
Divalent Metal Transporter 1 Knock-Down Modulates IL-1β Mediated Pancreatic Beta-Cell Pro-Apoptotic Signaling Pathways through the Autophagic Machinery


Abstract
1. Introduction
2. Results and Discussion
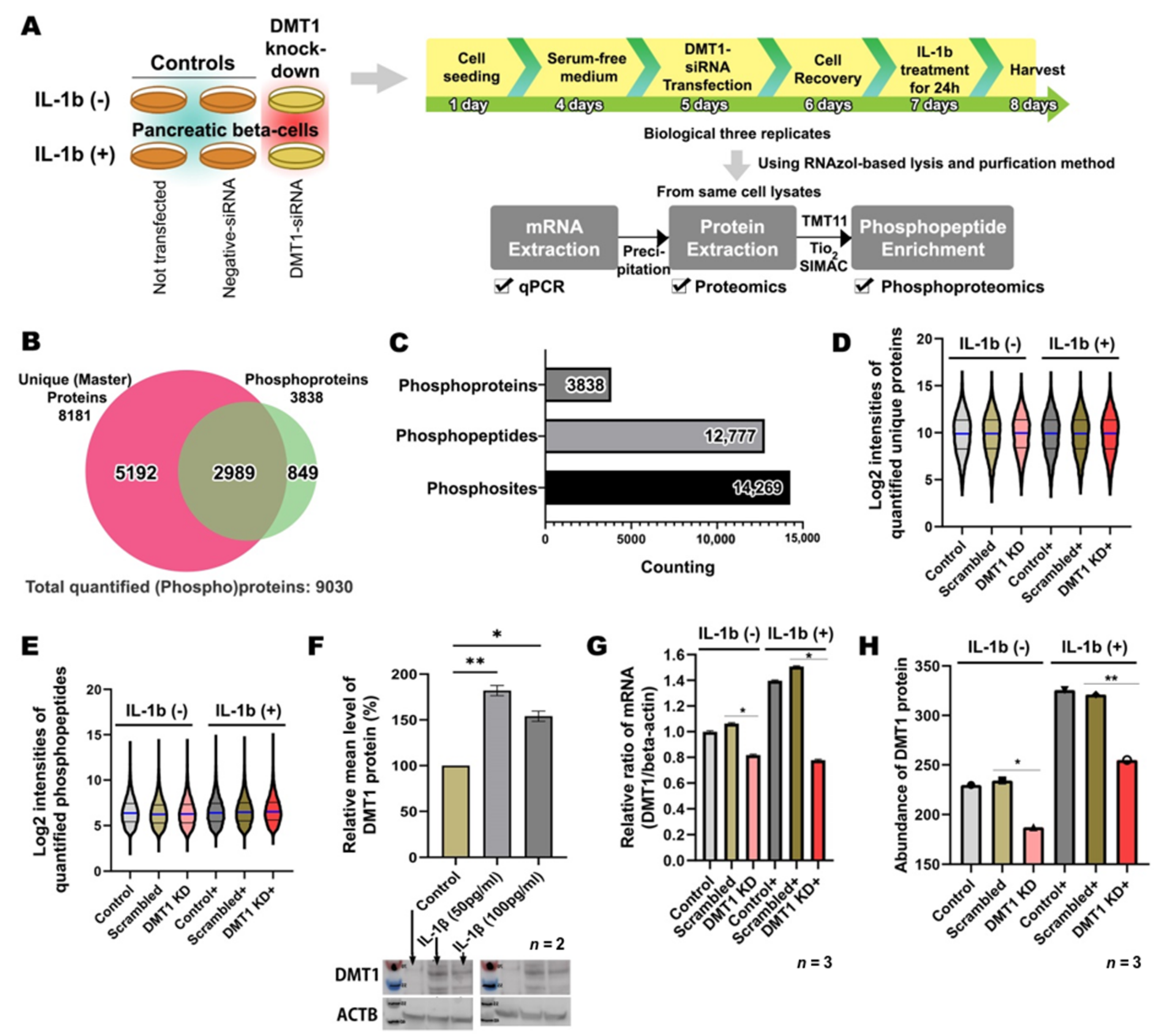
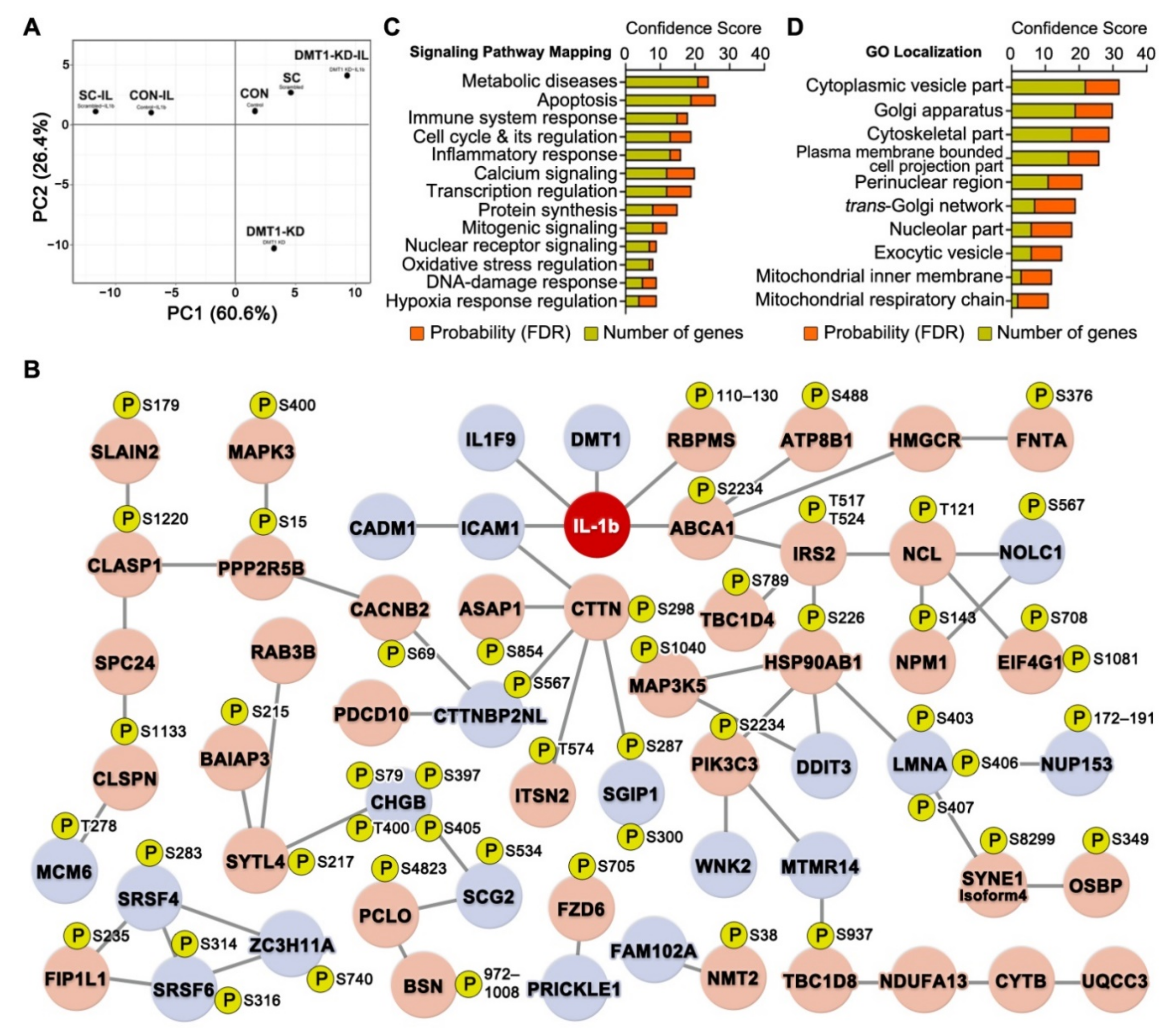
2.1. Overview of High-Throughput Analysis after DMT1 Knock-Down in Response to IL-1ß in INS-1E Cells
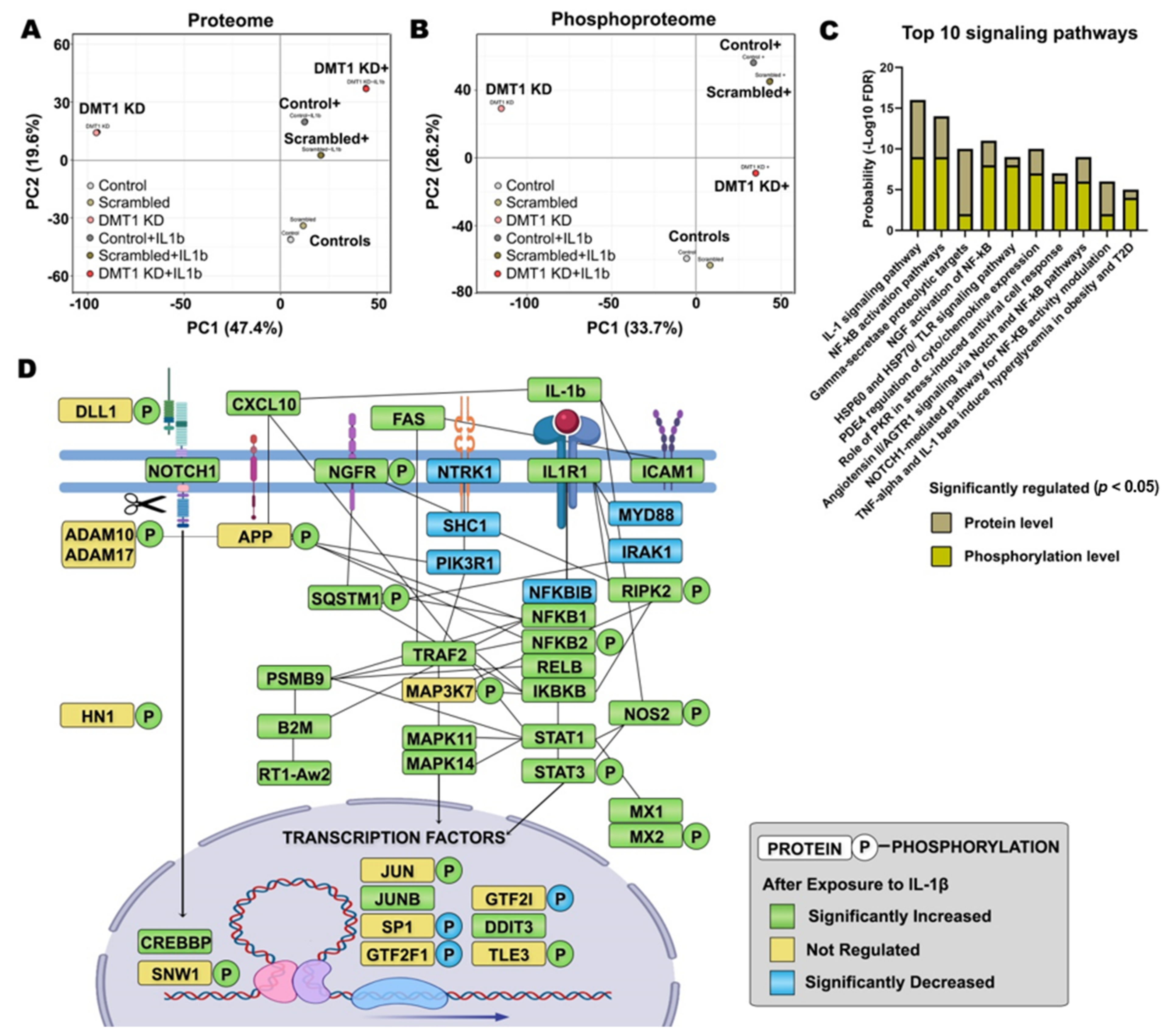
2.2. IL-1β Activates Inflammation and β-Cell Death through Pro-Apoptotic Signaling Pathways
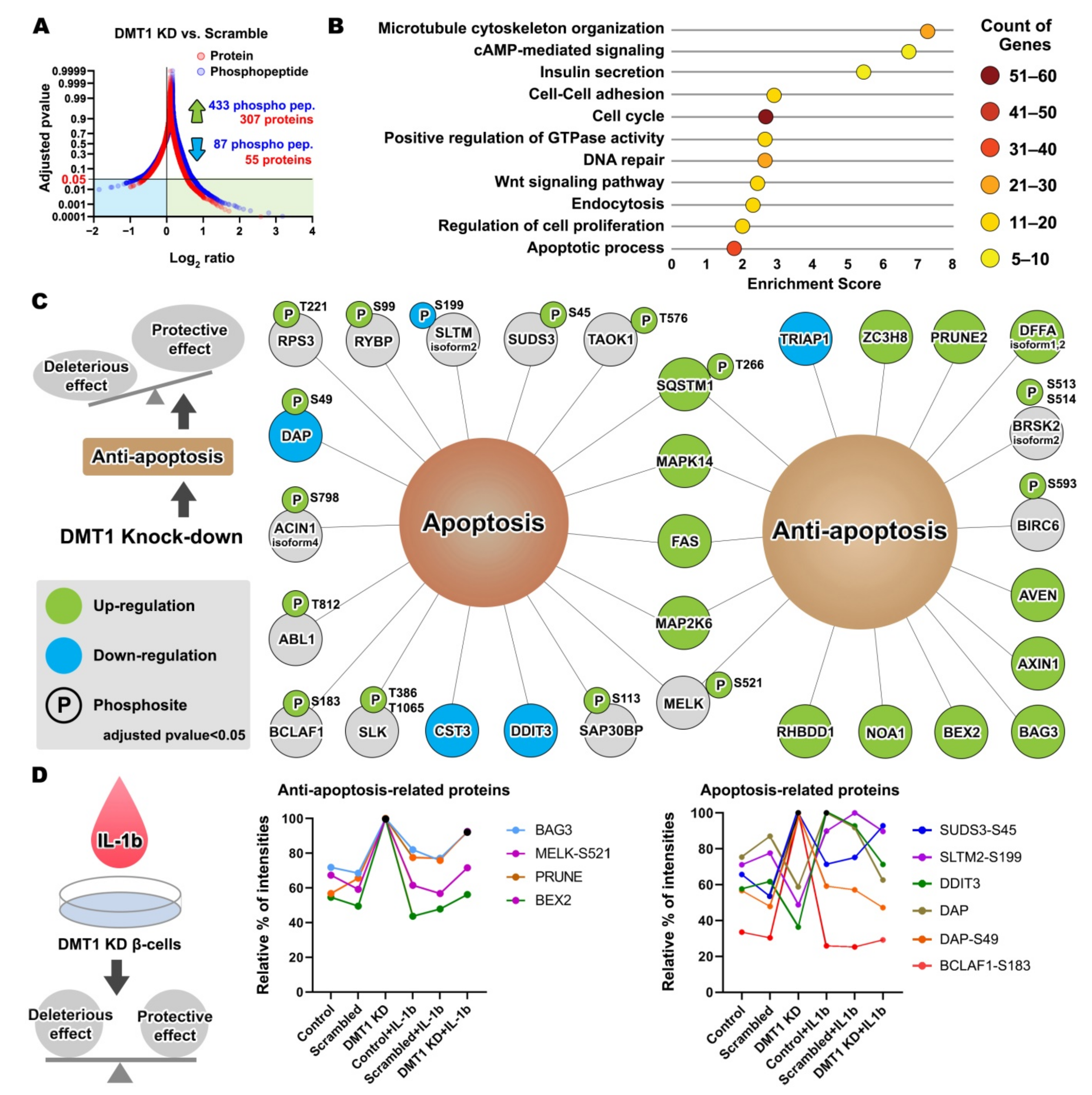
2.3. DMT1 Knock-Down Promotes Anti-Apoptotic Signaling in β-Cells
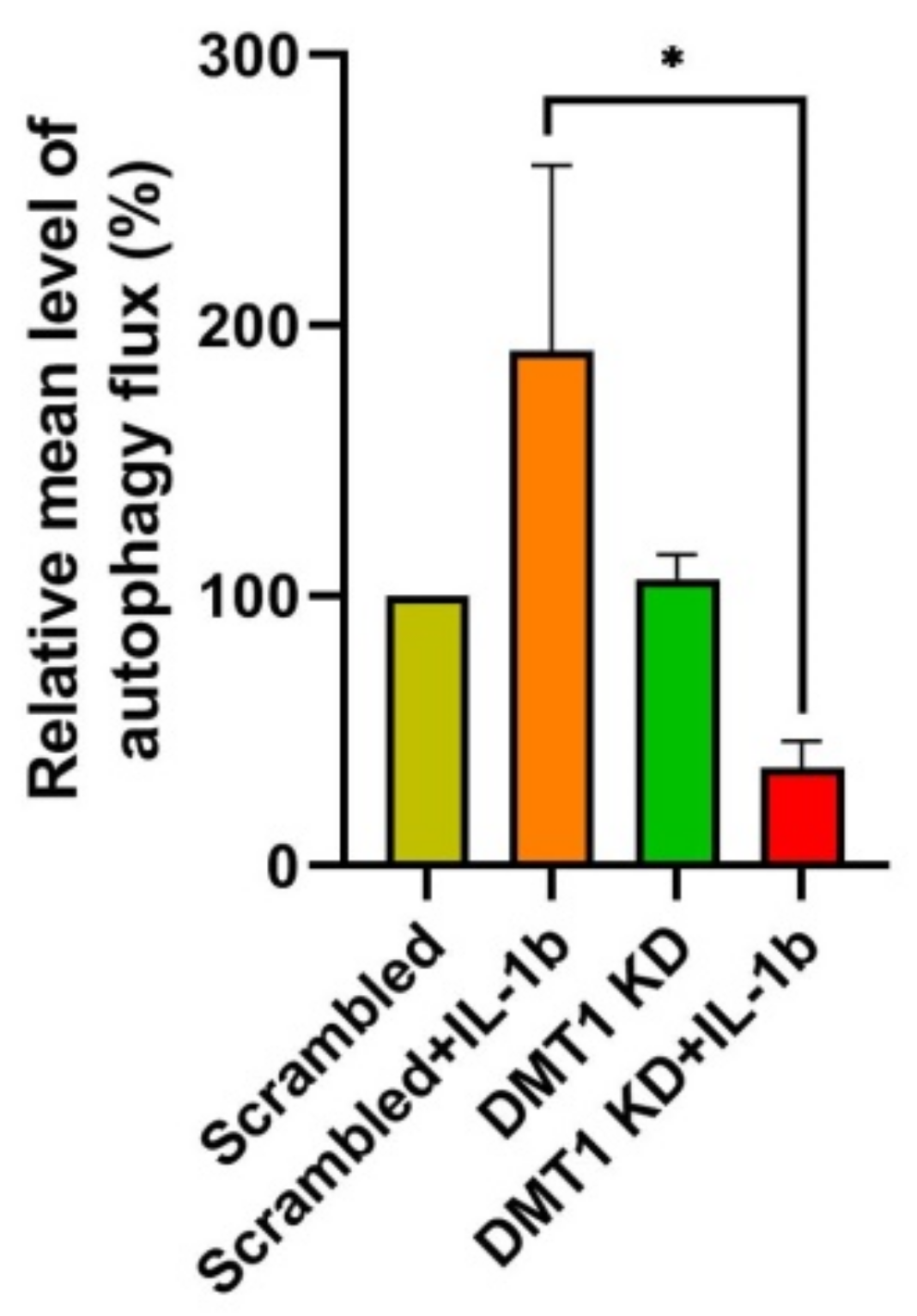
2.4. DMT1 Silencing Modulates the Balance between Apoptosis and Anti-Apoptosis Following Decreased Autophagic Flux in IL-1β Exposed β-Cells
2.5. DMT1 Silencing Reverts the IL-1β-Induced Responses towards the Normal State
2.6. Validation of Phosphoproteins Potentially Protective against β-Cell Inflammatory Response Using Parallel Reaction Monitoring (PRM)
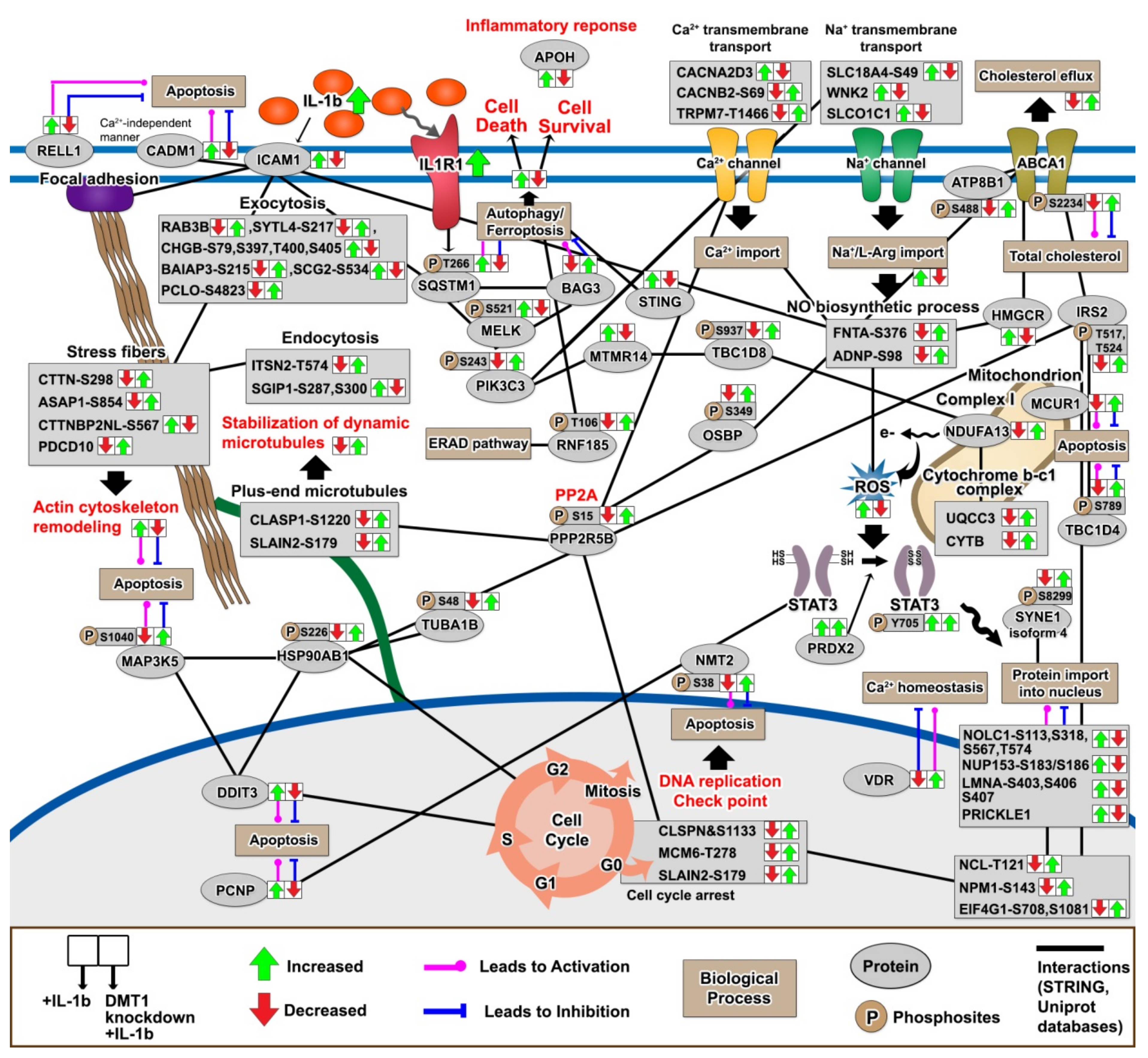
2.7. The Protective Protein Interaction Network Reveals the Alteration of New Phosphoproteins in Relation to β-Cell Anti-Inflammation and Anti-Apoptosis
3. Materials and Methods
3.1. Reagents
3.2. Cell Culture
3.3. mRNA Extraction
3.4. Real-Time PCR
3.5. Western Blot Analysis
3.6. Fluorescence Spectrophotometric Assay for Autophagic Flux
3.7. Protein Extraction, Sample Preparation, and TMT Labeling
3.8. Enrichment of Phosphorylated Peptides
3.9. Liquid Chromatography-Tandem Mass Spectrometry Analysis
3.10. Protein Identification and Quantification
3.11. Bioinformatics Analysis of Proteomic and Phosphoproteomic Data
3.12. Validation of Selected Protein Phosphorylation Using Parallel-Reaction Monitoring (PRM) Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| T1D | type 1 diabetes |
| T2D | type 2 diabetes |
| IL-1β | interleukin-1 beta |
| TNF | tumor necrosis factor |
| IFN | interferon |
| NF-κB | nuclear factor kappa B |
| ROS | reactive oxygen species |
| NGF | nerve growth factor |
| TLR | toll-like-receptor |
| ERAD | endoplasmic reticulum-associated degradation |
| JNK | c-Jun N-terminal kinase |
| siRNA | small interfering RNA |
| ER | endoplasmic reticulum |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| TMT | tandem mass tags |
| INS-1E | insulin-producing β-cell line |
| FDR | false discovery rate |
| mRNA | messenger RNA |
| ADAM | disintegrin and metalloproteinase domain-containing protein |
| CON | un-transfected control β-cells |
| SC | scrambled siRNA transfected control β-cells |
| DMT1-KD | control β-cells transfected with three different siRNAs targeting DMT1 |
| CON-IL | un-transfected β-cells exposed to IL-1β |
| SC-IL | scrambled siRNA transfected β-cells exposed to IL-1β |
| DMT1-KD-IL | β-cells exposed to IL-1β transfected with three different siRNAs targeting DMT1 |
References
- Hansen, J.B.; Tonnesen, M.F.; Madsen, A.N.; Hagedorn, P.H.; Friberg, J.; Grunnet, L.G.; Heller, R.S.; Nielsen, A.S.; Storling, J.; Baeyens, L.; et al. Divalent Metal Transporter 1 Regulates Iron-Mediated ROS and Pancreatic beta Cell Fate in Response to Cytokines. Cell Metab. 2012, 16, 449–461. [Google Scholar] [CrossRef]
- Backe, M.B.; Moen, I.W.; Ellervik, C.; Hansen, J.B.; Mandrup-Poulsen, T. Iron Regulation of Pancreatic Beta-Cell Functions and Oxidative Stress. Annu. Rev. Nutr. 2016, 36, 241–273. [Google Scholar] [CrossRef]
- Simcox, J.A.; McClain, D.A. Iron and diabetes risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef]
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Jones, D.L.; Kushner, J.P.; McClain, D.A. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology 2004, 145, 5305–5312. [Google Scholar] [CrossRef]
- Merkel, P.A.; Simonson, D.C.; Amiel, S.A.; Plewe, G.; Sherwin, R.S.; Pearson, H.A.; Tamborlane, W.V. Insulin resistance and hyperinsulinemia in patients with thalassemia major treated by hypertransfusion. N. Engl. J. Med. 1988, 318, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Dmochowski, K.; Finegood, D.T.; Francombe, W.; Tyler, B.; Zinman, B. Factors determining glucose tolerance in patients with thalassemia major. J. Clin. Endocrinol. Metab. 1993, 77, 478–483. [Google Scholar] [CrossRef]
- Wang, X.; Fang, X.; Wang, F. Pleiotropic actions of iron balance in diabetes mellitus. Rev. Endocr. Metab. Disord. 2015, 16, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Zinselmeyer, B.H.; Vomund, A.N.; Saunders, B.T.; Johnson, M.W.; Carrero, J.A.; Unanue, E.R. The resident macrophages in murine pancreatic islets are constantly probing their local environment, capturing beta cell granules and blood particles. Diabetologia 2018, 61, 1374–1383. [Google Scholar] [CrossRef]
- Donath, M.Y.; Storling, J.; Berchtold, L.A.; Billestrup, N.; Mandrup-Poulsen, T. Cytokines and beta-cell biology: From concept to clinical translation. Endocr. Rev. 2008, 29, 334–350. [Google Scholar] [CrossRef]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e455. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Lee, K.; Maxwell, E.L.; Liang, C.; Laybutt, D.R. Macrophage alterations in islets of obese mice linked to beta cell disruption in diabetes. Diabetologia 2019, 62, 993–999. [Google Scholar] [CrossRef]
- Azevedo-Martins, A.K.; Lortz, S.; Lenzen, S.; Curi, R.; Eizirik, D.L.; Tiedge, M. Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-kappaB activation in insulin-producing cells. Diabetes 2003, 52, 93–101. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.B.; Moen, I.W.; Mandrup-Poulsen, T. Iron: The hard player in diabetes pathophysiology. Acta Physiol. 2014, 210, 717–732. [Google Scholar] [CrossRef]
- Kang, T.; Jensen, P.; Huang, H.; Lund Christensen, G.; Billestrup, N.; Larsen, M.R. Characterization of the Molecular Mechanisms Underlying Glucose Stimulated Insulin Secretion from Isolated Pancreatic beta-cells Using Post-translational Modification Specific Proteomics (PTMomics). Mol. Cell. Proteom. 2018, 17, 95–110. [Google Scholar] [CrossRef]
- Kang, T.; Boland, B.B.; Alarcon, C.; Grimsby, J.S.; Rhodes, C.J.; Larsen, M.R. Proteomic Analysis of Restored Insulin Production and Trafficking in Obese Diabetic Mouse Pancreatic Islets Following Euglycemia. J. Proteome Res. 2019. [Google Scholar] [CrossRef]
- Kang, T.; Boland, B.B.; Jensen, P.; Alarcon, C.; Nawrocki, A.; Grimsby, J.S.; Rhodes, C.J.; Larsen, M.R. Characterization of signaling pathways associated with pancreatic beta-cell adaptive flexibility in compensation of obesity-linked diabetes in db/db mice. Mol. Cell. Proteom. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Jensen, P.; Solovyeva, V.; Brewer, J.R.; Larsen, M.R. Dynamic Changes in the Protein Localization in the Nuclear Environment in Pancreatic beta-Cell after Brief Glucose Stimulation. J. Proteome Res. 2018, 17, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Taus, T.; Kocher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and Confident Phosphorylation Site Localization Using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. [Google Scholar] [CrossRef]
- Bendtzen, K.; Mandruppoulsen, T.; Nerup, J.; Nielsen, J.H.; Dinarello, C.A.; Svenson, M. Cytotoxicity of Human Pi-7 Interleukin-1 for Pancreatic-Islets of Langerhans. Science 1986, 232, 1545–1547. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Silver, R.; Aldhahi, W.; Cai, D.S.; Tatro, E.; Lee, J.; Shoelson, S.E. Use of Salsalate to Target Inflammation in the Treatment of Insulin Resistance and Type 2 Diabetes. Clin. Transl. Sci. 2008, 1, 36–43. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, A.; Choksi, S.; Li, W.; Li, T.; Zhang, X.M.; Liu, Z.G. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016, 26, 886–900. [Google Scholar] [CrossRef] [PubMed]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. 2010, 45, 146–169. [Google Scholar] [CrossRef]
- Chami, L.; Buggia-Prevot, V.; Duplan, E.; Del Prete, D.; Chami, M.; Peyron, J.F.; Checler, F. Nuclear factor-kappaB regulates betaAPP and beta- and gamma-secretases differently at physiological and supraphysiological Abeta concentrations. J. Biol. Chem. 2012, 287, 24573–24584. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Marjaux, E.; Imhof, A.; De Strooper, B.; Haass, C.; Lichtenthaler, S.F. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J. Biol. Chem. 2007, 282, 11982–11995. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Denroche, H.C.; Ehses, J.A.; Verchere, C.B. Differential Activation of Innate Immune Pathways by Distinct Islet Amyloid Polypeptide (IAPP) Aggregates. J. Biol. Chem. 2016, 291, 8908–8917. [Google Scholar] [CrossRef]
- Abedini, A.; Plesner, A.; Cao, P.; Ridgway, Z.; Zhang, J.H.; Tu, L.H.; Middleton, C.T.; Chao, B.; Sartori, D.J.; Meng, F.L.; et al. Time-resolved studies define the nature of toxic IAPP intermediates, providing insight for anti-amyloidosis therapeutics. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Bash, J.; Zong, W.X.; Banga, S.; Rivera, A.; Ballard, D.W.; Ron, Y.; Gelinas, C. Rel/NF-kappaB can trigger the Notch signaling pathway by inducing the expression of Jagged1, a ligand for Notch receptors. EMBO J. 1999, 18, 2803–2811. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef]
- Valenti, L.; Mendoza, R.M.; Rametta, R.; Maggioni, M.; Kitajewski, C.; Shawber, C.J.; Pajvani, U.B. Hepatic Notch Signaling Correlates With Insulin Resistance and Nonalcoholic Fatty Liver Disease. Diabetes 2013, 62, 4052–4062. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Shawber, C.J.; Samuel, V.T.; Birkenfeld, A.L.; Shulman, G.I.; Kitajewski, J.; Accili, D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med. 2011, 17, 961–U978. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Qiang, L.; Kangsamaksin, T.; Kitajewski, J.; Ginsberg, H.N.; Accili, D. Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat. Med. 2013, 19, 1055. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, A.; Zhu, C.Y.; Sussel, L.; Pajvani, U.B. Notch signaling dynamically regulates adult beta cell proliferation and maturity. J. Clin. Investig. 2019, 129, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Bozkulak, E.C.; Weinmaster, G. Selective Use of ADAM10 and ADAM17 in Activation of Notch1 Signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [PubMed]
- Groot, A.J.; Vooijs, M.A. The Role of Adams in Notch Signaling. Notch Signal. Embryol. Cancer 2012, 727, 15–36. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Chung, K.W.; Oh, S.H.; Yun, J.W.; Im, S.H.; Lee, M.K.; Kim, K.W.; Lee, M.S. Essential role for signal transducer and activator of transcription-1 in pancreatic beta-cell death and autoimmune type 1 diabetes of nonobese diabetic mice. Diabetes 2007, 56, 2561–2568. [Google Scholar] [CrossRef][Green Version]
- Hu, H.X.; Nan, J.L.; Sun, Y.; Zhu, D.; Xiao, C.C.; Wang, Y.P.; Zhu, L.L.; Wu, Y.; Zhao, J.; Wu, R.R.; et al. Electron leak from NDUFA13 within mitochondrial complex I attenuates ischemia-reperfusion injury via dimerized STAT3. Proc. Natl. Acad. Sci. USA 2017, 114, 11908–11913. [Google Scholar] [CrossRef]
- Mattson, M.P.; Meffert, M.K. Roles for NF-kappa B in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006, 13, 852–860. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappa B collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Bollyky, P.L.; Bice, J.B.; Sweet, I.R.; Falk, B.A.; Gebe, J.A.; Clark, A.E.; Gersuk, V.H.; Aderem, A.; Hawn, T.R.; Nepom, G.T. The Toll-Like Receptor Signaling Molecule Myd88 Contributes to Pancreatic Beta-Cell Homeostasis in Response to Injury. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef]
- Look, D.C.; Pelletier, M.R.; Tidwell, R.M.; Roswit, W.T.; Holtzman, M.J. Stat1 depends on transcriptional synergy with Sp1. J. Biol. Chem. 1995, 270, 30264–30267. [Google Scholar] [CrossRef]
- Zhang, X.K.; Wrzeszczynska, M.H.; Horvath, C.M.; Darnell, J.E. Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol. Cell. Biol. 1999, 19, 7138–7146. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; He, L.Z.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [PubMed]
- Nejentsev, S.; Guja, C.; McCormack, R.; Cooper, J.; Howson, J.M.M.; Nutland, S.; Rance, H.; Walker, N.; Undlien, D.; Ronningen, K.S.; et al. Association of intercellular adhesion molecule-1 gene with type 1 diabetes. Lancet 2003, 362, 1723–1724. [Google Scholar] [CrossRef]
- Yagi, N.; Yokono, K.; Amano, K.; Nagata, M.; Tsukamoto, K.; Hasegawa, Y.; Yoneda, R.; Okamoto, N.; Moriyama, H.; Miki, M.; et al. Expression of Intercellular-Adhesion Molecule-1 on Pancreatic Beta-Cells Accelerates Beta-Cell Destruction by Cytotoxic T-Cells in Murine Autoimmune Diabetes. Diabetes 1995, 44, 744–752. [Google Scholar] [CrossRef]
- Vives-Pi, M.; Rodriguez-Fernandez, S.; Pujol-Autonell, I. How apoptotic beta-cells direct immune response to tolerance or to autoimmune diabetes: A review. Apoptosis 2015, 20, 263–272. [Google Scholar] [CrossRef]
- Chau, B.N.; Cheng, E.H.; Kerr, D.A.; Hardwick, J.M. Aven, a novel inhibitor of caspase activation, binds Bcl-xL and Apaf-1. Mol. Cell 2000, 6, 31–40. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, X.; Fok, K.L.; Li, S.; Zhang, X.; Miao, S.; Zong, S.; Koide, S.S.; Chan, H.C.; Wang, L. A novel member of the Rhomboid family, RHBDD1, regulates BIK-mediated apoptosis. Cell. Mol. Life Sci. 2008, 65, 3822–3829. [Google Scholar] [CrossRef]
- Zou, Q.; Gang, K.; Yang, Q.; Liu, X.; Tang, X.; Lu, H.; He, J.; Luo, L. The CCCH-type zinc finger transcription factor Zc3h8 represses NF-kappaB-mediated inflammation in digestive organs in zebrafish. J. Biol. Chem. 2018, 293, 11971–11983. [Google Scholar] [CrossRef]
- Heidler, J.; Al-Furoukh, N.; Kukat, C.; Salwig, I.; Ingelmann, M.E.; Seibel, P.; Kruger, M.; Holtz, J.; Wittig, I.; Braun, T.; et al. Nitric oxide-associated protein 1 (NOA1) is necessary for oxygen-dependent regulation of mitochondrial respiratory complexes. J. Biol. Chem. 2011, 286, 32086–32093. [Google Scholar] [CrossRef]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3 beta and beta-catenin and promotes GSK-3 beta-dependent phosphorylation of beta-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Biechele, T.L.; Kulikauskas, R.M.; Toroni, R.A.; Lucero, O.M.; Swift, R.D.; James, R.G.; Robin, N.C.; Dawson, D.W.; Moon, R.T.; Chien, A.J. Wnt/beta-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci. Signal. 2012, 5, ra3. [Google Scholar] [CrossRef]
- Rulifson, I.C.; Karnik, S.K.; Heiser, P.W.; Ten Berge, D.; Chen, H.N.; Gu, X.Y.; Taketo, M.M.; Nusse, R.; Hebrok, M.; Kim, S.K. Wnt signaling regulates pancreatic beta cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 6247–6252. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.L.; Ortolano, N.A.; Romero-Morales, A.I.; Gama, V. Wnt Signaling and Its Impact on Mitochondrial and Cell Cycle Dynamics in Pluripotent Stem Cells. Genes 2018, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Krishan, S.; Janson, P.J.; Gutierrez, E.; Lane, D.J.R.; Richardson, D.; Sahni, S. Iron Metabolism and Autophagy: A Poorly Explored Relationship That Has Important Consequences for Health and Disease. Nagoya J. Med. Sci. 2015, 77, 1–6. [Google Scholar] [PubMed]
- Yan, X.; Zhou, R.; Ma, Z. Autophagy-Cell Survival and Death. Adv. Exp. Med. Biol. 2019, 1206, 667–696. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.C.; Song, X.X.; Sun, X.F.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D.L. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.H.; Wang, J.; Wu, J.C.; He, D.; Zhang, C.F.; Duan, C.J.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, W.L.; Meng, H.Z.; Cai, Z.Y.; Yang, M.W. Regulation of DMT1 on autophagy and apoptosis in osteoblast. Int. J. Med. Sci. 2017, 14, 275–283. [Google Scholar] [CrossRef]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef]
- Trost, M.; English, L.; Lemieux, S.; Courcelles, M.; Desjardins, M.; Thibault, P. The phagosomal proteome in interferon-gamma-activated macrophages. Immunity 2009, 30, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, J.M.; Yan, J.; Zhang, D.L.; Liu, B.Q.; Jiang, J.Y.; Li, C.; Li, S.; Meng, X.N.; Wang, H.Q. BAG3 promotes autophagy and glutaminolysis via stabilizing glutaminase. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Rosati, A.; Graziano, V.; De Laurenzi, V.; Pascale, M.; Turco, M.C. BAG3: A multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, R.; Mohyeldin, A.; Thiel, J.; Kornblum, H.I.; Beullens, M.; Nakano, I. MELK-a conserved kinase: Functions, signaling, cancer, and controversy. Clin. Transl. Med. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Janostiak, R.; Rauniyar, N.; Lam, T.T.; Ou, J.; Zhu, L.J.; Green, M.R.; Wajapeyee, N. MELK Promotes Melanoma Growth by Stimulating the NF-kappaB Pathway. Cell Rep. 2017, 21, 2829–2841. [Google Scholar] [CrossRef]
- Mi, W.; Xia, Y.; Bian, Y. The influence of ICAM1 rs5498 on diabetes mellitus risk: Evidence from a meta-analysis. Inflamm. Res. 2019, 68, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Karimi, Z.; Kahe, F.; Jamil, A.; Marszalek, J.; Ghanbari, A.; Afarideh, M.; Khajeh, E.; Noshad, S.; Esteghamati, A.; Chi, G. Intercellular adhesion molecule-1 in diabetic patients with and without microalbuminuria. Diabetes Metab. Syndr. 2018, 12, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Shikata, K.; Hirata, K.; Akiyama, K.; Matsuda, M.; Kushiro, M.; Shikata, Y.; Miyatake, N.; Miyasaka, M.; Makino, H. Increased expression of intercellular adhesion molecule-1 (ICAM-1) in diabetic rat glomeruli: Glomerular hyperfiltration is a potential mechanism of ICAM-1 upregulation. Diabetes 1997, 46, 2075–2081. [Google Scholar] [CrossRef]
- Levy-Strumpf, N.; Kimchi, A. Death associated proteins (DAPs): From gene identification to the analysis of their apoptotic and tumor suppressive functions. Oncogene 1998, 17, 3331–3340. [Google Scholar] [CrossRef]
- Koren, I.; Reem, E.; Kimchi, A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr. Biol. 2010, 20, 1093–1098. [Google Scholar] [CrossRef]
- Yahiro, K.; Tsutsuki, H.; Ogura, K.; Nagasawa, S.; Moss, J.; Noda, M. DAP1, a negative regulator of autophagy, controls SubAB-mediated apoptosis and autophagy. Infect. Immun. 2014, 82, 4899–4908. [Google Scholar] [CrossRef]
- El Khouri, E.; Le Pavec, G.; Toledano, M.B.; Delaunay-Moisan, A. RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2013, 288, 31177–31191. [Google Scholar] [CrossRef]
- Kaneko, M.; Iwase, I.; Yamasaki, Y.; Takai, T.; Wu, Y.; Kanemoto, S.; Matsuhisa, K.; Asada, R.; Okuma, Y.; Watanabe, T.; et al. Genome-wide identification and gene expression profiling of ubiquitin ligases for endoplasmic reticulum protein degradation. Sci. Rep. UK 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Wang, B.; Li, N.; Wu, Y.F.; Jia, J.Y.; Suo, T.L.; Chen, Q.; Liu, Y.J.; Tang, J. RNF185, a Novel Mitochondrial Ubiquitin E3 Ligase, Regulates Autophagy through Interaction with BNIP1. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, H.; Wang, C.G.; Li, Y.J.; Tian, H.B.; Siraj, S.; Sehgal, S.A.; Wang, X.H.; Wang, J.; Shang, Y.L.; et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019, 26, 1735–1749. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.J.; Shi, P.Q.; Li, M.H.; Du, F.H.; Chen, Z.J.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262. [Google Scholar] [CrossRef]
- Reislander, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef]
- Offenbacher, S.; Jiao, Y.; Kim, S.J.; Marchesan, J.; Moss, K.L.; Jing, L.; Divaris, K.; Bencharit, S.; Agler, C.S.; Morelli, T.; et al. GWAS for Interleukin-1beta levels in gingival crevicular fluid identifies IL37 variants in periodontal inflammation. Nat. Commun. 2018, 9, 3686. [Google Scholar] [CrossRef]
- Gabay, C.; Towne, J.E. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J. Leukoc. Biol. 2015, 97, 645–652. [Google Scholar] [CrossRef]
- Shanmugaapriya, S.; van Caam, A.; de Kroon, L.; Vitters, E.L.; Walgreen, B.; van Beuningen, H.; Davidson, E.B.; van der Kraan, P.M. Expression of TGF-beta Signaling Regulator RBPMS (RNA-Binding Protein With Multiple Splicing) Is Regulated by IL-1beta and TGF-beta Superfamily Members, and Decreased in Aged and Osteoarthritic Cartilage. Cartilage 2016, 7, 333–345. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, L.; Zhang, H.; Han, J.; Yang, X.; Yan, J.; Zhu, Y.; Li, J.; Song, H.; Ye, Q. Potentiation of Smad-mediated transcriptional activation by the RNA-binding protein RBPMS. Nucleic Acids Res. 2006, 34, 6314–6326. [Google Scholar] [CrossRef] [PubMed]
- Haerian, B.S.; Haerian, M.S.; Roohi, A.; Mehrad-Majd, H. ABCA1 genetic polymorphisms and type 2 diabetes mellitus and its complications. Meta Gene 2017, 13, 104–114. [Google Scholar] [CrossRef]
- Li, C.Y.; Fan, D.J. Association between the ABCA1 rs1800977 polymorphism and susceptibility to type 2 diabetes mellitus in a Chinese Han population. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Rickels, M.R.; Goeser, E.S.; Fuller, C.; Lord, C.; Bowler, A.M.; Doliba, N.M.; Hegele, R.A.; Cuchel, M. Loss-of-function mutations in ABCA1 and enhanced beta-cell secretory capacity in young adults. Diabetes 2015, 64, 193–199. [Google Scholar] [CrossRef]
- Vergeer, M.; Brunham, L.R.; Koetsveld, J.; Kruit, J.K.; Verchere, C.B.; Kastelein, J.J.; Hayden, M.R.; Stroes, E.S. Carriers of loss-of-function mutations in ABCA1 display pancreatic beta-cell dysfunction. Diabetes Care 2010, 33, 869–874. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sato, S.; Imachi, H.; Lyu, J.; Miyai, Y.; Fukunaga, K.; Dong, T.; Ibata, T.; Kobayashi, T.; Yoshimoto, T.; Kikuchi, F.; et al. Effect of TNF-alpha on the expression of ABCA1 in pancreatic beta-cells. J. Mol. Endocrinol. 2018, 61, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Ibata, T.; Kobayashi, T.; Dong, T.; Yoshimoto, T.; Yonezaki, K.; Nagata, H.; et al. Angiotensin II induces cholesterol accumulation and impairs insulin secretion by regulating ABCA1 in beta cells. J. Lipid Res. 2018, 59, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Takei, S.; Nagashima, S.; Takei, A.; Yamamuro, D.; Wakabayashi, T.; Murakami, A.; Isoda, M.; Yamazaki, H.; Ebihara, C.; Takahashi, M.; et al. beta-Cell-Specific Deletion of HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A) Reductase Causes Overt Diabetes due to Reduction of beta-Cell Mass and Impaired Insulin Secretion. Diabetes 2020, 69, 2352–2363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Caldwell, T.A.; Mirbolooki, M.R.; Duong, D.; Park, E.J.; Chi, N.W.; Chessler, S.D. Extracellular CADM1 interactions influence insulin secretion by rat and human islet beta-cells and promote clustering of syntaxin-1. Am. J. Physiol. Endoc. Metab. 2016, 310, E874–E885. [Google Scholar] [CrossRef]
- Perelis, M.; Marcheva, B.; Ramsey, K.M.; Schipma, M.J.; Hutchison, A.L.; Taguchi, A.; Peek, C.B.; Hong, H.; Huang, W.Y.; Omura, C.; et al. Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 2015, 350. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef]
- Zhou, B.R.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D.L. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Kriel, J.; Loos, B. The good, the bad and the autophagosome: Exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 2019, 26, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Garcia, I.J.P.; Costa, T.G.F.; Silva, L.N.D.; Reno, C.O.; Oliveira, E.S.; Tilelli, C.Q.; Santos, L.L.; Cortes, V.F.; Santos, H.L.; et al. Effects of Iron Overload on the Activity of Na, K-ATPase and Lipid Profile of the Human Erythrocyte Membrane. PLoS ONE 2015, 10, e0132852. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.J.; Ginet, V.; Zhang, L.Y.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Sheng, J.; Yin, M.C.; Sun, Z.W.; Kang, X.; Liu, D.; Jiang, K.; Xu, J.; Zhao, F.X.; Guo, Q.F.; Zheng, W. SPC24 promotes osteosarcoma progression by increasing EGFR/MAPK signaling. Oncotarget 2017, 8, 105276–105283. [Google Scholar] [CrossRef]
- Yin, H.B.; Meng, T.; Zhou, L.; Chen, H.Y.; Song, D.W. SPC24 is critical for anaplastic thyroid cancer progression. Oncotarget 2017, 8, 21884–21891. [Google Scholar] [CrossRef]
- Naderi, A.; Liu, J.; Hughes-Davies, L. BEX2 has a functional interplay with c-Jun/JNK and p65/RelA in breast cancer. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Liu, J.; Bennett, I.C. BEX2 regulates mitochondrial apoptosis and G1 cell cycle in breast cancer. Int. J. Cancer 2010, 126, 1596–1610. [Google Scholar] [CrossRef]
- Shao, A.W.; Sun, H.; Geng, Y.; Peng, Q.; Wang, P.; Chen, J.; Xiong, T.; Cao, R.; Tang, J. Bclaf1 is an important NF-kappaB signaling transducer and C/EBPbeta regulator in DNA damage-induced senescence. Cell Death Differ. 2016, 23, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear Lamins. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Ivorra’, C.; Kubicek, M.; Gonzalez, J.M.; Sanz-Gonzalez, S.M.; Alvarez-Barrientos, A.; O’Connor, L.E.; Burke, B.; Andres, V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Gene Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef]
- BossyWetzel, E.; Bakiri, L.; Yaniv, M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997, 16, 1695–1709. [Google Scholar] [CrossRef]
- Brenner, D.A.; Ohara, M.; Angel, P.; Chojkier, M.; Karin, M. Prolonged Activation of Jun and Collagenase Genes by Tumor Necrosis Factor-Alpha. Nature 1989, 337, 661–663. [Google Scholar] [CrossRef]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Gene Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef]
- De Castro, S.C.P.; Malhas, A.; Leung, K.Y.; Gustavsson, P.; Vaux, D.J.; Copp, A.J.; Greene, N.D.E. Lamin B1 Polymorphism Influences Morphology of the Nuclear Envelope, Cell Cycle Progression, and Risk of Neural Tube Defects in Mice. PLoS Genet. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Likhite, N.; Warawdekar, U.M. A unique method for isolation and solubilization of proteins after extraction of RNA from tumor tissue using trizol. J. Biomol. Tech. 2011, 22, 37–44. [Google Scholar]
- Gekko, K.; Timasheff, S.N. Mechanism of protein stabilization by glycerol: Preferential hydration in glycerol-water mixtures. Biochemistry 1981, 20, 4667–4676. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Pathway | N | Gene Names |
|---|---|---|
| Autophagy | 22 | PIK3C3; RNF185; EIF4G1; CTTN; DDIT3; STING; BAG3; DAP; IRGFM1; HMGB1; MTCL1; SQSTM1; LZTS1; ATG4C; DNM1L; HSP90AB1; GPSM1; ABL1; MAPT; NUPR1; MTDH; SUPT5 |
| Macroautophagy | 6 | PIK3C3; STING; SQSTM1; LZTS1; GPSM1; SUPT5 |
| ERAD pathway | 4 | RNF185; RHBDD1; BRSK1; OS9 |
| Cell Cycle Arrest | 7 | PPP2R5B; DDIT3; TBRG1; HSP90AB1; BIN1; CAST; FOXM1 |
| Wnt Signaling | 28 | FZD6; DDIT3; PRICKLE1; WNK2; AXIN1; UBR5; MAPK14; SLC9A3R1; CD44; CSNK1G2; CTTNBIP1; RSPO4; CUSTOS; CSNK1D; HDAC1; CTNND1; CTTND2; LEO1; ABL1; HNF1A; TAX1BP3; CTR9; EMD; RF1; CSNK1G3; MARK2; CSNK1A1; SOX9 |
| STAT Signaling | 10 | SPHK2; SMPD3; CD44; FAS; STAT1; STAT3; STAT6; SOX9; HDAC1; CTR9 |
| JNK pathway | 7 | MAP3K5; IL1F9; AXIN1; HMGB1; PJA2; MINK1; TAOK1; |
| ROS biosynthetic/metabolic process | 9 | NDUFA13; SPHK2; SMPD3; MAPK14; COA8; SIRT3; HDAC4; PXN; FOXM1 |
| Apoptotic signaling pathway | 37 | LMNA; MAP3K5; CTTN; ICAM1; DDIT3; NDUFA13; PDCD10; VDR; SLC35F6; BAG3; DAP; TRIAP1; YBX3; HMGB2; ATP2A1; TRPS1; SLC9A3R1; CD44; COA8; ATAD5; MAPK7; FAS; BCLAF1; SCG2; ZC3HC1; DNM1L; BRSK2; RPS3; HDAC1; BIRC6; MAPT; NUPR1; MELK; PTPN1; VEGFA; CDK11B; FACTO |
| Inflammatory response | 17 | ICAM1; STING; IL1F9; HMGB1; HMGB2; MAPK14; CD44; CTNNBIP1; SMPDL3B; CDK19; PJA2; NPY; NUPR1; HCN2; RICTOR; SLC7A2; HNRNPA0 |
| Ubiquitin-proteasome system (UPS) | 32 | RNF185; DDIT3; PRICKLE1; PCNP; USP37; PSMA5; AXIN1; RNF4; OTUD5; PSMC2; RNF168; AURKA; UBE2C’ UBE2S; MTA1; SUMO3; PSMA7; RYBP; CSNK1D; NSFL1C; HSP90AB1; KAT7; BIRC6; TRIP12; UBA1; UHRF1; USP48; OS9; MAP1A; USP1; RNF4; CSNK1A1 |
| DNA repair | 23 | NPM1; CLSPN; UBR5; PTTG1; MCRS1; RNF168; RFC1; XRN2; TAOK1; RPS3; PAGR1; TRIP12; CHEK1; ABL1; BOD1L; UHRF1; TRIM28; UBR5; SMG1; USP1; FOXM1; SMC3; LIG3 |
| NF-Kb signaling | 7 | NPM1; ICAM1; DAP; PTMA; KRAS; RPS3; MTDH |
| Insulin secretion | 18 | IRS2; PCLO; BAIAP3; SYTL4; HMGCR; CPLX1; MAFA; RIMS2; ICA1; SIRT3; BMP8A; BRSK2; VGF; HDAC1; HNF1A; PTPRN2; NOC2; MYT1 |
| Glucose homeostasis | 8 | ALMS1; CACNA1A; NUCKS1; VGF; NCOR2; HNF1A; NOC2; MYT1 |
| Cholesterol homeostasis | 8 | ABCA1; OSBP; HMGCR; VDR; ALMS1; HMGCS1; HNF1A; RALY |
| Sodium ion transport | 11 | SLC38A4; WNK2; SLCO1C1; GNAS; PCP4; SLC9A3R1; NEFH; COMT; ANK3; HNRNPA1; HCN2 |
| Calcium ion transport | 15 | CACNB2; TRPM7; ICAM1; MCUR1; VDR; ATP2A1; CBARP; CACNA1A; LETM1; DNM1L; PANX1; CACNA1H; BIN1; STIM1; ANK2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, T.; Huang, H.; Mandrup-Poulsen, T.; Larsen, M.R. Divalent Metal Transporter 1 Knock-Down Modulates IL-1β Mediated Pancreatic Beta-Cell Pro-Apoptotic Signaling Pathways through the Autophagic Machinery. Int. J. Mol. Sci. 2021, 22, 8013. https://doi.org/10.3390/ijms22158013
Kang T, Huang H, Mandrup-Poulsen T, Larsen MR. Divalent Metal Transporter 1 Knock-Down Modulates IL-1β Mediated Pancreatic Beta-Cell Pro-Apoptotic Signaling Pathways through the Autophagic Machinery. International Journal of Molecular Sciences. 2021; 22(15):8013. https://doi.org/10.3390/ijms22158013
Chicago/Turabian StyleKang, Taewook, Honggang Huang, Thomas Mandrup-Poulsen, and Martin R. Larsen. 2021. "Divalent Metal Transporter 1 Knock-Down Modulates IL-1β Mediated Pancreatic Beta-Cell Pro-Apoptotic Signaling Pathways through the Autophagic Machinery" International Journal of Molecular Sciences 22, no. 15: 8013. https://doi.org/10.3390/ijms22158013
APA StyleKang, T., Huang, H., Mandrup-Poulsen, T., & Larsen, M. R. (2021). Divalent Metal Transporter 1 Knock-Down Modulates IL-1β Mediated Pancreatic Beta-Cell Pro-Apoptotic Signaling Pathways through the Autophagic Machinery. International Journal of Molecular Sciences, 22(15), 8013. https://doi.org/10.3390/ijms22158013