. Immun. 2017, 61, 117–126. [Google Scholar] [CrossRef]
- Loddick, S.A.; Rothwell, N.J. Neuroprotective effects of human recombinant Interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J. Cereb. Blood Flow Metab. 1996, 16, 932–940. [Google Scholar] [CrossRef]
- Clausen, B.H.; Lambertsen, K.L.; Dagnæs-Hansen, F.; Babcock, A.A.; von Linstow, C.U.; Meldgaard, M.; Kristensen, B.W.; Deierborg, T.; Finsen, B. Cell therapy centered on IL-1Ra is neuroprotective in experimental stroke. Acta Neuropathol. 2016, 131, 775–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulmond, S.; Rothwell, N.J. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995, 671, 261–266. [Google Scholar] [CrossRef]
- Newell, E.A.; Todd, B.P.; Mahoney, J.; Pieper, A.A.; Ferguson, P.J.; Bassuk, A.G. Combined blockade of Interleukin-1 α and β signaling protects mice from cognitive dysfunction after traumatic brain injury. eNeuro 2018, 5, 0385-17. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.P.; Woll, A.W.; Wu, S.; Todd, B.P.; Hehr, N.; Hedberg-Buenz, A.; Anderson, M.G.; Newell, E.A.; Ferguson, P.J.; Mahajan, V.B.; et al. Modulation of post-traumatic immune response using the IL-1 receptor antagonist Anakinra for improved visual outcomes. J. Neurotrauma 2020, 37, 1463–1480. [Google Scholar] [CrossRef]
- Galea, J.; Ogungbenro, K.; Hulme, S.; Patel, H.; Scarth, S.; Hoadley, M.; Illingworth, K.; McMahon, C.J.; Tzerakis, N.; King, A.T.; et al. Reduction of inflammation after administration of interleukin-1 receptor antagonist following aneurysmal subarachnoid hemorrhage: Results of the Subcutaneous Interleukin-1Ra in SAH (SCIL-SAH) study. J. Neurosurg. 2018, 128, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.; Hulme, S.; Vail, A.; Heal, C.; Parry-Jones, A.R.; Scarth, S.; Hopkins, K.; Hoadley, M.; Allan, S.M.; Rothwell, N.J.; et al. SCIL-STROKE (Subcutaneous Interleukin-1 receptor antagonist in ischemic stroke). Stroke 2018, 49, 1210–1216. [Google Scholar] [CrossRef] [Green Version]
- Emsley, H.C.; Smith, C.J.; Georgiou, R.F.; Vail, A.; Hopkins, S.J.; Rothwell, N.J.; Tyrrell, P.J. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1366–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-I.; Ju, W.-K.; Choi, J.-H.; Kim, J.; Choi, E.-K.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.-S. Expression of cytokine genes and increased nuclear factor-kappa B activity in the brains of scrapie-infected mice. Mol. Brain Res. 1999, 73, 17–27. [Google Scholar] [CrossRef]
- Campbell, I.L.; Eddleston, M.; Kemper, P.; Oldstone, M.B.; Hobbs, M.V. Activation of cerebral cytokine gene expression and its correlation with onset of reactive astrocyte and acute-phase response gene expression in scrapie. J. Virol. 1994, 68, 2383–2387. [Google Scholar] [CrossRef] [Green Version]
- Kordek, R.; Nerurkar, V.R.; Liberski, P.P.; Isaacson, S.; Yanagihara, R.; Gajdusek, D.C. Heightened expression of tumor necrosis factor alpha, interleukin 1 alpha, and glial fibrillary acidic protein in experimental Creutzfeldt-Jakob disease in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9754–9758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.R.; Webb, J.; Rebus, S.; Walker, R.; Williams, A.; Fazakerley, J.K. Inducible cytokine gene expression in the brain in the ME7/CV mouse model of scrapie is highly restricted, is at a strikingly low level relative to the degree of gliosis and occurs only late in disease. J. Gen. Virol. 2003, 84, 2605–2611. [Google Scholar] [CrossRef]
- Walsh, D.T.; Betmouni, S.; Perry, V.H. Absence of detectable IL-1β production in Murine Prion disease: A model of chronic neurodegeneration. J. Neuropathol. Exp. Neurol. 2001, 60, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.; Schwarz, A.; Neidhold, S.; Burwinkel, M.; Riemer, C.; Simon, D.; Kopf, M.; Otto, M.; Baier, M. Role of Interleukin-1 in Prion Disease-Associated astrocyte activation. Am. J. Pathol. 2004, 165, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Tamgüney, G.; Giles, K.; Glidden, D.V.; Lessard, P.; Wille, H.; Tremblay, P.; Groth, D.F.; Yehiely, F.; Korth, C.; Moore, R.C.; et al. Genes contributing to prion pathogenesis. J. Gen. Virol. 2008, 89, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Investig. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooff, Y.; Man, S.M.; Aggio-Bruce, R.; Natoli, R.; Fernando, N. IL-1 Family members mediate cell death, inflammation and angiogenesis in retinal degenerative diseases. Front. Immunol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Dabouz, R.; Cheng, C.W.H.; Abram, P.; Omri, S.; Cagnone, G.; Sawmy, K.V.; Joyal, J.-S.; Desjarlais, M.; Olson, D.; Weil, A.G.; et al. An allosteric interleukin-1 receptor modulator mitigates inflammation and photoreceptor toxicity in a model of retinal degeneration. J. Neuroinflamm. 2020, 17, 359. [Google Scholar] [CrossRef]
- Tomasoni, R.; Morini, R.; Lopez-Atalaya, J.P.; Corradini, I.; Canzi, A.; Rasile, M.; Mantovani, C.; Pozzi, D.; Garlanda, C.; Mantovani, A.; et al. Lack of IL-1R8 in neurons causes hyperactivation of IL-1 receptor pathway and induces MECP2-dependent synaptic defects. eLife 2017, 6, e21735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigers, G.P.A.; Anderson, L.J.; Caffes, P.; Brandhuber, B.J. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1β. Nature 1997, 386, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreuder, H.; Tardif, C.; Trump-Kallmeyer, S.; Soffientini, A.; Sarubbi, E.; Akeson, A.; Bowlin, T.; Yanofsky, S.; Barrett, R.W. A new cytokine-receptor binding mode revealed by the crystal structure of the IL-1 receptor with an antagonist. Nature 1997, 386, 194–200. [Google Scholar] [CrossRef]
- Hou, J.; Townson, S.A.; Kovalchin, J.T.; Masci, A.; Kiner, O.; Shu, Y.; King, B.M.; Schirmer, E.; Golden, K.; Thomas, C.; et al. Design of a superior cytokine antagonist for topical ophthalmic use. Proc. Natl. Acad. Sci. USA 2013, 110, 3913–3918. [Google Scholar] [CrossRef] [Green Version]
- Vigers, G.P.A.; Dripps, D.J.; Edwards, C.K.; Brandhuber, B.J. X-ray crystal structure of a small antagonist peptide bound to interleukin-1 receptor type 1. J. Biol. Chem. 2000, 275, 36927–36933. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, N.; Kimura, T.; Arita, K.; Ariyoshi, M.; Ohnishi, H.; Yamamoto, T.; Zuo, X.; Maenaka, K.; Park, E.Y.; Kondo, N.; et al. The structural basis for receptor recognition of human interleukin-18. Nat. Commun. 2014, 5, 5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Remesh, S.G.; Hammel, M.; Pan, S.; Mahan, A.D.; Wang, S.; Wang, X. Functional relevance of Interleukin-1 receptor inter-domain flexibility for cytokine binding and signaling. Structure 2019, 27, 1296–1307.e5. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, L.; Jobling, S.A.; Paik, L.S.; McDonald, B.; Rosenwasser, L.J.; Auron, P.E. A point mutation uncouples human interleukin-1 beta biological activity and receptor binding. J. Biol. Chem. 1990, 265, 5922–5925. [Google Scholar] [CrossRef]
- MacDonald, H.R.; Wingfield, P.; Schmeissner, U.; Shaw, A.; Clore, G.M.; Gronenborn, A.M. Point mutations of human interleukin-1 with decreased receptor binding affinity. FEBS Lett. 1986, 209, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.J.; Bray, J.; Childs, J.D.; Vigers, G.P.A.; Brandhuber, B.J.; Thompson, R.C.; Eisenberg, S.P.; Skalicky, J.J. Mapping receptor binding sites in Interleukin (IL)-1 receptor antagonist and IL-1β by site-directed mutagenesis. J. Biol. Chem. 1995, 270, 11477–11483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.Y. Identification of potential small molecule allosteric modulator sites on IL-1R1 ectodomain using accelerated conformational sampling method. PLoS ONE 2015, 10, e0118671. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Kamm, K.; VanderKolk, W.; Lawrence, C.; Jonker, M.; Davis, A.T. The effect of traumatic brain injury upon the concentration and expression of Interleukin-1 and Interleukin-10 in the rat. J. Trauma Inj. Infect. Crit. Care 2006, 60, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Dalgard, C.L.; Cole, J.T.; Kean, W.S.; Lucky, J.J.; Sukumar, G.; McMullen, D.C.; Pollard, H.B.; Watson, W.D. The cytokine temporal profile in rat cortex after controlled cortical impact. Front. Mol. Neurosci. 2012, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.-C.; Liao, Y.-E.; Yang, L.-Y.; Wang, J.-Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.-Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.H.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Investig. 2007, 117, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Matousek, S.B.; Ghosh, S.; Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; O’Banion, M.K. Chronic IL-1β-Mediated neuroinflammation mitigates amyloid pathology in a mouse model of Alzheimer’s disease without inducing overt neurodegeneration. J. Neuroimmune Pharmacol. 2012, 7, 156–164. [Google Scholar] [CrossRef]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained Interleukin-1 overexpression exacerbates Tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J.M. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2008, 132, 288–295. [Google Scholar] [CrossRef]
- Moore, A.H.; Wu, M.; Shaftel, S.S.; Graham, K.A.; O’Banion, M.K. Sustained expression of interleukin-1β in mouse hippocampus impairs spatial memory. Neuroscience 2009, 164, 1484–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2013, 9, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Town, T.; Jeng, D.; Alexopoulou, L.; Tan, J.; Flavell, R.A. Microglia recognize double-stranded RNA via TLR3. J. Immunol. 2006, 176, 3804–3812. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Taylor, N.; Yao, X.; Bhattacharya, A. Mouse primary microglia respond differently to LPS and poly(I:C) in vitro. Sci. Rep. 2021, 11, 10447. [Google Scholar] [CrossRef]
- Kloss, C.U.A.; Bohatschek, M.; Kreutzberg, G.W.; Raivich, G. Effect of lipopolysaccharide on the morphology and integrin immunoreactivity of ramified microglia in the mouse brain and in cell culture. Exp. Neurol. 2001, 168, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 74. [Google Scholar] [CrossRef] [Green Version]
- Furst, D.E. Anakinra: Review of recombinant human interleukin-I receptor antagonist in the treatment of rheumatoid arthritis. Clin. Ther. 2004, 26, 1960–1975. [Google Scholar] [CrossRef]
- So, A.; De Smedt, T.; Revaz, S.; Tschopp, J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 2007, 9, R28. [Google Scholar] [CrossRef] [Green Version]
- Koné-Paut, I.; Galeotti, C. Anakinra for cryopyrin-associated periodic syndrome. Expert Rev. Clin. Immunol. 2014, 10, 7–18. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1–receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of Interleukin-1 blockade with Anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Kron, J.; Crawford, T.; Mihalick, V.; Bogun, F.; Jordan, J.H.; Koelling, T.; Syed, H.; Syed, A.; Iden, T.; Polly, K.; et al. Interleukin-1 blockade in cardiac sarcoidosis: Study design of the multimodality assessment of granulomas in cardiac sarcoidosis: Anakinra Randomized Trial (MAGiC-ART). J. Transl. Med. 2021, 19, 460. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.L.; Imazio, M.; Cremer, P.; Brucato, A.; Abbate, A.; Fang, F.; Insalaco, A.; LeWinter, M.; Lewis, B.S.; Lin, D.; et al. Phase 3 trial of Interleukin-1 Trap Rilonacept in recurrent pericarditis. N. Engl. J. Med. 2021, 384, 31–41. [Google Scholar] [CrossRef]
- Hoffman, H.M. Rilonacept for the treatment of cryopyrin-associated periodic syndromes (CAPS). Expert Opin. Biol. Ther. 2009, 9, 519–531. [Google Scholar] [CrossRef]
- Terkeltaub, R.; Sundy, J.S.; Schumacher, H.R.; Murphy, F.; Bookbinder, S.; Biedermann, S.; Wu, R.; Mellis, S.; Radin, A. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: Results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann. Rheum. Dis. 2009, 68, 1613–1617. [Google Scholar] [CrossRef]
- Lachmann, H.J.; Kone-Paut, I.; Kuemmerle-Deschner, J.B.; Leslie, K.S.; Hachulla, E.; Quartier, P.; Gitton, X.; Widmer, A.; Patel, N.; Hawkins, P.N. Use of Canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 2009, 360, 2416–2425. [Google Scholar] [CrossRef] [Green Version]
- Gattorno, M.; Obici, L.; Cattalini, M.; Tormey, V.; Abrams, K.; Davis, N.; Speziale, A.; Bhansali, S.G.; Martini, A.; Lachmann, H.J. Canakinumab treatment for patients with active recurrent or chronic TNF receptor-associated periodic syndrome (TRAPS): An open-label, phase II study. Ann. Rheum. Dis. 2017, 76, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Manubens, J.; Iglesias, E.; Anton, J. Canakinumab for the treatment of hyperimmunoglobulin D syndrome. Expert Rev. Clin. Immunol. 2019, 15, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Ozdogan, H.; Ugurlu, S. Canakinumab for the treatment of familial Mediterranean fever. Expert Rev. Clin. Immunol. 2017, 13, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, A.; Howard, C.P.; Botha, J.; Thuren, T. Effect of anti-IL-1β antibody (canakinumab) on insulin secretion rates in impaired glucose tolerance or type 2 diabetes: Results of a randomized, placebo-controlled trial. Diabetes Obes. Metab. 2012, 14, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Sfriso, P.; Bindoli, S.; Doria, A.; Feist, E.; Galozzi, P. Canakinumab for the treatment of adult-onset Still’s disease. Expert Rev. Clin. Immunol. 2020, 16, 129–138. [Google Scholar] [CrossRef]
- Tugal-Tutkun, I.; Pavesio, C.; De Cordoue, A.; Bernard-Poenaru, O.; Gül, A. Use of Gevokizumab in patients with Behçet’s disease Uveitis: An international, randomized, double-masked, placebo-controlled study and open-label extension study. Ocul. Immunol. Inflamm. 2018, 26, 1023–1033. [Google Scholar] [CrossRef]
- Sloan-Lancaster, J.; Abu-Raddad, E.; Polzer, J.; Miller, J.W.; Scherer, J.C.; De Gaetano, A.; Berg, J.K.; Landschulz, W.H. Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1 antibody, in patients with type 2 diabetes. Diabetes Care 2013, 36, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieber, T. Atopic dermatitis: An expanding therapeutic pipeline for a complex disease. Nat. Rev. Drug Discov. 2021, 21, 21–40. [Google Scholar] [CrossRef]
- Calverley, P.M.A.; Sethi, S.; Dawson, M.; Ward, C.K.; Finch, D.K.; Penney, M.; Newbold, P.; van der Merwe, R. A randomised, placebo-controlled trial of anti–interleukin-1 receptor 1 monoclonal antibody MEDI8968 in chronic obstructive pulmonary disease. Respir. Res. 2017, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.H.; Martel, J.R.; Sall, K.; Goldberg, D.F.; Abrams, M.; Rubin, J.; Sheppard, J.; Tauber, J.; Korenfeld, M.; Agahigian, J.; et al. Multicenter study of a novel topical Interleukin-1 receptor inhibitor, Isunakinra, in subjects with moderate to severe dry eye disease. Eye Contact Lens Sci. Clin. Pract. 2017, 43, 287–296. [Google Scholar] [CrossRef]
- Chauhan, D.; Vande Walle, L.; Lamkanfi, M. Therapeutic modulation of inflammasome pathways. Immunol. Rev. 2020, 297, 123–138. [Google Scholar] [CrossRef]
- Wannamaker, W.; Davies, R.; Namchuk, M.; Pollard, J.; Ford, P.; Ku, G.; Decker, C.; Charifson, P.; Weber, P.; Germann, U.A.; et al. (S)-1-((S)-2-{[1-(4-Amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R, 3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective Interleukin (IL)-converting Enzyme/Caspa. J. Pharmacol. Exp. Ther. 2007, 321, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Maroso, M.; Balosso, S.; Ravizza, T.; Iori, V.; Wright, C.I.; French, J.; Vezzani, A. Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 2011, 8, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, C.; Knight, A.; Nordström, D.; Pettersson, T.; Fransson, J.; Florin-Robertsson, E.; Pilström, B. Injection-site reactions upon Kineret (anakinra) administration: Experiences and explanations. Rheumatol. Int. 2012, 32, 295–299. [Google Scholar] [CrossRef] [Green Version]
- Sjöström, E.O.; Culot, M.; Leickt, L.; Åstrand, M.; Nordling, E.; Gosselet, F.; Kaiser, C. Transport study of interleukin-1 inhibitors using a human in vitro model of the blood-brain barrier. Brain Behav. Immun.-Health 2021, 16, 100307. [Google Scholar] [CrossRef]
- Cohen, S.B.; Proudman, S.; Kivitz, A.J.; Burch, F.X.; Donohue, J.P.; Burstein, D.; Sun, Y.-N.; Banfield, C.; Vincent, M.S.; Ni, L.; et al. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 2011, 13, R125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, M.; Bachmann, M.F.; Marsland, B.J. Averting inflammation by targeting the cytokine environment. Nat. Rev. Drug Discov. 2010, 9, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Yanofsky, S.D.; Baldwin, D.N.; Butler, J.H.; Holden, F.R.; Jacobs, J.W.; Balasubramanian, P.; Chinn, J.P.; Cwirla, S.E.; Peters-Bhatt, E.; Whitehorn, E.A.; et al. High affinity type I interleukin 1 receptor antagonists discovered by screening recombinant peptide libraries. Proc. Natl. Acad. Sci. USA 1996, 93, 7381–7386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiniou, C.; Sapieha, P.; Lahaie, I.; Hou, X.; Brault, S.; Beauchamp, M.; Leduc, M.; Rihakova, L.; Joyal, J.-S.; Nadeau, S.; et al. Development of a novel noncompetitive antagonist of IL-1 receptor. J. Immunol. 2008, 180, 6977–6987. [Google Scholar] [CrossRef] [PubMed]
- Klementiev, B.; Li, S.; Korshunova, I.; Dmytriyeva, O.; Pankratova, S.; Walmod, P.S.; Kjær, L.K.; Dahllöf, M.S.; Lundh, M.; Christensen, D.P.; et al. Anti-inflammatory properties of a novel peptide interleukin 1 receptor antagonist. J. Neuroinflamm. 2014, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock, S.A.; Pennington, L.D. Structure−brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Sarabu, R.; Cooper, J.P.; Cook, C.M.; Gillespie, P.; Perrotta, A.V.; Olson, G.L. Design and synthesis of small molecule interleukin-1 receptor antagonists based on a benzene template. Drug Des. Discov. 1998, 15, 191–198. [Google Scholar]
- Ahn, S.-H.; Lee, J.-K.; Kim, N.; Kim, S.-H.; Lee, S.; Jung, S.; Chay, K.-O.; Lee, T.-H. DPIE [2-(1,2-diphenyl-1H-indol-3-yl)ethanamine] augments pro-inflammatory cytokine production in IL-1β-stimulated primary human oral cells. Int. J. Mol. Sci. 2018, 19, 1835. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Jiang, S.; Pan, H.; Xu, A.; Wang, G.; Ma, C.; Shi, Z. Short hairpin RNA interference targeting interleukin 1 receptor type I in the paraventricular nucleus attenuates hypertension in rats. Pflügers Arch.-Eur. J. Physiol. 2018, 470, 439–448. [Google Scholar] [CrossRef]
- Lin, W.-P.; Lin, J.-H.; Cai, B.; Shi, J.-X.; Li, W.-J.; Choudhury, G.R.; Wu, S.-Q.; Wu, J.-Z.; Wu, H.-P.; Ke, Q.-F. Effect of adenovirus-mediated RNA interference of IL-1β expression on spinal cord injury in rats. Spinal Cord 2016, 54, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhu, S.; Zou, Y.; Wang, T.; Fu, X. Knockdown of IL-1β improves hypoxia–ischemia brain associated with IL-6 Up-regulation in cell and animal models. Mol. Neurobiol. 2015, 51, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Min, L.; Duan, H.; Shi, R.; Zhang, W.; Hong, S.; Tu, C. Short hairpin RNA (shRNA) of type 2 interleukin-1 receptor (IL1R2) inhibits the proliferation of human osteosarcoma U-2 OS cells. Med. Oncol. 2015, 32, 364. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Shiroshima, T.; Lee, S.-J.; Yasumura, M.; Uemura, T.; Chen, X.; Iwakura, Y.; Mishina, M. Interleukin-1 receptor accessory protein organizes neuronal synaptogenesis as a cell adhesion molecule. J. Neurosci. 2012, 32, 2588–2600. [Google Scholar] [CrossRef]
- Lianxu, C.; Hongti, J.; Changlong, Y. NF-κBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1β-induced and TNF-α-induced chondrocytes. Osteoarthr. Cartil. 2006, 14, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Zhao, J.; Zhu, G.; Huang, Y.; Jin, L. SiRNA directed against NF-κB inhibits mononuclear macrophage cells releasing proinflammatory cytokines in vitro. Mol. Med. Rep. 2017, 16, 9060–9066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, J.W.; Fleischman, A.; Al-Fayoumi, S.; Mascarenhas, J.O.; Yu, Q.; Agarwal, A. Inhibition of interleukin-1 receptor-associated kinase 1 (IRAK1) as a therapeutic strategy. Oncotarget 2018, 9, 33416–33439. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Entry | Resolution (Å) | R-Value (Work) | Pub Year | Description |
|---|---|---|---|---|
| 1G0Y | 3.00 | 0.223 | 2000 | IL-1R1 complexed with antagonist peptide AF10847 |
| 1IRA | 2.70 | 0.213 | 1997 | IL-1R1 complexed with IL-1Ra |
| 1ITB | 2.50 | 0.229 | 1997 | IL-1R1 complexed with IL-1β |
| 4DEP2 chains | 3.10 | 0.210 | 2012 | IL-1R1 complexed with IL-1β and IL-1RAcP |
| 4GAF | 2.15 | 0.215 | 2013 | IL-1R1 complexed EBI-005, a chimera of human IL-1β and IL-1Ra |
| Volume (Å3) | Surface (Å2) | Depth (Å) | Nº Residues | Hydrophobicity Ratio | HBA/HBD | Drug Score | |
|---|---|---|---|---|---|---|---|
| PO_0 | 1209.78 | 1641.79 | 26.31 | 55 | 0.37 | 86/28 | 0.82 |
| PO_1 | 575.29 | 1161.23 | 15.32 | 25 | 0.36 | 48/27 | 0.71 |
| PO_2 | 490.77 | 605.27 | 22.21 | 27 | 0.38 | 29/16 | 0.84 |
| PO_3 | 191.44 | 398.74 | 11.78 | 10 | 0.51 | 18/4 | 0.43 |
| PO_4 | 179.47 | 464.04 | 9.93 | 9 | 0.42 | 20/11 | 0.29 |
| Therapeutic Agent | Type | Target | Company | Therapeutic Indication |
|---|---|---|---|---|
| Anakinra | Recombinant form of human IL-1Ra | IL-1R1 | Amgen (now Swedish Orphan Biovitrum) | Rheumatoid arthritis 1 [217] Gout [218] CAPS 1 [219] Type 2 Diabetes [220] Cardiovascular disease [221,222] Giant Cell Arthritis (NCT02902731) 2 Subarachnoid Hemorrhage (NCT03249207) 2 Intracerebral Hemorrhage (NCT03737344, NCT04834388) 2 Multiple Sclerosis (NCT04025554) 2 |
| Rilonacept | Fusion protein of IL-1RAcP, IL-1RI and IgG-Fc | IL-1β IL-1α IL-1Ra | Regeneron | Recurrent Pericarditis 1 [223] CAPS 1 [224] Gout [225] |
| Canakinumab | Human IgG1 monoclonal antibody | IL-1β | Novartis | CAPS 1 [226] TRAPS 1 [227] HIDS/MKD 1 [228] FMF 1 [229] Type 2 Diabetes [230] Still’s disease 1 [231] AD (NCT04795466) 2 |
| Gevokizumab | Human IgG2 monoclonal antibody | IL-1β | Xoma | Behcet’s Uveitis [232] |
| LY2189102 | Human IgG1 monoclonal antibody | IL-1β | Lilly | Type 2 diabetes [233] Rheumatoid arthritis (NCT00380744) 2 |
| Bermekimab (MABp1) | Human IgG1 monoclonal antibody | IL-1α | XBiotech | Atopic Dermatitis [234] |
| MEDI-8968 (AMG108) | Human IgG2 monoclonal antibody | IL-1R1 | MedImmune | COPD [235] |
| Isunakinra (EBI-005) | Human IL-1β and IL-1Ra chimeric protein | IL-1R1 | Eleven Biotherapeutics | Dry eye disease [236] Solid Tumors (NCT04121442) 2 |
| AF10847 | peptide | IL-1R1 | Array BioPharma | -- |
| rytvela | peptide | IL-1R1 | Elim Biopharmaceuticals | -- |
| Inzomelid | Small molecule | NLRP3 inflammasome | Roche (previously Inflazome) | CAPS [237] |
| Belnacasan (VX-765) | Small molecule | Caspase-1 | Vertex Pharmaceuticals | Rheumatoid arthritis [238] Epilepsy [239] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luís, J.P.; Simões, C.J.V.; Brito, R.M.M. The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders. Int. J. Mol. Sci. 2022, 23, 1731. https://doi.org/10.3390/ijms23031731
Luís JP, Simões CJV, Brito RMM. The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders. International Journal of Molecular Sciences. 2022; 23(3):1731. https://doi.org/10.3390/ijms23031731
Chicago/Turabian StyleLuís, João P., Carlos J. V. Simões, and Rui M. M. Brito. 2022. "The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders" International Journal of Molecular Sciences 23, no. 3: 1731. https://doi.org/10.3390/ijms23031731
APA StyleLuís, J. P., Simões, C. J. V., & Brito, R. M. M. (2022). The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders. International Journal of Molecular Sciences, 23(3), 1731. https://doi.org/10.3390/ijms23031731