
SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19



Abstract
:1. Introduction
- (1)
- Recognition by the virus by cellular receptors, which can be divided into three functional groups:
- (a)
- Receptors that enable the virus to penetrate the target cell. To implement this strategy, viruses strive to increase their binding affinity as well as expand the repertoire of these receptors and their coreceptors [6].
- (b)
- Receptors that transmit to the target cell information useful for the virus (combinations of properties A and B are possible in one receptor).
- (c)
- Cellular receptors which, after recognizing a virus, initiate an antiviral response. In this case, the virus strategy is to inhibit these receptors and their signaling pathways [6].
- (2)
- Suppression of the antiviral response, from both the infected target cells and the immune system of the host organism. This virus strategy can also be subdivided into several components:
- (a)
- Inhibition of early antiviral effects of interferons (IFNs) type 1 (INF-I) and type 3 (IFN-III).
- (b)
- Disruption of universal cellular stress signaling pathways or specific immune pathways.
- (c)
- Protection of the virus from the direct action of antiviral response factors.
- (3)
- The ability of the virus to provoke immune system aggression against its tissues in the form of an autoimmune and autoinflammatory process is a separate strategy for viral survival in the host body.
2. General Characteristics of SARS-CoV-2 Infection
3. SARS-CoV-2 Receptors
- Ubiquitination of proteins; this process is important for the regulation of function and proteolysis of many intracellular proteins [138];
- Mitochondrial stress, in particular, is dependent on the RLR/MAVS signaling pathway [139];
- Endoplasmic reticulum (ER) stress [140];
- The formation of a pro-inflammatory cell secretary phenotype, which can manifest itself as a cytokine storm syndrome when generalized to the whole organism.
4. Pathogenicity Factors of SARS-CoV-2 Overcoming the IFN-Response at the Initial Stages of Viral Invasion
- (1)
- Inhibition of the formation and function of PRRs that recognize SARS-CoV-2 RNA.
- (A)
- Non-structural proteins nsp14 (N-7-methyltransferase) and nsp16 (2-Oʹ-methyltransferase) β-CoV can modify viral RNA to prevent it from being recognized by PRR and IFIT1 (interferon-induced protein with tetratricopeptide repeats 1, which inhibits viral replication and translation initiation) [152,153]. The nsp16 factor, in conjunction with nsp10, methylates the viral mRNA’s 5′ end to mimic the host’s mRNA [154,155].
- (B)
- Factor nsp1 blocks several cell proteins, including antiviral PRRs, from being synthesized on ribosomes, while N and M proteins bind to and inhibit RIG-I. (Table 2).
- (2)
- Inhibition of IRF3 activation and translocation into the cell nucleus or cleavage, which initiates IFN-I-III production. Many of the SARS-CoV-2 proteins (nsp1, nsp3, nsp5, nsp6, nsp9, nsp13, nsp14, nsp15, orf3b, orf6, orf9b, N, and M) can block a variety of signaling pathways, including RLR/MAVS/TRAF3/(IKK and TBK1)/(IRF3, NF-κB)/ (Figure 3).
- (3)
- Inhibition of IFNs synthesis on ribosomes (nsp1, Table 2).
- (4)
- (5)
- Inhibition of IFNAR1–IFN-IR chain (orf3a), inhibition of Tyk2 kinase associated with IFN-R (nsp1) (Table 2, Figure 4) [156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220].
- (6)
- Direct inhibition of ISG expression by blocking the activation and translocation into the nucleus of the ISGF3 complex, which initiates ISG transcription. For example, nsp1, nsp3, nsp6, nsp13, orf3a, orf6, orf7a, orf7b, orf8, and M (Table 2) have pathogenic capabilities for blocking several signaling pathways, including IFN-I-III/IFN-R (Tyk2, Jak1)/ISGF3 (STAT1, STAT2, IRF9)/ISG/antiviral response, cellular stress (Figure 4).
5. Dysfunction of the Adaptive Antiviral Response. The Alleged Role of the Autoimmune Process in the Pathogenesis of COVID-19
5.1. The Main Features of Immune Response Development and Its Dysfunctions in COVID-19
- Migration of cells to the focus of inflammation;
- Direct infection of lymphocytes expressing ACE2 and other SARS-CoV-2 receptors, followed by their apoptosis or cytolysis;
- Apoptosis and pyroptosis of hyperactivated cells not infected with SARS-CoV-2;
- Metabolic and neuroendocrine changes caused by a viral infection lead to depletion and impairment of lymphoid cell regeneration.
- Inhibition of MHC-I in infected cells, which makes it difficult for CTL to recognize them (orf8, Table 2);
- Binding of orf8 to the IL-17 receptor and its activation on immunocompetent cells, which can contribute to immune imbalances [201];
- Possible APC dysfunctions associated with the action of the SARS-CoV-2 S-protein on PRR, SR, and other receptors on these cells (Table 1);
- Disturbance in immunocompetent cells of the balance between various signaling pathways and mechanisms of cellular stress by many factors of SARS-CoV-2 pathogenicity.
5.2. Possible Causes of the Autoimmune and Anti-Inflammatory Process in COVID-19
- (1)
- Molecular mimicry of viral proteins.
- (2)
- “Bystander activation”–the release of autoantigens from tissue damaged by viruses and inflammation.
- (3)
- Violation of biological barriers in immunoprivileged organs (central nervous system, eye, testes, placenta), providing accessibility for adoptive immunity of potential autoantigens.
- (4)
- Polyclonal activation of lymphocytes as a result of the action of superantigens (SAg) or for other reasons (potentially autoreactive clones of T and B lymphocytes can also be activated).
- (5)
- “Epitope spreading”–when the targets of autoimmune responses do not remain fixed, but can be expanded by including other epitopes on the same protein or in other proteins in the same tissue.
6. Conclusions
- (1)
- Invasion and forced replication of the virus in integumentary tissues, primarily in the epithelium of the respiratory tract and intestines. The key mechanisms of this stage are the recognition of target cells by the virus through receptor structures and blockade of the primary (early) IFN-I-III response.
- (2)
- Virus-induced loss of the barrier functions at the focus of inflammation through the dysregulation of the innate and then the adaptive immune response controlled by the pathogenicity factors. The selective suppression of specific antiviral mechanisms, the indiscriminate polyclonal activation of lymphocytes, the development of hypoinflammation according to the variant of an autoinflammatory process, and in some cases, a latent or even clinically manifest autoimmune process are all factors that assist viral expansion during this period.
- (3)
- Generalization of the virus in the body, systemic changes in the immune status, and other parameters of homeostasis with a high risk of critical complications.
- (4)
- The stage of long-term consequences of a viral infection associated with long-term persistence of the virus in the body, the consequences of primary (viral) and secondary (autogenous) tissue damage, including possible breach of the BBB integrity, endothelial dysfunction, hypercoagulation, and persistent changes in the immune status.
- The presence, in some cases, of long-term persistence of SARS-CoV-2 in the body in immunodeficient patients [324];
- The presence, in about every fifth case, of long-term consequences caused by the transferred infection in the form of a long COVID;
- Longer (in comparison with SARS-CoV-1 and MERS-CoV) retention of SARS-CoV-2 RNA in biological fluids in COVID-19 patients (up to 1–2 months) [325], as well as the detection of SARS-CoV-2 RNA in more than one out of ten patients with relapse within 60 days after recovery from primary infection [326];
- The development of a latent and, in some cases, clinically significant auto-immune response in COVID-19;
- The potential ability of the virus to actively overcome the factors of not only innate but also adaptive immune response.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Malik, Y.S.; Sircar, S.; Bhat, S.; Sharun, K.; Dhama, K.; Dadar, M.; Tiwari, R.; Chaicumpa, W. Emerging novel coronavirus (2019-nCoV)—current scenario, evolutionary perspective based on genome analysis and recent developments. Vet. Q. 2020, 40, 68–76. [Google Scholar] [CrossRef]
- Dhama, K.; Khan, S.; Tiwari, R.; Sircar, S.; Bhat, S.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Coronavirus Disease 2019–COVID-19. Clin. Microbiol. Rev. 2020, 33. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.-M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Shell disorder analysis predicts greater resilience of the SARS-CoV-2 (COVID-19) outside the body and in body fluids. Microb. Pathog. 2020, 144, 104177. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A.; Hu, D.; Chereshnev, V. Problems of Pathogenesis and Pathogenetic Therapy of COVID-19 from the Perspective of the General Theory of Pathological Systems (General Pathological Processes). Int. J. Mol. Sci. 2021, 22, 7582. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.; Solomatina, L.; Zhuravleva, Y.; Sarapultsev, A. The Pathogenesis of End-Stage Renal Disease from the Standpoint of the Theory of General Pathological Processes of Inflammation. Int. J. Mol. Sci. 2021, 22, 11453. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.R.; Shrestha, P.; Yang, B.; Chen, Y.; Liu, S.; Maarouf, M.; Chen, J.-L. Acute Infection of Viral Pathogens and Their Innate Immune Escape. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Rehman, S.U.; Shafique, L.; Ihsan, A.; Liu, Q. Evolutionary Trajectory for the Emergence of Novel Coronavirus SARS-CoV-2. Pathogens 2020, 9, 240. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Li, X.; Ge, C.; Ding, Y.; Zhang, T.; Cao, S.; Meng, L.; Lu, S. Molecular detection of SARS-CoV-2 being challenged by virus variation and asymptomatic infection. J. Pharm. Anal. 2021. [Google Scholar] [CrossRef]
- Chilamakuri, R.; Agarwal, S. COVID-19: Characteristics and Therapeutics. Cells 2021, 10, 206. [Google Scholar] [CrossRef]
- Peng, Q.; Peng, R.; Yuan, B.; Zhao, J.; Wang, M.; Wang, X.; Wang, Q.; Sun, Y.; Fan, Z.; Qi, J.; et al. Structural and Biochemical Characterization of the nsp12-nsp7-nsp8 Core Polymerase Complex from SARS-CoV-2. Cell Rep. 2020, 31, 107774. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Verma, H.; Singhvi, N.; Sood, U.; Gupta, V.; Singh, M.; Kumari, R.; Hira, P.; Nagar, S.; Talwar, C.; et al. Comparative Genomic Analysis of Rapidly Evolving SARS-CoV-2 Reveals Mosaic Pattern of Phylogeographical Distribution. mSystems 2020, 5, e00505-20. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J.; Mayer, C.; Poch, O.; Thompson, J.D. Characterization of accessory genes in coronavirus genomes. Virol. J. 2020, 17, 131. [Google Scholar] [CrossRef] [PubMed]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and Function of Major SARS-CoV-2 and SARS-CoV Proteins. Bioinform. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, R.; Huo, S.; Zhou, Y.; Jiang, S.; Wang, Q.; Yu, F. Molecular mechanism of interaction between SARS-CoV-2 and host cells and interventional therapy. Signal Transduct. Target. Ther. 2021, 6, 233. [Google Scholar] [CrossRef]
- Hassan, S.S.; Attrish, D.; Ghosh, S.; Choudhury, P.P.; Uversky, V.N.; Aljabali, A.A.; Lundstrom, K.; Uhal, B.D.; Rezaei, N.; Seyran, M.; et al. Notable sequence homology of the ORF10 protein introspects the architecture of SARS-CoV-2. Int. J. Biol. Macromol. 2021, 181, 801–809. [Google Scholar] [CrossRef]
- Liu, T.; Jia, P.; Fang, B.; Zhao, Z. Differential Expression of Viral Transcripts From Single-Cell RNA Sequencing of Moderate and Severe COVID-19 Patients and Its Implications for Case Severity. Front. Microbiol. 2020, 11, 603509. [Google Scholar] [CrossRef]
- Mena, E.L.; Donahue, C.J.; Vaites, L.P.; Li, J.; Rona, G.; O’Leary, C.; Lignitto, L.; Miwatani-Minter, B.; Paulo, J.A.; Dhabaria, A.; et al. ORF10–Cullin-2–ZYG11B complex is not required for SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2023157118. [Google Scholar] [CrossRef]
- Pancer, K.; Milewska, A.; Owczarek, K.; Dabrowska, A.; Kowalski, M.; Łabaj, P.P.; Branicki, W.; Sanak, M.; Pyrc, K. The SARS-CoV-2 ORF10 is not essential in vitro or in vivo in humans. PLoS Pathog. 2020, 16, e1008959. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, M.G.; Bouhaddou, M.; Krogan, N.J. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. bioXriv 2020. [Google Scholar] [CrossRef]
- Lam, J.-Y.; Yuen, C.-K.; Ip, J.D.; Wong, W.-M.; To, K.K.-W.; Yuen, K.-Y.; Kok, K.-H. Loss of orf3b in the circulating SARS-CoV-2 strains. Emerg. Microbes Infect. 2020, 9, 2685–2696. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldaais, E.A.; Yegnaswamy, S.; Albahrani, F.; Alsowaiket, F.; Alramadan, S. Sequence and structural analysis of COVID-19 E and M proteins with MERS virus E and M proteins—A comparative study. Biochem. Biophys. Rep. 2021, 26, 101023. [Google Scholar] [CrossRef] [PubMed]
- Bakhshandeh, B.; Jahanafrooz, Z.; Abbasi, A.; Goli, M.B.; Sadeghi, M.; Mottaqi, M.S.; Zamani, M. Mutations in SARS-CoV-2; Consequences in structure, function, and pathogenicity of the virus. Microb. Pathog. 2021, 154, 104831. [Google Scholar] [CrossRef] [PubMed]
- Camporota, L.; Chiumello, D.; Busana, M.; Gattinoni, L.; Marini, J.J. Pathophysiology of COVID-19-associated acute respiratory distress syndrome. Lancet Respir. Med. 2020, 9, 1201–1208. [Google Scholar] [CrossRef]
- Asakura, H.; Ogawa, H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int. J. Hematol. 2020, 113, 45–57. [Google Scholar] [CrossRef]
- Petersen, E.; Koopmans, M.; Go, U.; Hamer, D.H.; Petrosillo, N.; Castelli, F.; Storgaard, M.; Al Khalili, S.; Simonsen, L. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020, 20, e238–e244. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Hasan, S.S.; Capstick, T.; Ahmed, R.; Kow, C.S.; Mazhar, F.; Merchant, H.A.; Zaidi, S.T.R. Mortality in COVID-19 patients with acute respiratory distress syndrome and corticosteroids use: A systematic review and meta-analysis. Expert Rev. Respir. Med. 2020, 14, 1149–1163. [Google Scholar] [CrossRef]
- Acter, T.; Uddin, N.; Das, J.; Akhter, A.; Choudhury, T.R.; Kim, S. Evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as coronavirus disease 2019 (COVID-19) pandemic: A global health emergency. Sci. Total Environ. 2020, 730, 138996. [Google Scholar] [CrossRef]
- Shoaib, M.H.; Ahmed, F.R.; Sikandar, M.; Yousuf, R.I.; Saleem, M.T. A Journey From SARS-CoV-2 to COVID-19 and Beyond: A Comprehensive Insight of Epidemiology, Diagnosis, Pathogenesis, and Overview of the Progress into Its Therapeutic Management. Front. Pharmacol. 2021, 12, 576448. [Google Scholar] [CrossRef]
- Okamoto, M.; Tsukamoto, H.; Kouwaki, T.; Seya, T.; Oshiumi, H. Recognition of Viral RNA by Pattern Recognition Receptors in the Induction of Innate Immunity and Excessive Inflammation During Respiratory Viral Infections. Viral Immunol. 2017, 30, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Prabhudas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [Green Version]
- Gusev, E.Y.; Zotova, N.V.; Zhuravleva, Y.A.; Chereshnev, V. Physiological and pathogenic role of scavenger receptors in humans. Med Immunol. 2020, 22, 7–48. [Google Scholar] [CrossRef]
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.-L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell. Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAK-STAT 2013, 2, e23931. [Google Scholar] [CrossRef] [Green Version]
- Hilleman, M.R. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc. Natl. Acad. Sci. USA 2004, 101, 14560–14566. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Renner, K.; Schwittay, T.; Chaabane, S.; Gottschling, J.; Müller, C.; Tiefenböck, C.; Salewski, J.-N.; Winter, F.; Buchtler, S.; Balam, S.; et al. Severe T cell hyporeactivity in ventilated COVID-19 patients correlates with prolonged virus persistence and poor outcomes. Nat. Commun. 2021, 12, 3006. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-W.; Wang, S.-F. SARS-CoV-2 Entry Related Viral and Host Genetic Variations: Implications on COVID-19 Severity, Immune Escape, and Infectivity. Int. J. Mol. Sci. 2021, 22, 3060. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, L.; Charitos, I.A.; Carretta, D.M.; De Nitto, E.; Lovero, R. The human coronaviruses (HCoVs) and the molecular mechanisms of SARS-CoV-2 infection. Klin. Wochenschr. 2020, 99, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- National Center for Immunization and Respiratory Diseases (NCIRD), Division of Viral Diseases. Science Brief: Omicron (B.1.1.529) Variant. In CDC COVID-19 Science Briefs; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2020. [Google Scholar]
- Kandeel, M.; Mohamed, M.E.M.; El-Lateef, H.M.A.; Venugopala, K.N.; El-Beltagi, H.S. Omicron variant genome evolution and phylogenetics. J. Med. Virol. 2021. [Google Scholar] [CrossRef]
- Dhawan, M.; Priyanka, O.P.C. Omicron SARS-CoV-2 variant: Reasons of emergence and lessons learnt-Correspondence. Int. J. Surg. 2021, 97, 106198. [Google Scholar] [CrossRef]
- Del Rio, C.; Omer, S.B.; Malani, P.N. Winter of Omicron—The Evolving COVID-19 Pandemic. JAMA 2022, 327, 319. [Google Scholar] [CrossRef]
- Burki, T.K. Omicron variant and booster COVID-19 vaccines. Lancet Respir. Med. 2021, 10, E17. [Google Scholar] [CrossRef]
- Wang, X.; Powell, C.A. How to translate the knowledge of COVID-19 into the prevention of Omicron variants. Clin. Transl. Med. 2021, 11, e680. [Google Scholar] [CrossRef]
- Torjesen, I. COVID-19: Omicron may be more transmissible than other variants and partly resistant to existing vaccines, scientists fear. BMJ 2021, 375, n2943. [Google Scholar] [CrossRef] [PubMed]
- Dyer, O. COVID-19: Omicron is causing more infections but fewer hospital admissions than delta, South African data show. BMJ 2021, 375, n3104. [Google Scholar] [CrossRef] [PubMed]
- Christie, B. COVID-19: Early studies give hope omicron is milder than other variants. BMJ 2021, 375, n3144. [Google Scholar] [CrossRef] [PubMed]
- SeyedAlinaghi, S.; Mirzapour, P.; Dadras, O.; Pashaei, Z.; Karimi, A.; MohsseniPour, M.; Soleymanzadeh, M.; Barzegary, A.; Afsahi, A.M.; Vahedi, F.; et al. Characterization of SARS-CoV-2 different variants and related morbidity and mortality: A systematic review. Eur. J. Med. Res. 2021, 26, 51. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, O.M.; Ata, O.; Turgut-Balik, D. An updated analysis of variations in SARS-CoV-2 genome. Turk. J. Biol. 2020, 44, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Simões e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, N.Z.; Grandvaux, N. ACE2: Evidence of role as entry receptor for SARS-CoV-2 and implications in comorbidities. eLife 2020, 9, e61390. [Google Scholar] [CrossRef] [PubMed]
- Masre, S.F.; Jufri, N.F.; Ibrahim, F.W.; Raub, S.H.A. Classical and alternative receptors for SARS-CoV-2 therapeutic strategy. Rev. Med. Virol. 2020, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.-L.; Zhao, R.; Matalon, S.; Matthay, M.A. Elevated Plasmin(ogen) as a Common Risk Factor for COVID-19 Susceptibility. Physiol. Rev. 2020, 100, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.P.; Liu, S.-L. Role of host factors in SARS-CoV-2 entry. J. Biol. Chem. 2021, 297, 100847. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatmal, M.M.; Alshaer, W.; Al-Hatamleh, M.A.I.; Hatmal, M.; Smadi, O.; Taha, M.O.; Oweida, A.J.; Boer, J.C.; Mohamud, R.; Plebanski, M. Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2. Cells 2020, 9, 2638. [Google Scholar] [CrossRef]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4240–4245. [Google Scholar] [CrossRef] [Green Version]
- Prabhakara, C.; Godbole, R.; Sil, P.; Jahnavi, S.; Gulzar, S.-E.; van Zanten, T.S.; Sheth, D.; Subhash, N.; Chandra, A.; Shivaraj, A.; et al. Strategies to target SARS-CoV-2 entry and infection using dual mechanisms of inhibition by acidification inhibitors. PLOS Pathog. 2021, 17, e1009706. [Google Scholar] [CrossRef]
- Shang, C.; Zhuang, X.; Zhang, H.; Li, Y.; Zhu, Y.; Lu, J.; Ge, C.; Cong, J.; Li, T.; Tian, M.; et al. Inhibitors of endosomal acidification suppress SARS-CoV-2 replication and relieve viral pneumonia in hACE2 transgenic mice. Virol. J. 2021, 18, 46. [Google Scholar] [CrossRef]
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural Modeling of the SARS-CoV-2 Spike/Human ACE2 Complex Interface can Identify High-Affinity Variants Associated with Increased Transmissibility. J. Mol. Biol. 2021, 433, 167051. [Google Scholar] [CrossRef]
- Ou, J.; Zhou, Z.; Dai, R.; Zhang, J.; Zhao, S.; Wu, X.; Lan, W.; Ren, Y.; Cui, L.; Lan, Q.; et al. V367F Mutation in SARS-CoV-2 Spike RBD Emerging during the Early Transmission Phase Enhances Viral Infectivity through Increased Human ACE2 Receptor Binding Affinity. J. Virol. 2021, 95, JVI0061721. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Yuan, M.; Ma, Q.; Wang, S.; Tan, Y.; Xu, Y.; Ye, J.; Gao, Y.; Sun, X.; Yang, Z.; et al. Landscape of SARS-CoV-2 spike protein-interacting cells in human tissues. Int. Immunopharmacol. 2021, 95, 107567. [Google Scholar] [CrossRef] [PubMed]
- Lokhande, K.B.; Apte, G.R.; Shrivastava, A.; Singh, A.; Pal, J.K.; Swamy, K.V.; Gupta, R.K. Sensing the interactions between carbohydrate-binding agents and N-linked glycans of SARS-CoV-2 spike glycoprotein using molecular docking and simulation studies. J. Biomol. Struct. Dyn. 2020, 1–19. [Google Scholar] [CrossRef]
- Verma, J.; Subbarao, N. A comparative study of human betacoronavirus spike proteins: Structure, function and therapeutics. Arch. Virol. 2021, 166, 697–714. [Google Scholar] [CrossRef] [PubMed]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemost. 2020, 120, 1700–1715. [Google Scholar] [CrossRef]
- Kim, C.-H. SARS-CoV-2 Evolutionary Adaptation toward Host Entry and Recognition of Receptor O-Acetyl Sialylation in Virus–Host Interaction. Int. J. Mol. Sci. 2020, 21, 4549. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef] [Green Version]
- Seyran, M.; Takayama, K.; Uversky, V.N.; Lundstrom, K.; Palù, G.; Sherchan, S.P.; Attrish, D.; Rezaei, N.; Aljabali, A.A.A.; Ghosh, S.; et al. The structural basis of accelerated host cell entry by SARS-CoV-2†. FEBS J. 2020, 288, 5010–5020. [Google Scholar] [CrossRef]
- Dakal, T.C. SARS-CoV-2 attachment to host cells is possibly mediated via RGD-integrin interaction in a calcium-dependent manner and suggests pulmonary EDTA chelation therapy as a novel treatment for COVID 19. Immunobiology 2020, 226, 152021. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M. Molecular Simulations and Network Modeling Reveal an Allosteric Signaling in the SARS-CoV-2 Spike Proteins. J. Proteome Res. 2020, 19, 4587–4608. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Wu, L.-A.; Wang, Q.; Qi, J.; Gao, G.F. Cell entry by SARS-CoV-2. Trends Biochem. Sci. 2021, 46, 848–860. [Google Scholar] [CrossRef]
- Mayi, B.S.; Leibowitz, J.A.; Woods, A.T.; Ammon, K.A.; Liu, A.E.; Raja, A. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021, 17, e1009153. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.; Lechien, J.R.; Saussez, S. More that ACE2? NRP1 may play a central role in the underlying pathophysiological mechanism of olfactory dysfunction in COVID-19 and its association with enhanced survival. Med. Hypotheses 2020, 146, 110406. [Google Scholar] [CrossRef]
- Kyrou, I.; Randeva, H.S.; Spandidos, D.A.; Karteris, E. Not only ACE2—the quest for additional host cell mediators of SARS-CoV-2 infection: Neuropilin-1 (NRP1) as a novel SARS-CoV-2 host cell entry mediator implicated in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef]
- Fenizia, C.; Galbiati, S.; Vanetti, C.; Vago, R.; Clerici, M.; Tacchetti, C.; Daniele, T. SARS-CoV-2 Entry: At the Crossroads of CD147 and ACE2. Cells 2021, 10, 1434. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Elfiky, A.A.; Ibrahim, I.M.; Amin, F.G.; Ismail, A.M.; Elshemey, W.M. COVID-19 and Cell Stress. Adv. Exp. Med. Biol. 2021, 1318, 169–178. [Google Scholar] [CrossRef]
- Felordi, M.S.; Memarnejadian, A.; Najimi, M.; Vosough, M. Is There any Alternative Receptor for SARS-CoV-2? Cell J. 2021, 23, 247–250. [Google Scholar] [CrossRef]
- Gonzalez-Gronow, M.; Gopal, U.; Austin, R.C.; Pizzo, S.V. Glucose-regulated protein (GRP78) is an important cell surface receptor for viral invasion, cancers, and neurological disorders. IUBMB Life 2021, 73, 843–854. [Google Scholar] [CrossRef]
- Carlos, A.J.; Ha, D.P.; Yeh, D.W.; Van Krieken, R.; Tseng, C.C.; Zhang, P.; Gill, P.; Machida, K.; Lee, A.S. The chaperone GRP78 is a host auxiliary factor for SARS-CoV-2 and GRP78 depleting antibody blocks viral entry and infection. J. Biol. Chem. 1007, 296, 100759. [Google Scholar] [CrossRef] [PubMed]
- Weigel, P. Systemic Glycosaminoglycan Clearance by HARE/Stabilin-2 Activates Intracellular Signaling. Cells 2020, 9, 2366. [Google Scholar] [CrossRef]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Central Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N. C-type Lectin CD209L/L-SIGN and CD209/DC-SIGN: Cell Adhesion Molecules Turned to Pathogen Recognition Receptors. Biology 2020, 10, 1. [Google Scholar] [CrossRef]
- Khoo, U.-S.; Chan, K.Y.K.; Chan, V.S.F.; Lin, C.L.S. DC-SIGN and L-SIGN: The SIGNs for infection. Klin. Wochenschr. 2008, 86, 861–874. [Google Scholar] [CrossRef]
- Gadanec, L.; McSweeney, K.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? Int. J. Mol. Sci. 2021, 22, 992. [Google Scholar] [CrossRef]
- Krejner-Bienias, A.; Grzela, K.; Grzela, T. DPP4 Inhibitors and COVID-19–Holy Grail or Another Dead End? Arch. Immunol. Ther. Exp. 2021, 69, 1. [Google Scholar] [CrossRef]
- Pinheiro, M.M.; Fabbri, A.; Infante, M. Cytokine storm modulation in COVID-19: A proposed role for vitamin D and DPP-4 inhibitor combination therapy (VIDPP-4i). Immunotherapy 2021, 13, 753–765. [Google Scholar] [CrossRef]
- Elrashdy, F.; Redwan, E.M.; Uversky, V.N. Why COVID-19 Transmission Is More Efficient and Aggressive Than Viral Transmission in Previous Coronavirus Epidemics? Biomolecules 2020, 10, 1312. [Google Scholar] [CrossRef]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020, 526, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.P.; Steer, C.J. Binding of the SARS-CoV-2 Spike Protein to the Asialoglycoprotein Receptor on Human Primary Hepatocytes and Immortalized Hepatocyte-Like Cells by Confocal Analysis. Hepatic Med. Évid. Res. 13, 37–44. [CrossRef] [PubMed]
- Mori, Y.; Fink, C.; Ichimura, T.; Sako, K.; Mori, M.; Lee, N.N.; Aschauer, P.; Padmanabha Das, K.M.; Hong, S.; Song, M.; et al. KIM-1/TIM-1 is a Receptor for SARS-CoV-2 in Lung and Kidney. medRxiv [Preprint]. 2022. [Google Scholar] [CrossRef]
- Altan-Bonnet, N. Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 2016, 32, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Hassanpour, M.; Rezaie, J.; Nouri, M.; Panahi, Y. The role of extracellular vesicles in COVID-19 virus infection. Infect. Genet. Evol. 2020, 85, 104422. [Google Scholar] [CrossRef]
- Elrashdy, F.; Aljaddawi, A.A.; Redwan, E.M.; Uversky, V.N. On the potential role of exosomes in the COVID-19 reinfection/reactivation opportunity. J. Biomol. Struct. Dyn. 2020, 39, 5831–5842. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Yuan, P.; Liu, Y.; Wang, Y.; Cao, W.; Zheng, J.C. Emerging roles of extracellular vesicles in COVID-19, a double-edged sword? Immunology 2021, 163, 416–430. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Kim, J.-H. Diverse Effects of Exosomes on COVID-19: A Perspective of Progress From Transmission to Therapeutic Developments. Front. Immunol. 2021, 12, 716407. [Google Scholar] [CrossRef]
- Kočar, E.; Režen, T.; Rozman, D. Cholesterol, lipoproteins, and COVID-19: Basic concepts and clinical applications. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1866, 158849. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.W.; Kemper, C.; Woodruff, T.M. COVID-19: Complement, Coagulation, and Collateral Damage. J. Immunol. 2020, 205, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Benigni, A.; Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020, 98, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2020, 17, 46–64. [Google Scholar] [CrossRef]
- Polycarpou, A.; Howard, M.; Farrar, C.A.; Greenlaw, R.; Fanelli, G.; Wallis, R.; Klavinskis, L.S.; Sacks, S. Rationale for targeting complement in COVID-19. EMBO Mol. Med. 2020, 12, e12642. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Mallick, B.; Sharma, G.; Lee, S.-S.; Chakraborty, C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect. Genet. Evol. 2020, 85, 104587. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Moreno-Eutimio, M.A.; López-Macías, C.; Pastelin-Palacios, R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect. 2020, 22, 226–229. [Google Scholar] [CrossRef]
- De Marcken, M.; Dhaliwal, K.; Danielsen, A.C.; Gautron, A.S.; Dominguez-Villar, M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci. Signal. 2019, 12, eaaw1347. [Google Scholar] [CrossRef] [PubMed]
- Kouwaki, T.; Nishimura, T.; Wang, G.; Oshiumi, H. RIG-I-Like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape From the Host Innate Immune Responses. Front. Immunol. 2021, 12, 700926. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ghonime, M.G.; Wang, R.; Cassady, K.A. The Antiviral Apparatus: STING and Oncolytic Virus Restriction. Mol. Ther. Oncolytics 2019, 13, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, R.; Jiang, Q.; Zhou, X.; Wang, C.; Guan, Y.; Tao, J.; Xi, J.; Feng, J.-M.; Jiang, Z. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017, 13, e1006720. [Google Scholar] [CrossRef]
- Jiang, Q.-X. Structural Variability in the RLR-MAVS Pathway and Sensitive Detection of Viral RNAs. Med. Chem. 2019, 15, 443–458. [Google Scholar] [CrossRef]
- Mdkhana, B.; Sharif-Askari, N.S.; Ramakrishnan, R.K.; Goel, S.; Hamid, Q.; Halwani, R. Nucleic Acid-Sensing Pathways During SARS-CoV-2 Infection: Expectations versus Reality. J. Inflamm. Res. 2021, 14, 199–216. [Google Scholar] [CrossRef]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Verhelst, K.; Verstrepen, L.; Carpentier, I.; Beyaert, R. IκB kinase ɛ (IKKɛ): A therapeutic target in inflammation and cancer. Biochem. Pharmacol. 2013, 85, 873–880. [Google Scholar] [CrossRef]
- Abe, H.; Satoh, J.; Shirasaka, Y.; Kogure, A.; Kato, H.; Ito, S.; Fujita, T. Priming Phosphorylation of TANK-Binding Kinase 1 by IκB Kinase β Is Essential in Toll-Like Receptor 3/4 Signaling. Mol. Cell. Biol. 2020, 40, e00509-19. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2020, 218, e20201707. [Google Scholar] [CrossRef]
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannatà, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Agents Actions 2020, 70, 7–10. [Google Scholar] [CrossRef]
- Gusev, E.Y. Cellular Stress and General Pathological Processes. Curr. Pharm. Des. 2019, 25, 251–297. [Google Scholar] [CrossRef] [PubMed]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. J. Protein Chem. 2020, 39, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Heras, N.D.L.; Giménez, V.M.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2020, 56, 427–442.e5. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, K.; Mao, B.; Liu, S.; Trilling, M.; Huang, A.; Lu, M.; Lin, Y. The interplay between emerging human coronavirus infections and autophagy. Emerg. Microbes Infect. 2021, 10, 196–205. [Google Scholar] [CrossRef]
- Haas, P.; Muralidharan, M.; Krogan, N.J.; Kaake, R.M.; Hüttenhain, R. Proteomic Approaches to Study SARS-CoV-2 Biology and COVID-19 Pathology. J. Proteome Res. 2021, 20, 1133–1152. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS Regulation at the Mitochondrial Membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef] [Green Version]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-level inhibition of coronavirus replication by chemical ER stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef]
- Chauhan, N.; Jaggi, M.; Chauhan, S.C.; Yallapu, M.M. COVID-19: Fighting the invisible enemy with microRNAs. Expert Rev. Anti-Infect. Ther. 2021, 19, 137–145. [Google Scholar] [CrossRef]
- Abedi, F.; Rezaee, R.; Hayes, A.W.; Nasiripour, S.; Karimi, G. MicroRNAs and SARS-CoV-2 life cycle, pathogenesis, and mutations: Biomarkers or therapeutic agents? Cell Cycle 2020, 20, 143–153. [Google Scholar] [CrossRef]
- Abu-Izneid, T.; AlHajri, N.; Ibrahim, A.M.; Javed, N.; Salem, K.M.; Pottoo, F.H.; Kamal, M.A. Micro-RNAs in the regulation of immune response against SARS CoV-2 and other viral infections. J. Adv. Res. 2020, 30, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Houshmandfar, S.; Saeedi-Boroujeni, A.; Rashno, M.; Khodadadi, A.; Mahmoudian-Sani, M.-R. miRNA-223 as a regulator of inflammation and NLRP3 inflammasome, the main fragments in the puzzle of immunopathogenesis of different inflammatory diseases and COVID-19. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Danladi, J.; Sabir, H. Innate immunity, inflammation activation and heat-shock protein in COVID-19 pathogenesis. J. Neuroimmunol. 2021, 358, 577632. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, C.M.; Hainaut, P. Viral strategies for circumventing p53: The case of severe acute respiratory syndrome coronavirus. Curr. Opin. Oncol. 2021, 33, 149–158. [Google Scholar] [CrossRef]
- Victor, J.; Deutsch, J.; Whitaker, A.; Lamkin, E.N.; March, A.; Zhou, P.; Botten, J.W.; Chatterjee, N. SARS-CoV-2 triggers DNA damage response in Vero E6 cells. Biochem. Biophys. Res. Commun. 2021, 579, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, K.; Grandvaux, N. STAT2 and IRF9. JAK-STAT 2013, 2, e27521. [Google Scholar] [CrossRef] [Green Version]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and Type III Interferons – Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef] [Green Version]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.-Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, T.; Arya, S.; Chan, S.-H.; Qi, S.; Dai, N.; Misra, A.; Park, J.-G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Nakagawa, K.; Makino, S. Mechanisms of Coronavirus Nsp1-Mediated Control of Host and Viral Gene Expression. Cells 2021, 10, 300. [Google Scholar] [CrossRef]
- Kumar, A.; Ishida, R.; Strilets, T.; Cole, J.; Lopez-Orozco, J.; Fayad, N.; Felix-Lopez, A.; Elaish, M.; Evseev, D.; Magor, K.E.; et al. SARS-CoV-2 Nonstructural Protein 1 Inhibits the Interferon Response by Causing Depletion of Key Host Signaling Factors. J. Virol. 2021, 95, e0026621. [Google Scholar] [CrossRef]
- Díaz, J. SARS-CoV-2 Molecular Network Structure. Front. Physiol. 2020, 11, 870. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, Y.; Wu, W.; Chen, Z. Structure and Function of N-Terminal Zinc Finger Domain of SARS-CoV-2 NSP2. Virol. Sin. 2021, 36, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Verba, K.; Gupta, M.; Azumaya, C.; Moritz, M.; Pourmal, S.; Diallo, A.; Merz, G.; Jang, G.; Bouhaddou, M.; Fossati, A.; et al. CryoEM and AI reveal a structure of SARS-CoV-2 Nsp2, a multifunctional protein in-volved in key host processes. Res. Sq. 2021, rs.3.rs-515215, [Preprint]. [Google Scholar] [CrossRef]
- Zheng, Y.-X.; Wang, L.; Kong, W.-S.; Chen, H.; Wang, X.-N.; Meng, Q.; Zhang, H.-N.; Guo, S.-J.; Jiang, H.-W.; Tao, S.-C. Nsp2 has the potential to be a drug target revealed by global identification of SARS-CoV-2 Nsp2-interacting proteins. Acta Biochim. Biophys. Sin. 2021, 53, 1134–1141. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Mielech, A.M.; Chen, Y.; Mesecar, A.D.; Baker, S.C. Nidovirus papain-like proteases: Multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014, 194, 184–190. [Google Scholar] [CrossRef]
- Kim, M.-J.; Hwang, S.-Y.; Imaizumi, T.; Yoo, J.-Y. Negative Feedback Regulation of RIG-I-Mediated Antiviral Signaling by Interferon-Induced ISG15 Conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef] [Green Version]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Shcherbik, N.; Sawaya, B.E. Why do SARS-CoV-2 NSPs rush to the ER? J. Neurol. 2020, 268, 2013–2022. [Google Scholar] [CrossRef]
- Sundar, S.; Thangamani, L.; Piramanayagam, S.; Rahul, C.N.; Aiswarya, N.; Sekar, K.; Natarajan, J. Screening of FDA-approved compound library identifies potential small-molecule inhibitors of SARS-CoV-2 non-structural proteins NSP1, NSP4, NSP6 and NSP13: Molecular modeling and molecular dynamics studies. J. Proteins Proteom. 2021, 12, 161–175. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, Z.; Wu, Y.; Shi, X.; Zhang, Y.; Zhang, M.; Zhang, M.; Wang, L.; Liu, W. SARS-CoV-2 3C-like protease antagonizes interferon-beta production by facilitating the degradation of IRF3. Cytokine 2021, 148, 155697. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.-M. SARS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Biswal, M.; Diggs, S.; Xu, D.; Khudaverdyan, N.; Lu, J.; Fang, J.; Blaha, G.; Hai, R.; Song, J. Two conserved oligomer interfaces of NSP7 and NSP8 underpin the dynamic assembly of SARS-CoV-2 RdRP. Nucleic Acids Res. 2021, 49, 5956–5966. [Google Scholar] [CrossRef]
- Yan, L.; Ge, J.; Zheng, L.; Zhang, Y.; Gao, Y.; Wang, T.; Huang, Y.; Yang, Y.; Gao, S.; Li, M.; et al. Cryo-EM Structure of an Extended SARS-CoV-2 Replication and Transcription Complex Reveals an Intermediate State in Cap Synthesis. Cell 2020, 184, 184–193.e10. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis. Med 2021, 2, 99–112. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Gadhave, K.; Kumar, P.; Kumar, A.; Bhardwaj, T.; Garg, N.; Giri, R. Conformational dynamics of 13 amino acids long NSP11 of SARS-CoV-2 under membrane mimetics and different solvent conditions. Microb. Pathog. 2021, 158, 105041. [Google Scholar] [CrossRef]
- Eskier, D.; Suner, A.; Oktay, Y.; Karakülah, G. Mutations of SARS-CoV-2 nsp14 exhibit strong association with increased genome-wide mutation load. PeerJ 2020, 8, e10181. [Google Scholar] [CrossRef]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV -2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef]
- Deng, X.; Baker, S.C. An “Old” protein with a new story: Coronavirus endoribonuclease is important for evading host antiviral defenses. Virology 2018, 517, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Case, J.B.; Ashbrook, A.W.; Dermody, T.S.; Denison, M.R. Mutagenesis of S -Adenosyl- l -Methionine-Binding Residues in Coronavirus nsp14 N7-Methyltransferase Demonstrates Differing Requirements for Genome Translation and Resistance to Innate Immunity. J. Virol. 2016, 90, 7248–7256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalapbutr, P.; Kongtaworn, N.; Rungrotmongkol, T. Structural insight into the recognition of S-adenosyl-L-homocysteine and sinefungin in SARS-CoV-2 Nsp16/Nsp10 RNA cap 2′-O-Methyltransferase. Comput. Struct. Biotechnol. J. 2020, 18, 2757–2765. [Google Scholar] [CrossRef]
- Vithani, N.; Ward, M.D.; Zimmerman, M.I.; Novak, B.; Borowsky, J.H.; Singh, S.; Bowman, G.R. SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential. Biophys. J. 2021, 120, 2880–2889. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a Protein Causes Endoplasmic Reticulum Stress and Induces Ligand-Independent Downregulation of the Type 1 Interferon Receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef]
- Tan, Y.; Schneider, T.; Shukla, P.K.; Chandrasekharan, M.B.; Aravind, L.; Zhang, D. Unification and extensive diversification of M/Orf3-related ion channel proteins in coronaviruses and other nidoviruses. Virus Evol. 2021, 7, veab014. [Google Scholar] [CrossRef]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; Nakagawa, S.; Sato, K. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Hachim, A.; Kavian, N.; Cohen, C.A.; Chin, A.W.H.; Chu, D.K.W.; Mok, C.K.P.; Tsang, O.T.Y.; Yeung, Y.C.; Perera, R.A.P.M.; Poon, L.L.M.; et al. ORF8 and ORF3b antibodies are accurate serological markers of early and late SARS-CoV-2 infection. Nat. Immunol. 2020, 21, 1293–1301. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- Pandey, P.; Prasad, K.; Prakash, A.; Kumar, V. Insights into the biased activity of dextromethorphan and haloperidol towards SARS-CoV-2 NSP6: In silico binding mechanistic analysis. Klin. Wochenschr. 2020, 98, 1659–1673. [Google Scholar] [CrossRef]
- Nizamudeen, Z.A.; Xu, E.-R.; Karthik, V.; Halawa, M.; Arkill, K.P.; Jackson, A.M.; Bates, D.O.; Elmsley, J. Structural assessment of SARS-CoV2 accessory protein ORF7a predicts LFA-1 and Mac-1 binding potential. Biosci. Rep. 2021, 41, BSR20203837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Huang, C.; Zhou, Z.; Huang, Z.; Su, L.; Kang, S.; Chen, X.; Chen, Q.; He, S.; Rong, X.; et al. Structural insight reveals SARS-CoV-2 ORF7a as an immunomodulating factor for human CD14+ monocytes. iScience 2021, 24, 102187. [Google Scholar] [CrossRef]
- Ramasamy, S.; Subbian, S. Critical Determinants of Cytokine Storm and Type I Interferon Response in COVID-19 Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e00299-20. [Google Scholar] [CrossRef]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2021785118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Ι. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Liao, C.-H.; Wang, Q.; Tan, Y.-J.; Luo, R.; Qiu, Y.; Ge, X.-Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Fu, B.; Yin, S.; Li, Z.; Liu, H.; Zhang, H.; Xing, N.; Wang, Y.; Xue, W.; Xiong, Y.; et al. ORF8 contributes to cytokine storm during SARS-CoV-2 infection by activating IL-17 pathway. iScience 2021, 24, 102293. [Google Scholar] [CrossRef]
- Barrantes, F.J. Structural biology of coronavirus ion channels. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-W.; Zhang, H.-N.; Meng, Q.-F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.-X.; Wang, X.-N.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Brandherm, L.; Kobaš, A.M.; Klöhn, M.; Brüggemann, Y.; Pfaender, S.; Rassow, J.; Kreimendahl, S. Phosphorylation of SARS-CoV-2 Orf9b Regulates Its Targeting to Two Binding Sites in TOM70 and Recruitment of Hsp90. Int. J. Mol. Sci. 2021, 22, 9233. [Google Scholar] [CrossRef]
- Han, L.; Zhuang, M.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.; Zhang, X.; Gao, C.; Wang, P. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Shepley-McTaggart, A.; Sagum, C.A.; Oliva, I.; Rybakovsky, E.; DiGuilio, K.; Liang, J.; Bedford, M.T.; Cassel, J.; Sudol, M.; Mullin, J.M.; et al. SARS-CoV-2 Envelope (E) protein interacts with PDZ-domain-2 of host tight junction protein ZO1. PLoS ONE 2021, 16, e0251955. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Guardeno, J.M.J.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV -2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130. [Google Scholar] [CrossRef]
- Xia, B.; Shen, X.; He, Y.; Pan, X.; Liu, F.-L.; Wang, Y.; Yang, F.; Fang, S.; Wu, Y.; Duan, Z.; et al. SARS-CoV-2 envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target. Cell Res. 2021, 31, 847–860. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhuang, M.-W.; Han, L.; Zhang, J.; Nan, M.-L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.-H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 2020, 5, 299. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Shin, O. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Troya, J.; Bastard, P.; Planas-Serra, L.; Ryan, P.; Ruiz, M.; de Carranza, M.; Torres, J.; Martínez, A.; Abel, L.; Casanova, J.-L.; et al. Neutralizing Autoantibodies to Type I IFNs in >10% of Patients with Severe COVID-19 Pneumonia Hospitalized in Madrid, Spain. J. Clin. Immunol. 2021, 41, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Chauvineau-Grenier, A.; Bastard, P.; Servajean, A.; Gervais, A.; Rosain, J.; Jouanguy, E.; Cobat, A.; Casanova, J.-L.; Rossi, B. Autoantibodies Neutralizing Type I Interferons in 20% of COVID-19 Deaths in a French Hospital. J. Clin. Immunol. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Solanich, X.; Rigo-Bonnin, R.; Gumucio, V.-D.; Bastard, P.; Rosain, J.; Philippot, Q.; Perez-Fernandez, X.-L.; Fuset-Cabanes, M.-P.; Gordillo-Benitez, M.; Suarez-Cuartin, G.; et al. Pre-existing Autoantibodies Neutralizing High Concentrations of Type I Interferons in Almost 10% of COVID-19 Patients Admitted to Intensive Care in Barcelona. J. Clin. Immunol. 2021, 41, 1733–1744. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Marchand, B.; Velthuis, A.J.W.T.; Snijder, E.J.; Weiss, S.; Eoff, R.L.; Singh, K.; Sarafianos, S.G. Mechanism of Nucleic Acid Unwinding by SARS-CoV Helicase. PLoS ONE 2012, 7, e36521. [Google Scholar] [CrossRef] [Green Version]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Tangye, S.G.; Bucciol, G.; Meyts, I. Mechanisms underlying host defense and disease pathology in response to severe acute respiratory syndrome (SARS)-CoV2 infection: Insights from inborn errors of immunity. Curr. Opin. Allergy Clin. Immunol. 2021, 21, 515–524. [Google Scholar] [CrossRef]
- Sposito, B.; Broggi, A.; Pandolfi, L.; Crotta, S.; Clementi, N.; Ferrarese, R.; Sisti, S.; Criscuolo, E.; Spreafico, R.; Long, J.M.; et al. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell 2021, 184, 4953–4968.e16. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, I.; Fernbach, S.; Pohl, M.O.; Karakus, U.; Huber, M.; Trkola, A.; Stertz, S.; Hale, B.G. Antiviral Activity of Type I, II, and III Interferons Counterbalances ACE2 Inducibility and Restricts SARS-CoV-2. mBio 2020, 11, e01928-20. [Google Scholar] [CrossRef] [PubMed]
- Khosroshahi, L.M.; Rokni, M.; Mokhtari, T.; Noorbakhsh, F. Immunology, immunopathogenesis and immunotherapeutics of COVID-19; an overview. Int. Immunopharmacol. 2021, 93, 107364. [Google Scholar] [CrossRef]
- Liu, W.J.; Zhao, M.; Liu, K.; Xu, K.; Wong, G.; Tan, W.; Gao, G.F. T-cell immunity of SARS-CoV: Implications for vaccine development against MERS-CoV. Antivir. Res. 2016, 137, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.I.; Westerhof, L.; MacLeod, M.K.L. The roles of resident, central and effector memory CD4 T-cells in protective immunity following infection or vaccination. Immunology 2018, 154, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchan, J. Conserved HLA binding peptides from five non-structural proteins of SARS-CoV-2—An in silico glance. Hum. Immunol. 2020, 81, 588–595. [Google Scholar] [CrossRef]
- Nielsen, S.C.; Yang, F.; Jackson, K.J.; Hoh, R.A.; Röltgen, K.; Jean, G.H.; Stevens, B.A.; Lee, J.-Y.; Rustagi, A.; Rogers, A.J.; et al. Human B Cell Clonal Expansion and Convergent Antibody Responses to SARS-CoV-2. Cell Host Microbe 2020, 28, 516–525.e5. [Google Scholar] [CrossRef]
- Chung, J.Y.; Thone, M.N.; Kwon, Y.J. COVID-19 vaccines: The status and perspectives in delivery points of view. Adv. Drug Deliv. Rev. 2020, 170, 1–25. [Google Scholar] [CrossRef]
- Dos Santos, W.G. Impact of virus genetic variability and host immunity for the success of COVID-19 vaccines. Biomed. Pharmacother. 2021, 136, 111272. [Google Scholar] [CrossRef]
- Lumley, S.F.; O’Donnell, D.; Stoesser, N.E.; Matthews, P.C.; Howarth, A.; Hatch, S.B.; Marsden, B.D.; Cox, S.; James, T.; Warren, F.; et al. Antibody Status and Incidence of SARS-CoV-2 Infection in Health Care Workers. N. Engl. J. Med. 2021, 384, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Fakhroo, A.; AlKhatib, H.A.; Al Thani, A.A.; Yassine, H.M. Reinfections in COVID-19 Patients: Impact of Virus Genetic Variability and Host Immunity. Vaccines 2021, 9, 1168. [Google Scholar] [CrossRef] [PubMed]
- Abu-Raddad, L.J.; Chemaitelly, H.; Malek, J.A.; Ahmed, A.A.; Mohamoud, Y.A.; Younuskunju, S.; Ayoub, H.H.; Al Kanaani, Z.; Al Khal, A.; Al Kuwari, E.; et al. Assessment of the Risk of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Reinfection in an Intense Reexposure Setting. Clin. Infect. Dis. 2020, 73, e1830–e1840. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, L.; Kong, X.; Geng, J.; Xiao, D.; Ma, C.; Jiang, X.-M.; Wang, P.-H. Long-term coexistence of SARS-CoV-2 with antibody response in COVID-19 patients. J. Med. Virol. 2020, 92, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-F.; Tseng, S.-P.; Yen, C.-H.; Yang, J.-Y.; Tsao, C.-H.; Shen, C.-W.; Chen, K.-H.; Liu, F.-T.; Liu, W.-T.; Chen, Y.-M.A.; et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem. Biophys. Res. Commun. 2014, 451, 208–214. [Google Scholar] [CrossRef]
- Ulrich, H.; Pillat, M.M.; Tárnok, A. Dengue Fever, COVID-19 (SARS-CoV-2), and Antibody-Dependent Enhancement (ADE): A Perspective. Cytom. Part A 2020, 97, 662–667. [Google Scholar] [CrossRef]
- Tetro, J.A. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020, 22, 72–73. [Google Scholar] [CrossRef]
- Wen, J.; Cheng, Y.; Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Zhang, S.; Jiang, Y. Antibody-dependent enhancement of coronavirus. Int. J. Infect. Dis. 2020, 100, 483–489. [Google Scholar] [CrossRef]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Gangaev, A.; Ketelaars, S.L.C.; Isaeva, O.I.; Patiwael, S.; Dopler, A.; Hoefakker, K.; De Biasi, S.; Gibellini, L.; Mussini, C.; Guaraldi, G.; et al. Identification and characterization of a SARS-CoV-2 specific CD8+ T cell response with immunodominant features. Nat. Commun. 2021, 12, 2593. [Google Scholar] [CrossRef]
- Wang, M.; Wang, C.; Zhang, L. Inflammatory endotypes of CRSwNP and responses to COVID-19. Curr. Opin. Allergy Clin. Immunol. 2020, 21, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, Y.; Novelli, L.; Rojas, M.; De Santis, M.; Acosta-Ampudia, Y.; Monsalve, D.M.; Ramírez-Santana, C.; Costanzo, A.; Ridgway, W.M.; Ansari, A.A.; et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J. Autoimmun. 2020, 114, 102506. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alzahrani, K.J.; Cruz-Martins, N.; Batiha, G.E.-S. The potential role of neopterin in Covid-19: A new perspective. Mol. Cell. Biochem. 2021, 476, 4161–4166. [Google Scholar] [CrossRef]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; The NU SCRIPT Study Investigators; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef]
- Zhang, F.; Mears, J.R.; Shakib, L.; Beynor, J.; Shanaj, S.; Korsunsky, I.; Nathan, A.; Donlin, L.T.; Raychaudhuri, S.; Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med. 2021, 13, 64. [Google Scholar] [CrossRef]
- Ward, J.D.; Cornaby, C.; Schmitz, J.L. Indeterminate QuantiFERON Gold Plus Results Reveal Deficient Interferon Gamma Responses in Severely Ill COVID-19 Patients. J. Clin. Microbiol. 2021, 59, JCM0081121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wherry, E.J. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020, 20, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-J.; Xu, J.; Yin, J.-M.; Li, L.; Hou, W.; Zhang, L.-L.; Zhou, Z.; Yu, Y.-Z.; Li, H.-J.; Feng, Y.-M.; et al. Lower Circulating Interferon-Gamma Is a Risk Factor for Lung Fibrosis in COVID-19 Patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. J. Infect. Dis. 2020, 221, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.-Q.; Wang, Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef]
- Esmaeilzadeh, A.; Elahi, R. Immunobiology and immunotherapy of COVID-19: A clinically updated overview. J. Cell. Physiol. 2020, 236, 2519–2543. [Google Scholar] [CrossRef]
- Haroun, R.A.-H.; Osman, W.H.; Eessa, A.M. Interferon-γ-induced protein 10 (IP-10) and serum amyloid A (SAA) are excellent biomarkers for the prediction of COVID-19 progression and severity. Life Sci. 2021, 269, 119019. [Google Scholar] [CrossRef] [PubMed]
- Rokni, M.; Ahmadikia, K.; Asghari, S.; Mashaei, S.; Hassanali, F. Comparison of clinical, para-clinical and laboratory findings in survived and deceased patients with COVID-19: Diagnostic role of inflammatory indications in determining the severity of illness. BMC Infect. Dis. 2020, 20, 869. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Zhang, Z.; Prajapati, M.; Li, Y. Lymphopenia Caused by Virus Infections and the Mechanisms Beyond. Viruses 2021, 13, 1876. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.T.; Park, L.P.; Nicholson, B.; Veldman, T.; Zaas, A.K.; Turner, R.; Lambkin-Williams, R.; Gilbert, A.S.; Ginsburg, G.S.; Woods, C.W. Longitudinal analysis of leukocyte differentials in peripheral blood of patients with acute respiratory viral infections. J. Clin. Virol. 2013, 58, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Cizmecioglu, A.; Cizmecioglu, H.A.; Goktepe, M.H.; Emsen, A.; Korkmaz, C.; Tasbent, F.E.; Colkesen, F.; Artac, H. Apoptosis-induced T-cell lymphopenia is related to COVID-19 severity. J. Med. Virol. 2020, 93, 2867–2874. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Jafarzadeh, S.; Nozari, P.; Mokhtari, P.; Nemati, M. Lymphopenia an important immunological abnormality in patients with COVID-19: Possible mechanisms. Scand. J. Immunol. 2020, 93, e12967. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Martonik, D.; Parfieniuk-Kowerda, A.; Rogalska, M.; Flisiak, R. The Role of Th17 Response in COVID-19. Cells 2021, 10, 1550. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, H.; Dauphars, D.J.; He, Y.-W. A Potential Role of Interleukin 10 in COVID-19 Pathogenesis. Trends Immunol. 2020, 42, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, A.; Kurver, L.; Overheul, G.J.; Kooistra, E.J.; Abdo, W.F.; van Crevel, R.; Duivenvoorden, R.; Kox, M.; Oever, J.T.; Schouten, J.; et al. Interferon gamma immunotherapy in five critically ill COVID-19 patients with impaired cellular immunity: A case series. Med 2021, 2, 1163–1170.e2. [Google Scholar] [CrossRef] [PubMed]
- Kacar, M.; Cortes-Acevedo, P.; Patel, V.; Carter, C.; Hughes, P.; McGann, H.P.; Gkrania-Klotsas, E.; Baxendale, H.E.; Barcenas-Morales, G.; Doffinger, R.; et al. Neutralizing Anti-interferon-γ Autoantibodies: An Ameliorating Factor in COVID-19 Infection? J. Clin. Immunol. 2021, 41, 1531–1535. [Google Scholar] [CrossRef]
- Heuberger, J.; Trimpert, J.; Vladimirova, D.; Goosmann, C.; Lin, M.; Schmuck, R.; Mollenkopf, H.; Brinkmann, V.; Tacke, F.; Osterrieder, N.; et al. Epithelial response to IFN-γ promotes SARS-CoV-2 infection. EMBO Mol. Med. 2021, 13, e13191. [Google Scholar] [CrossRef]
- Rha, M.-S.; Jeong, H.W.; Ko, J.-H.; Choi, S.J.; Seo, I.-H.; Lee, J.S.; Sa, M.; Kim, A.R.; Joo, E.-J.; Ahn, J.Y.; et al. PD-1-Expressing SARS-CoV-2-Specific CD8+ T Cells Are Not Exhausted, but Functional in Patients with COVID-19. Immunity 2020, 54, 44–52.e3. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Doria, A.; Zen, M.; Bettio, S.; Gatto, M.; Bassi, N.; Nalotto, L.; Ghirardello, A.; Iaccarino, L.; Punzi, L. Autoinflammation and autoimmunity: Bridging the divide. Autoimmun. Rev. 2012, 12, 22–30. [Google Scholar] [CrossRef]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020, 16, 413–414. [Google Scholar] [CrossRef]
- Gusev, E.Y.; Zhuravleva, Y.A.; Zotova, N.V. Correlation of the Evolution of Immunity and Inflammation in Vertebrates. Biol. Bull. Rev. 2019, 9, 358–372. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Gratz, I.K.; Paw, J.S.; Abbas, A.K. Treating Human Autoimmunity: Current Practice and Future Prospects. Sci. Transl. Med. 2012, 4, 125sr1. [Google Scholar] [CrossRef] [Green Version]
- Arleevskaya, M.I.; Manukyan, G.; Inoue, R.; Aminov, R. Editorial: Microbial and Environmental Factors in Autoimmune and Inflammatory Diseases. Front. Immunol. 2017, 8, 243. [Google Scholar] [CrossRef] [Green Version]
- Costenbader, K.H.; Gay, S.; Alarcón-Riquelme, M.E.; Iaccarino, L.; Doria, A. Genes, epigenetic regulation and environmental factors: Which is the most relevant in developing autoimmune diseases? Autoimmun. Rev. 2012, 11, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.S.; Vanderlugt, C.J.; Hashimoto, T.; Nishikawa, T.; Zone, J.J.; Black, M.M.; Wojnarowska, F.; Stevens, S.R.; Chen, M.; Fairley, J.; et al. Epitope Spreading: Lessons From Autoimmune Skin Diseases. J. Investig. Dermatol. 1998, 110, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Tuohy, V.K.; Kinkel, R.P. Epitope Spreading: A Mechanism for Progression of Autoimmune Disease. Autoimmunity 2001, 48, 39–48. [Google Scholar] [CrossRef]
- Siloşi, I.; Siloşi, C.A.; Boldeanu, M.V.; Cojocaru, M.; Biciuşcă, V.; Avrămescu, C.S.; Cojocaru, I.M.; Bogdan, M.; Folcuţi, R.M. The role of autoantibodies in health and disease. Romanian J. Morphol. Embryol. 2016, 57, 633–638. [Google Scholar]
- Khavinson, V.; Terekhov, A.; Kormilets, D.; Maryanovich, A. Homology between SARS CoV-2 and human proteins. Sci. Rep. 2021, 11, 17199. [Google Scholar] [CrossRef]
- Kasperkiewicz, M. COVID-19, heat shock proteins, and autoimmune bullous diseases: A potential link deserving further attention. Cell Stress Chaperones 2020, 26, 1–2. [Google Scholar] [CrossRef]
- Marino Gammazza, A.; Légaré, S.; Lo Bosco, G.; Fucarino, A.; Angileri, F.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. Human molecular chaperones share with SARS-CoV-2 antigenic epitopes potentially capable of eliciting autoimmunity against endothelial cells: Possible role of molecular mimicry in COVID-19. Cell Stress Chaperones 2020, 25, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Winchester, N.E.; Calabrese, C.; Calabrese, L. The intersection of COVID-19 and autoimmunity: What is our current understanding? Pathog. Immun. 2021, 6, 31–54. [Google Scholar] [CrossRef]
- Krakauer, T. Staphylococcal Superantigens: Pyrogenic Toxins Induce Toxic Shock. Toxins 2019, 11, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alouf, J.E.; Müller-Alouf, H. Staphylococcal and streptococcal superantigens: Molecular, biological and clinical aspects. Int. J. Med. Microbiol. 2003, 292, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Scaglioni, V.; Soriano, E.R. Are superantigens the cause of cytokine storm and viral sepsis in severe COVID-19? Observations and hypothesis. Scand. J. Immunol. 2020, 92, e12944. [Google Scholar] [CrossRef] [PubMed]
- Lappin, E.; Ferguson, A. Gram-positive toxic shock syndromes. Lancet Infect. Dis. 2009, 9, 281–290. [Google Scholar] [CrossRef]
- Moreews, M.; Le Gouge, K.; Khaldi-Plassart, S.; Pescarmona, R.; Mathieu, A.-L.; Malcus, C.; Djebali, S.; Bellomo, A.; Dauwalder, O.; Perret, M.; et al. Polyclonal expansion of TCR Vb 21.3 + CD4 + and CD8 + T cells is a hallmark of multisystem inflammatory syndrome in children. Sci. Immunol. 2021, 6, eabh1516. [Google Scholar] [CrossRef]
- Bukulmez, H. Current Understanding of Multisystem Inflammatory Syndrome (MIS-C) Following COVID-19 and Its Distinction from Kawasaki Disease. Curr. Rheumatol. Rep. 2021, 23, 58. [Google Scholar] [CrossRef]
- Morris, S.B.; Schwartz, N.G.; Patel, P.; Abbo, L.; Beauchamps, L.; Balan, S.; Lee, E.H.; Paneth-Pollak, R.; Geevarughese, A.; Lash, M.K.; et al. Case Series of Multisystem Inflammatory Syndrome in Adults Associated with SARS-CoV-2 Infection — United Kingdom and United States, March–August 2020. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 1450–1456. [Google Scholar] [CrossRef]
- Rivas, M.N.; Porritt, R.A.; Cheng, M.H.; Bahar, I.; Arditi, M. COVID-19–associated multisystem inflammatory syndrome in children (MIS-C): A novel disease that mimics toxic shock syndrome—the superantigen hypothesis. J. Allergy Clin. Immunol. 2020, 147, 57–59. [Google Scholar] [CrossRef]
- Brown, M.; Bhardwaj, N. Super(antigen) target for SARS-CoV-2. Nat. Rev. Immunol. 2021, 21, 72. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Porritt, R.A.; Rivas, M.N.; Krieger, J.M.; Ozdemir, A.B.; Garcia, G.; Arumugaswami, V.; Fries, B.C.; Arditi, M.; Bahar, I. A monoclonal antibody against staphylococcal enterotoxin B superantigen inhibits SARS-CoV-2 entry in vitro. Structure 2021, 29, 951–962.e3. [Google Scholar] [CrossRef] [PubMed]
- Kunkl, M.; Amormino, C.; Caristi, S.; Tedeschi, V.; Fiorillo, M.T.; Levy, R.; Popugailo, A.; Kaempfer, R.; Tuosto, L. Binding of Staphylococcal Enterotoxin B (SEB) to B7 Receptors Triggers TCR- and CD28-Mediated Inflammatory Signals in the Absence of MHC Class II Molecules. Front. Immunol. 2021, 12, 3316. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Zhang, S.; Porritt, R.A.; Rivas, M.N.; Paschold, L.; Willscher, E.; Binder, M.; Arditi, M.; Bahar, I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 25254–25262. [Google Scholar] [CrossRef]
- Porritt, R.A.; Paschold, L.; Rivas, M.N.; Cheng, M.H.; Yonker, L.M.; Chandnani, H.; Lopez, M.; Simnica, D.; Schultheiß, C.; Santiskulvong, C.; et al. HLA class I-associated expansion of TRBV11-2 T cells in Multisystem Inflammatory Syndrome in Children. J. Clin. Investig. 2021, 131, 10. [Google Scholar] [CrossRef] [PubMed]
- Porritt, R.A.; Binek, A.; Paschold, L.; Rivas, M.N.; McArdle, A.; Yonker, L.M.; Alter, G.; Chandnani, H.K.; Lopez, M.; Fasano, A.; et al. The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J. Clin. Investig. 2021, 131, 20. [Google Scholar] [CrossRef]
- Ramaswamy, A.; Brodsky, N.N.; Sumida, T.S.; Comi, M.; Asashima, H.; Hoehn, K.B.; Li, N.; Liu, Y.; Shah, A.; Ravindra, N.G.; et al. Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 2021, 54, 1083–1095.e7. [Google Scholar] [CrossRef]
- Kouo, T.; Chaisawangwong, W. SARS-CoV-2 as a superantigen in multisystem inflammatory syndrome in children (MIS-C). J. Clin. Investig. 2021, 131, 10. [Google Scholar] [CrossRef]
- Zotova, N.V.; Chereshnev, V.A.; Gusev, E.Y. Systemic Inflammation: Methodological Approaches to Identification of the Common Pathological Process. PLoS ONE 2016, 11, e0155138. [Google Scholar] [CrossRef]
- Zotova, N.V.; Zhuravleva, Y.A.; Zubova, T.E.; Gusev, E.Y. Integral estimation of systemic inflammatory response under sepsis. Gen. Physiol. Biophys. 2020, 39, 13–26. [Google Scholar] [CrossRef]
- Rea, I.M.; Alexander, H.D. Triple jeopardy in ageing: COVID-19, co-morbidities and inflamm-ageing. Ageing Res. Rev. 2021, 73, 101494. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J. Persistent SARS-2 infections contribute to long COVID-19. Med Hypotheses 2021, 149, 110538. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. COVID-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2020, 33, 155–162. [Google Scholar] [CrossRef]
- Costello, F.; Dalakas, M.C. Cranial neuropathies and COVID-19. Neurology 2020, 95, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Lucchese, G.; Flöel, A. Molecular mimicry between SARS-CoV-2 and respiratory pacemaker neurons. Autoimmun. Rev. 2020, 19, 102556. [Google Scholar] [CrossRef]
- Pascolini, S.; Vannini, A.; Deleonardi, G.; Ciordinik, M.; Sensoli, A.; Carletti, I.; Veronesi, L.; Ricci, C.; Pronesti, A.; Mazzanti, L.; et al. COVID-19 and Immunological Dysregulation: Can Autoantibodies be Useful? Clin. Transl. Sci. 2020, 14, 502–508. [Google Scholar] [CrossRef]
- Vink, M.; Vink-Niese, A. Could Cognitive Behavioural Therapy Be an Effective Treatment for Long COVID and Post COVID-19 Fatigue Syndrome? Lessons from the Qure Study for Q-Fever Fatigue Syndrome. Healthcare 2020, 8, 552. [Google Scholar] [CrossRef]
- El Sayed, S.; Shokry, D.; Gomaa, S.M. Post-COVID-19 fatigue and anhedonia: A cross-sectional study and their correlation to post-recovery period. Neuropsychopharmacol. Rep. 2020, 41, 50–55. [Google Scholar] [CrossRef]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Raveendran, A.V.; Jayadevan, R.; Sashidharan, S. Long COVID: An overview. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 869–875. [Google Scholar] [CrossRef]
- Dani, M.; Dirksen, A.; Taraborrelli, P.; Torocastro, M.; Panagopoulos, D.; Sutton, R.; Lim, P.B. Autonomic dysfunction in ‘long COVID’: Rationale, physiology and management strategies. Clin. Med. 2020, 21, e63–e67. [Google Scholar] [CrossRef] [PubMed]
- Kashif, A.; Chaudhry, M.; Fayyaz, T.; Abdullah, M.; Malik, A.; Anwer, J.M.A.; Inam, S.H.A.; Fatima, T.; Iqbal, N.; Shoaib, K. Follow-up of COVID-19 recovered patients with mild disease. Sci. Rep. 2021, 11, 13414. [Google Scholar] [CrossRef] [PubMed]
- Alquisiras-Burgos, I.; Peralta-Arrieta, I.; Alonso-Palomares, L.A.; Zacapala-Gómez, A.E.; Salmerón-Bárcenas, E.G.; Aguilera, P. Neurological Complications Associated with the Blood-Brain Barrier Damage Induced by the Inflammatory Response During SARS-CoV-2 Infection. Mol. Neurobiol. 2020, 58, 520–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Z.; Zhou, J.; Han, S.; Kang, Z.; Chuang, H.; Fan, H.; Zhao, H.; Wang, L.; Ning, Y.; et al. Next-Generation Sequencing and Proteomics of Cerebrospinal Fluid From COVID-19 Patients With Neurological Manifestations. Front. Immunol. 2021, 12, 782731. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood–Brain Barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef]
- Alam, S.B.; Willows, S.; Kulka, M.; Sandhu, J.K. Severe acute respiratory syndrome coronavirus 2 may be an underappreciated pathogen of the central nervous system. Eur. J. Neurol. 2020, 27, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Verma, A.; Ransohoff, R.M. The blood-brain barrier. Handb. Clin. Neurol. 2016, 133, 39–59. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Setti, L.; Passarini, F.; De Gennaro, G.; Barbieri, P.; Pallavicini, A.; Ruscio, M.; Piscitelli, P.; Colao, A.; Miani, A. Searching for SARS-COV-2 on Particulate Matter: A Possible Early Indicator of COVID-19 Epidemic Recurrence. Int. J. Environ. Res. Public Health 2020, 17, 2986. [Google Scholar] [CrossRef]
- Dembrovszky, F.; Váncsa, S.; Farkas, N.; Erőss, B.; Szakó, L.; Teutsch, B.; Bunduc, S.; Nagy, R.; Dohos, D.; Kiss, S.; et al. Immunoglobulin Response and Prognostic Factors in Repeated SARS-CoV-2 Positive Patients: A Systematic Review and Meta-Analysis. Viruses 2021, 13, 809. [Google Scholar] [CrossRef]
- Khoshkam, Z.; Aftabi, Y.; Stenvinkel, P.; Lawrence, B.P.; Rezaei, M.H.; Ichihara, G.; Fereidouni, S. Recovery scenario and immunity in COVID-19 disease: A new strategy to predict the potential of reinfection. J. Adv. Res. 2021, 31, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Xiao, X.; Li, B.; Zhu, W.; Li, Y.; Wu, J.; Huang, X.; Jin, J.; Chen, D.; Jin, J.; et al. Low Humoral Immune Response and Ineffective Clearance of SARS-Cov-2 in a COVID-19 Patient With CLL During a 69-Day Follow-Up. Front. Oncol. 2020, 10, 1272. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xiao, J.; Sun, R.; Tang, X.; Liang, C.; Lin, H.; Zeng, L.; Hu, J.; Yuan, R.; Zhou, P.; et al. Prolonged Persistence of SARS-CoV-2 RNA in Body Fluids. Emerg. Infect. Dis. 2020, 26, 1834–1838. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzi, C.; Henry, B.M.; Sanchis-Gomar, F.; Lippi, G. SARS-CoV-2 recurrent RNA positivity after recovering from coronavirus disease 2019 (COVID-19): A meta-analysis. Acta Biomed. 2020, 91, e2020014. [Google Scholar] [CrossRef] [PubMed]
- Brehm, T.; Pfefferle, S.; von Possel, R.; Kobbe, R.; Nörz, D.; Schmiedel, S.; Grundhoff, A.; Olearo, F.; Emmerich, P.; Robitaille, A.; et al. SARS-CoV-2 Reinfection in a Healthcare Worker Despite the Presence of Detectable Neutralizing Antibodies. Viruses 2021, 13, 661. [Google Scholar] [CrossRef] [PubMed]
- Wang, P. Recurrent presence of SARS-CoV-2 RNA in a 33-year-old man. J. Med. Virol. 2020, 93, 592–594. [Google Scholar] [CrossRef]
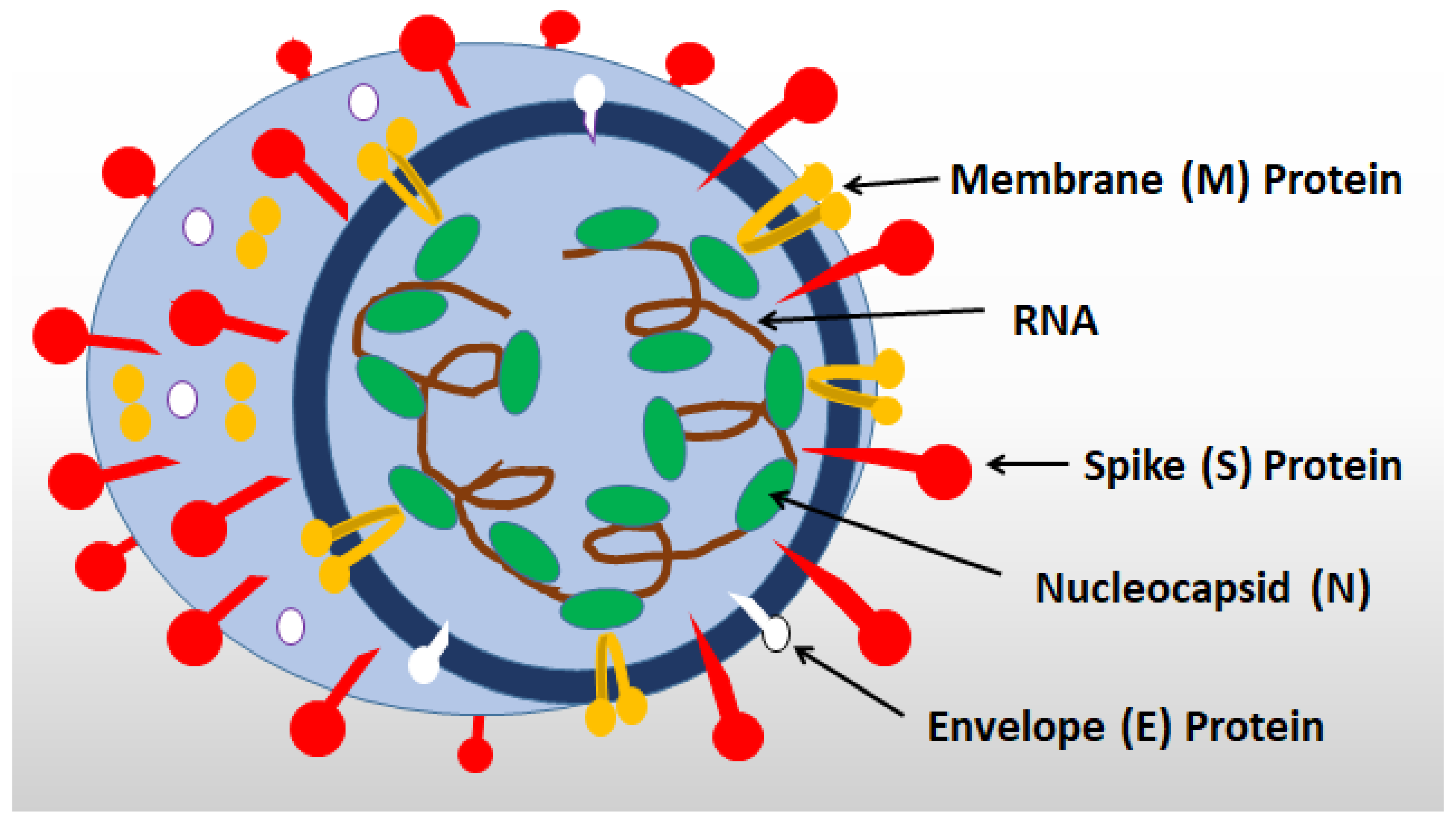
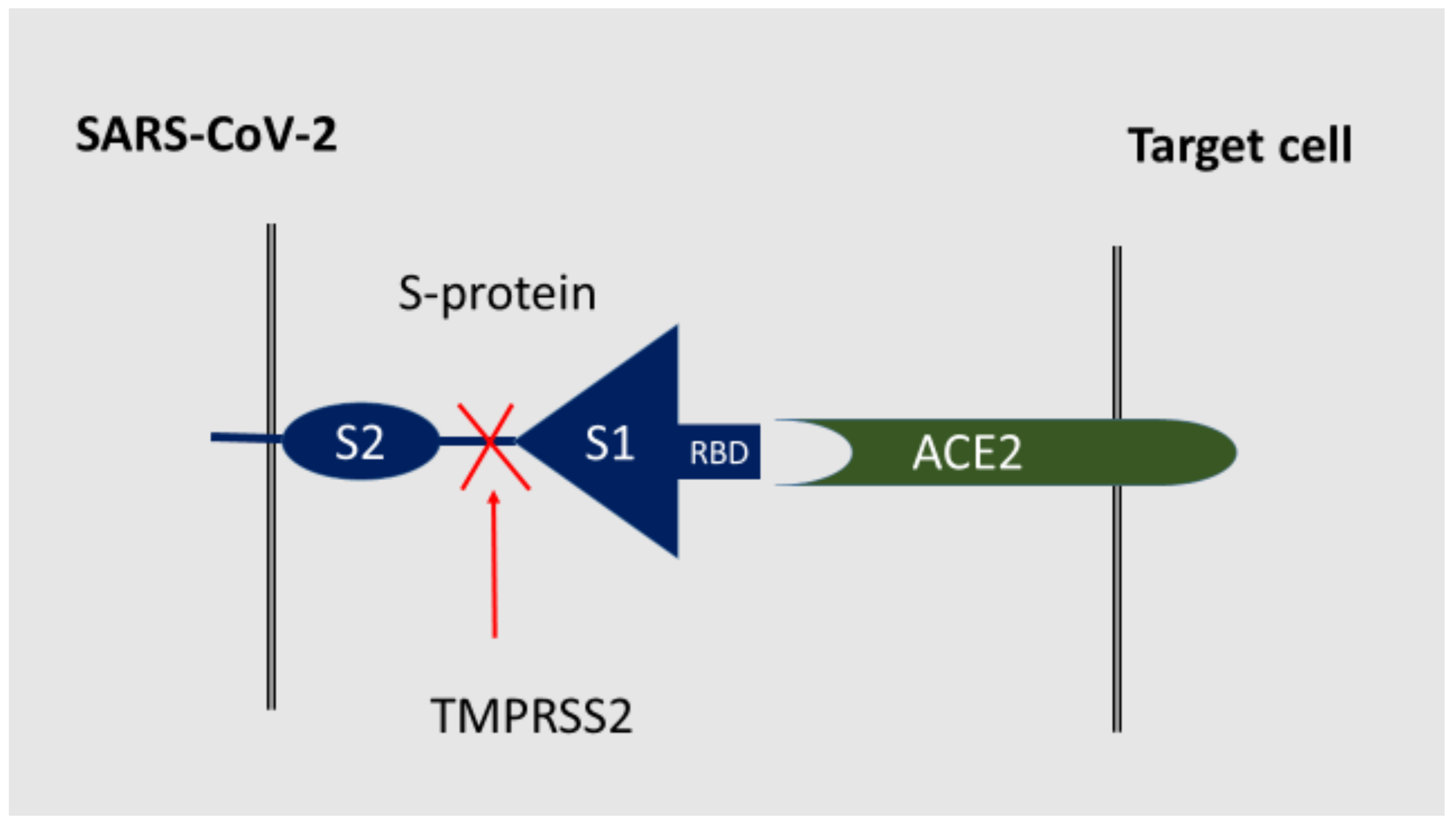
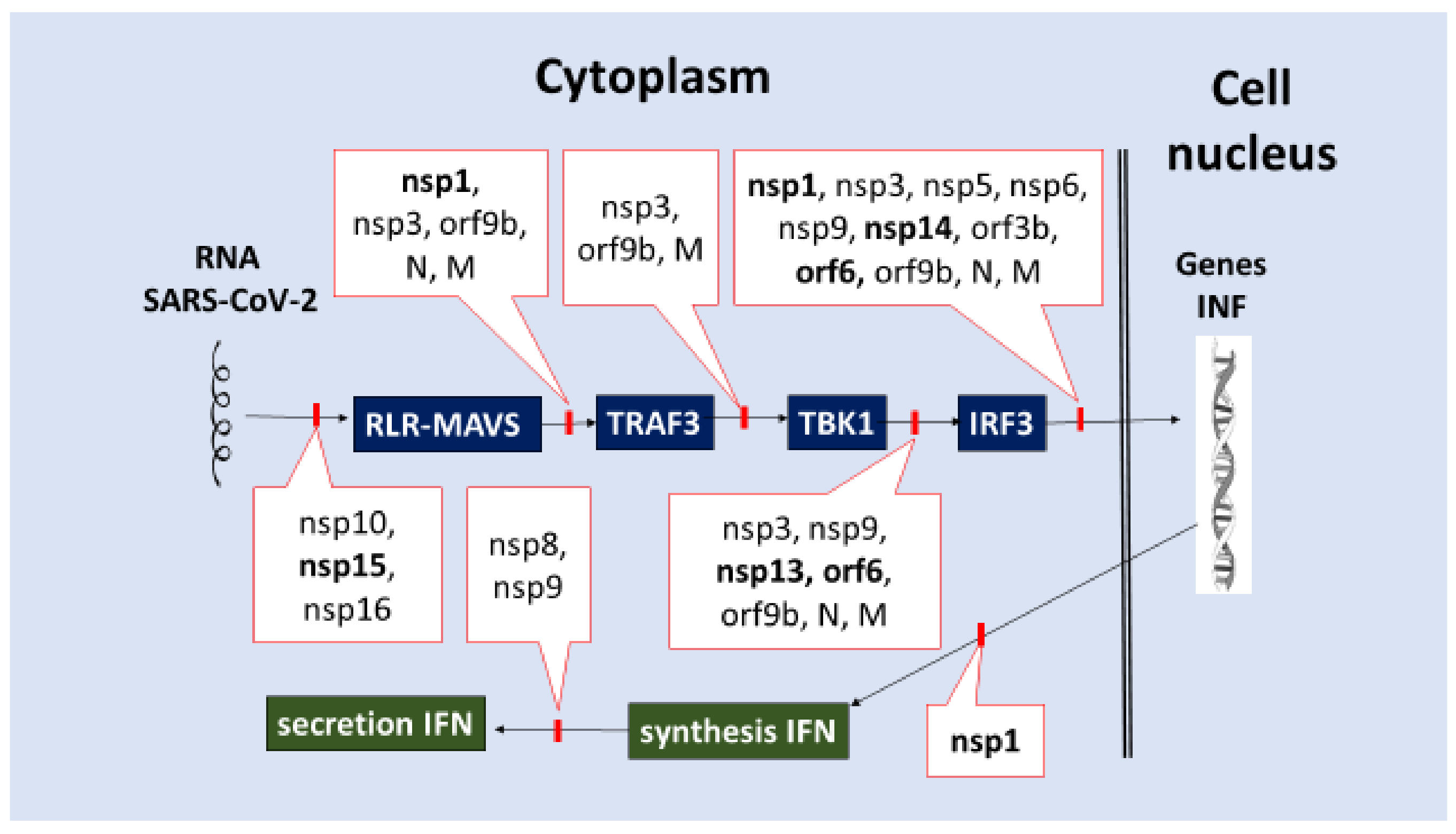
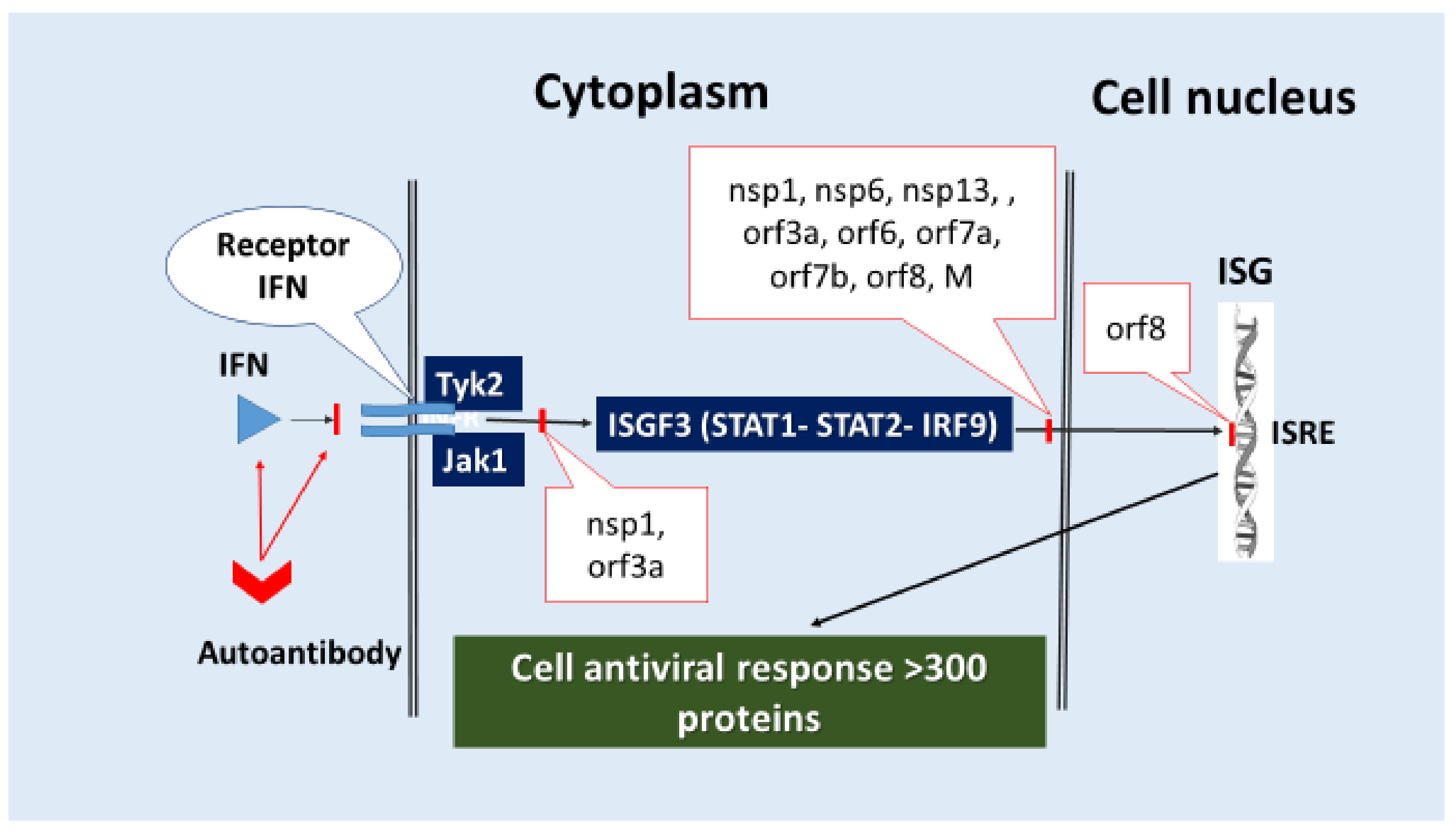
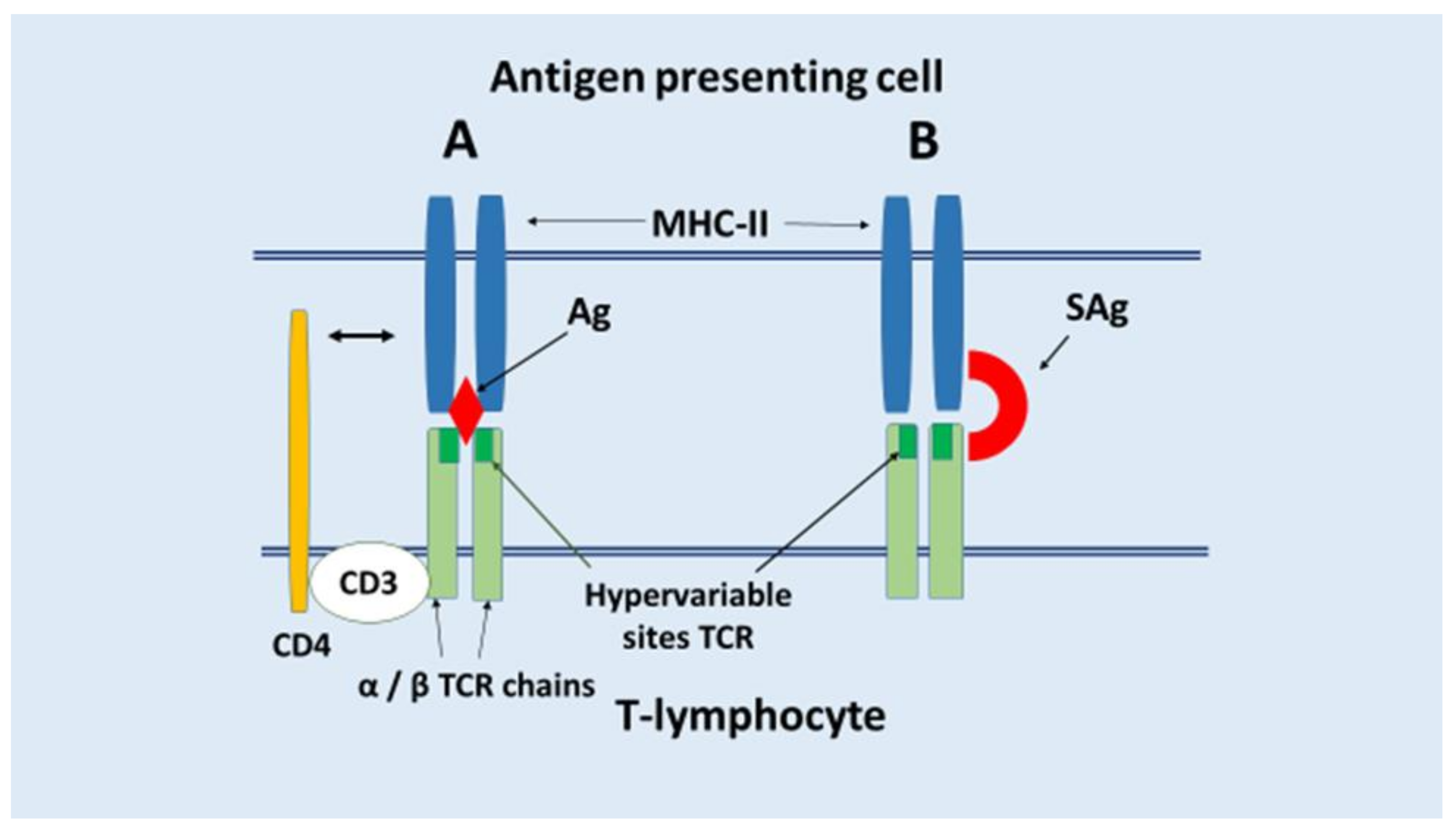
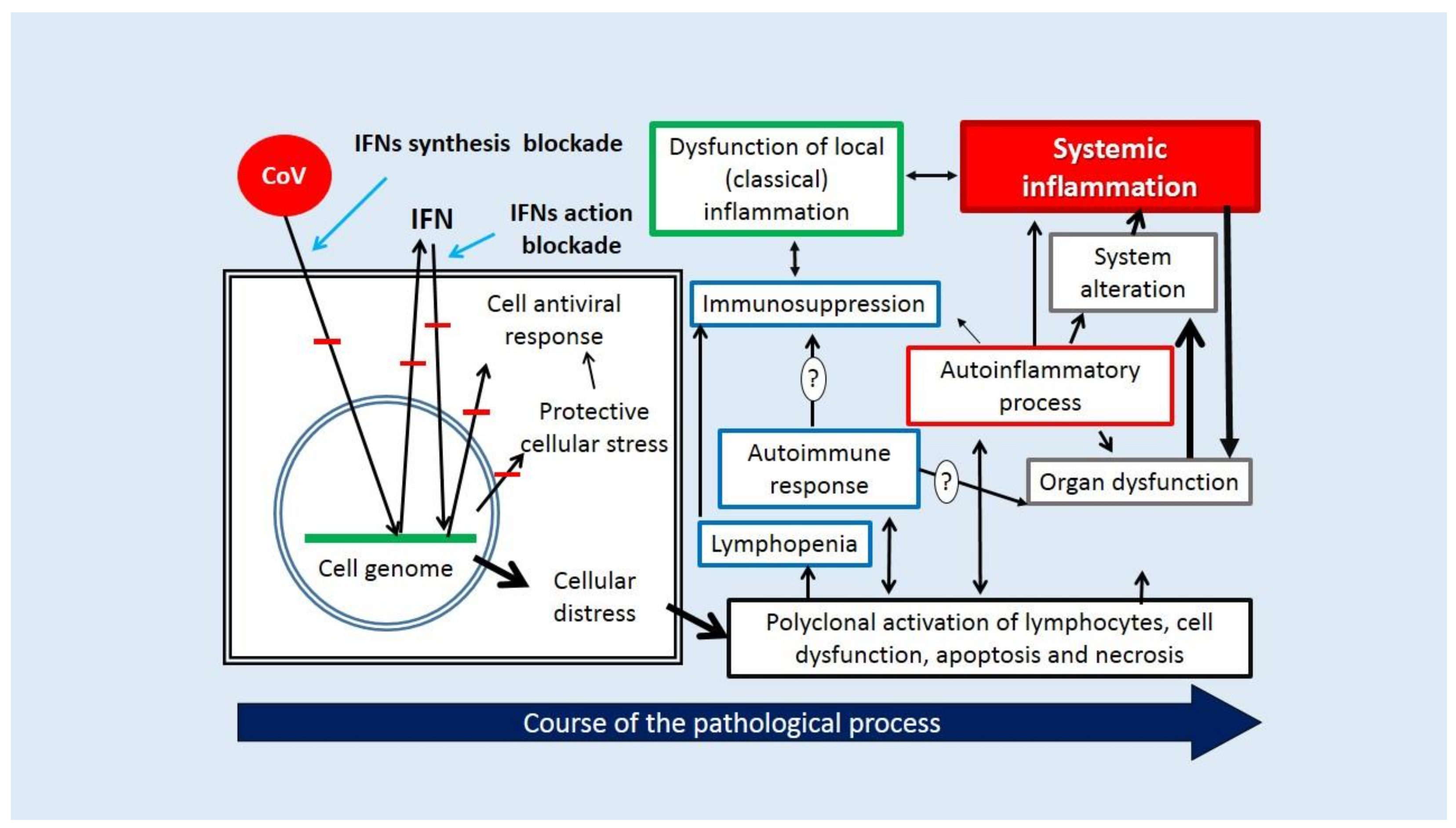

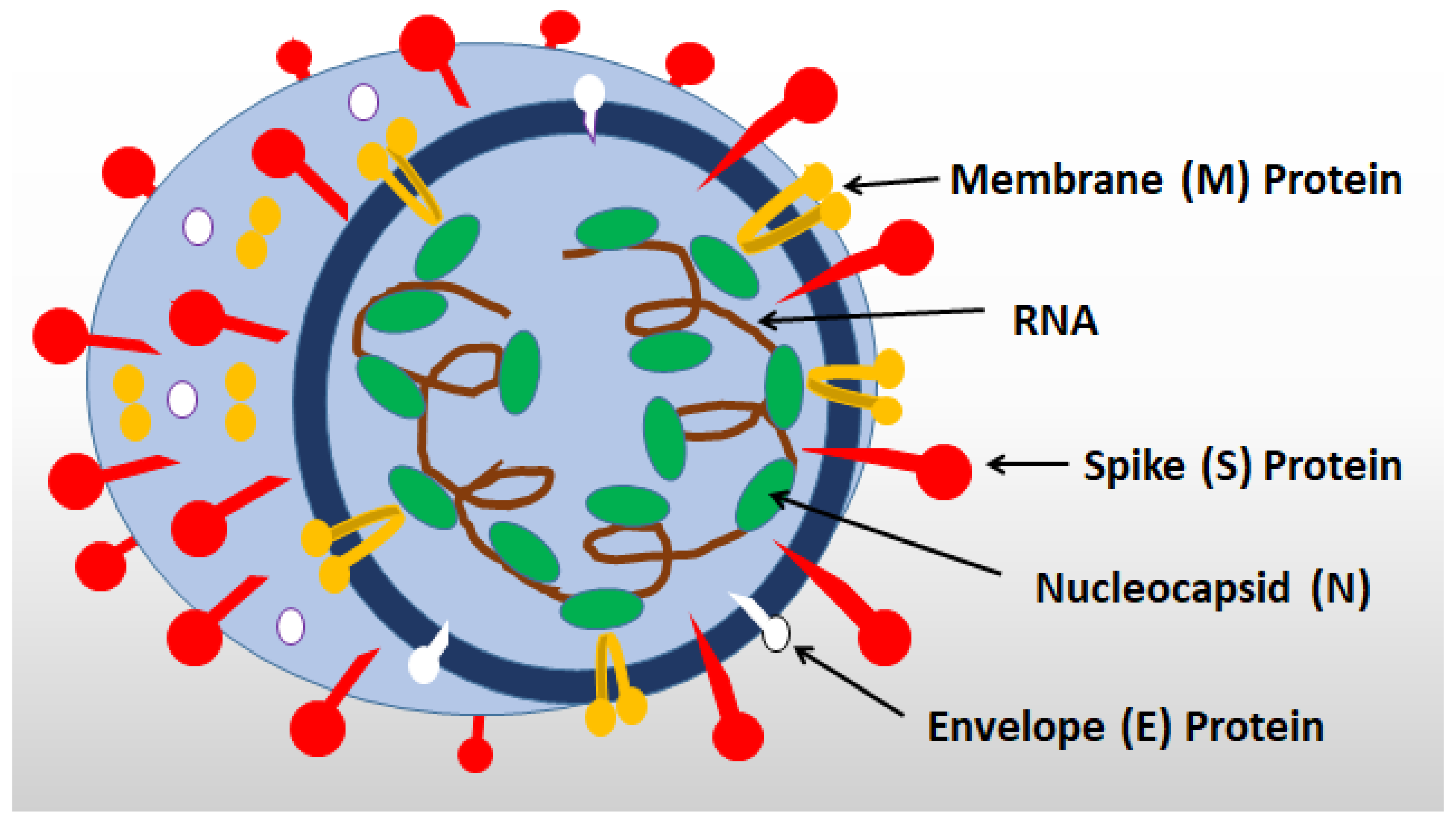{kind=link}
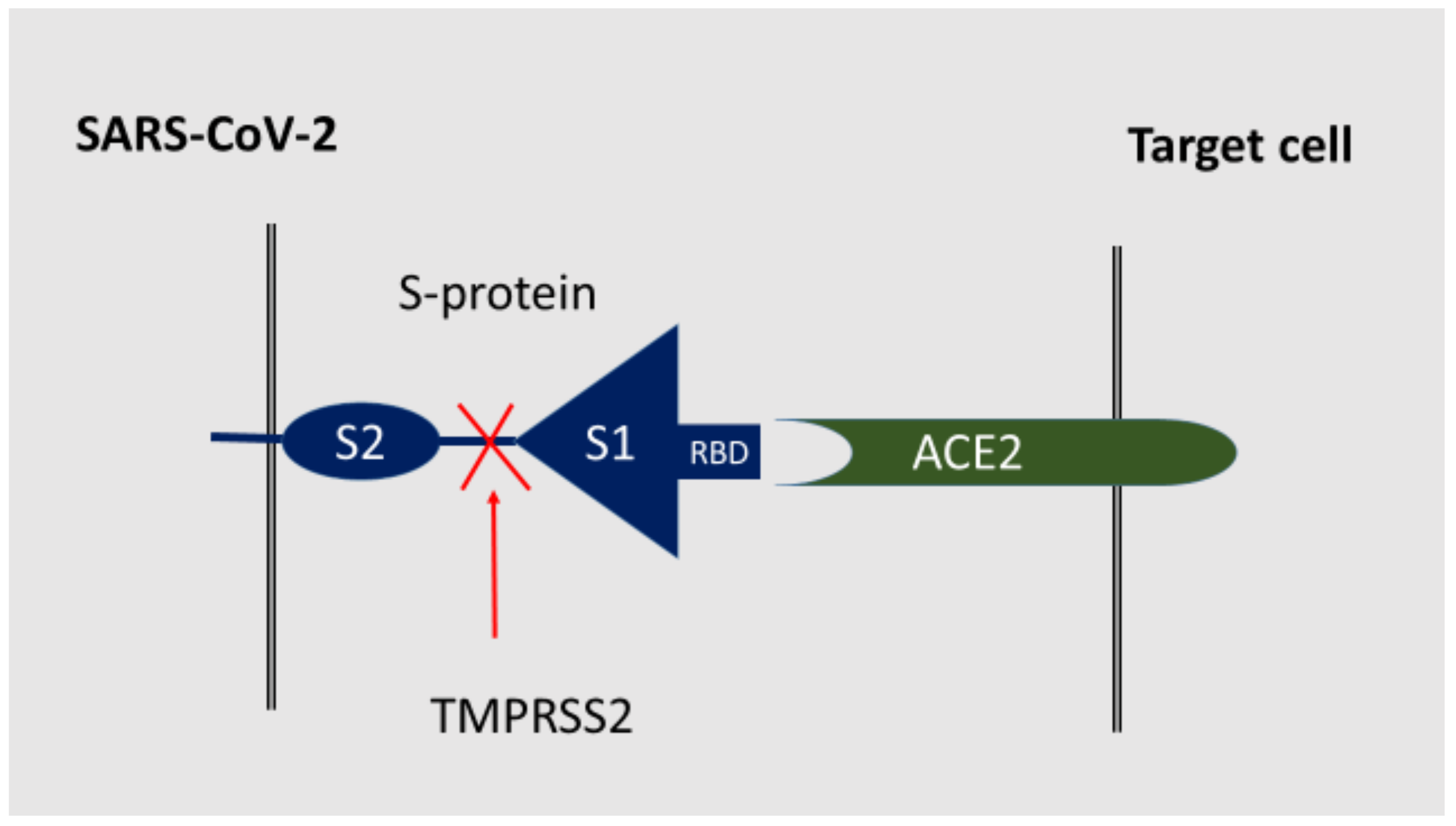{kind=link}
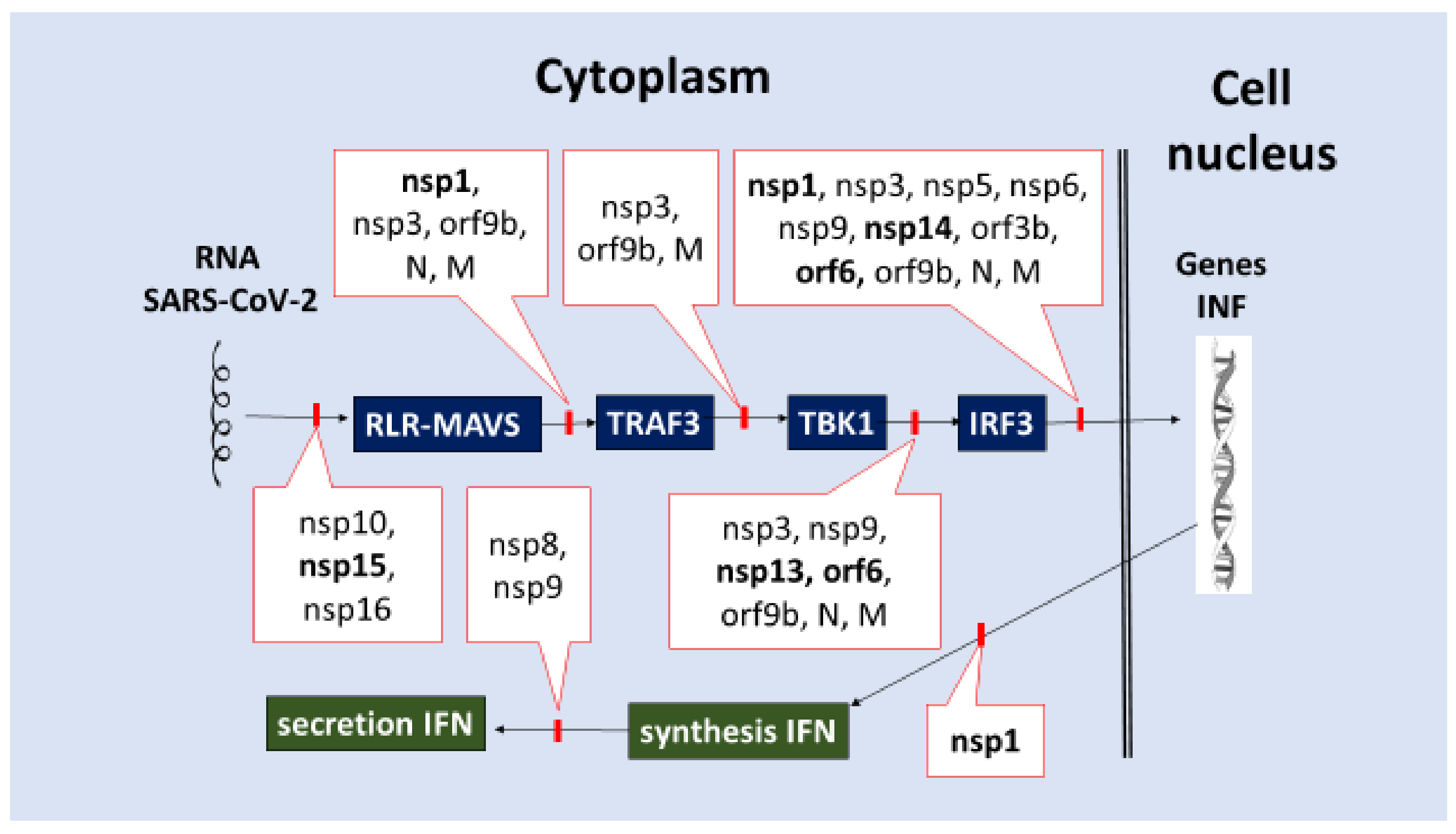{kind=link}
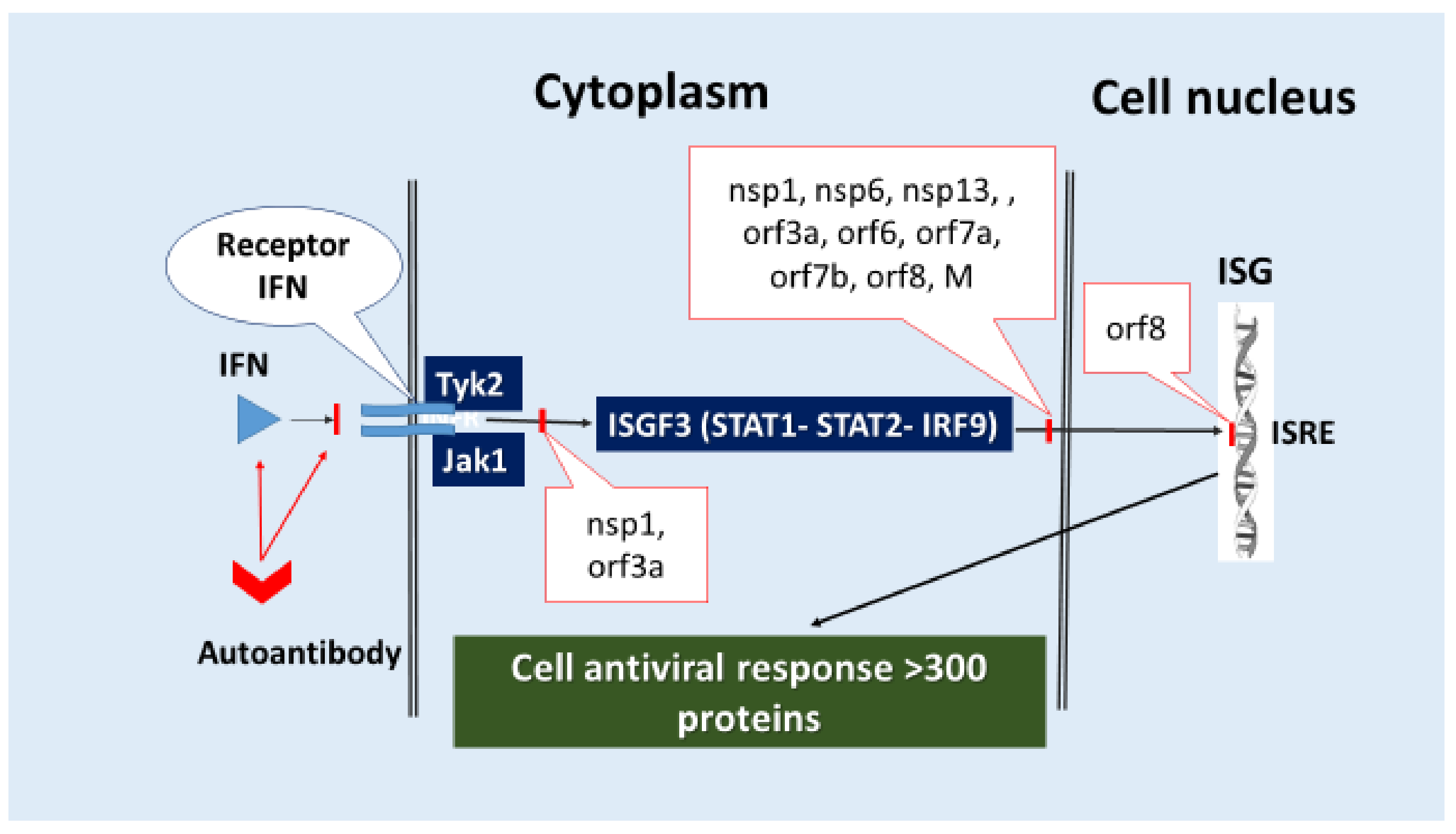{kind=link}
{kind=link}
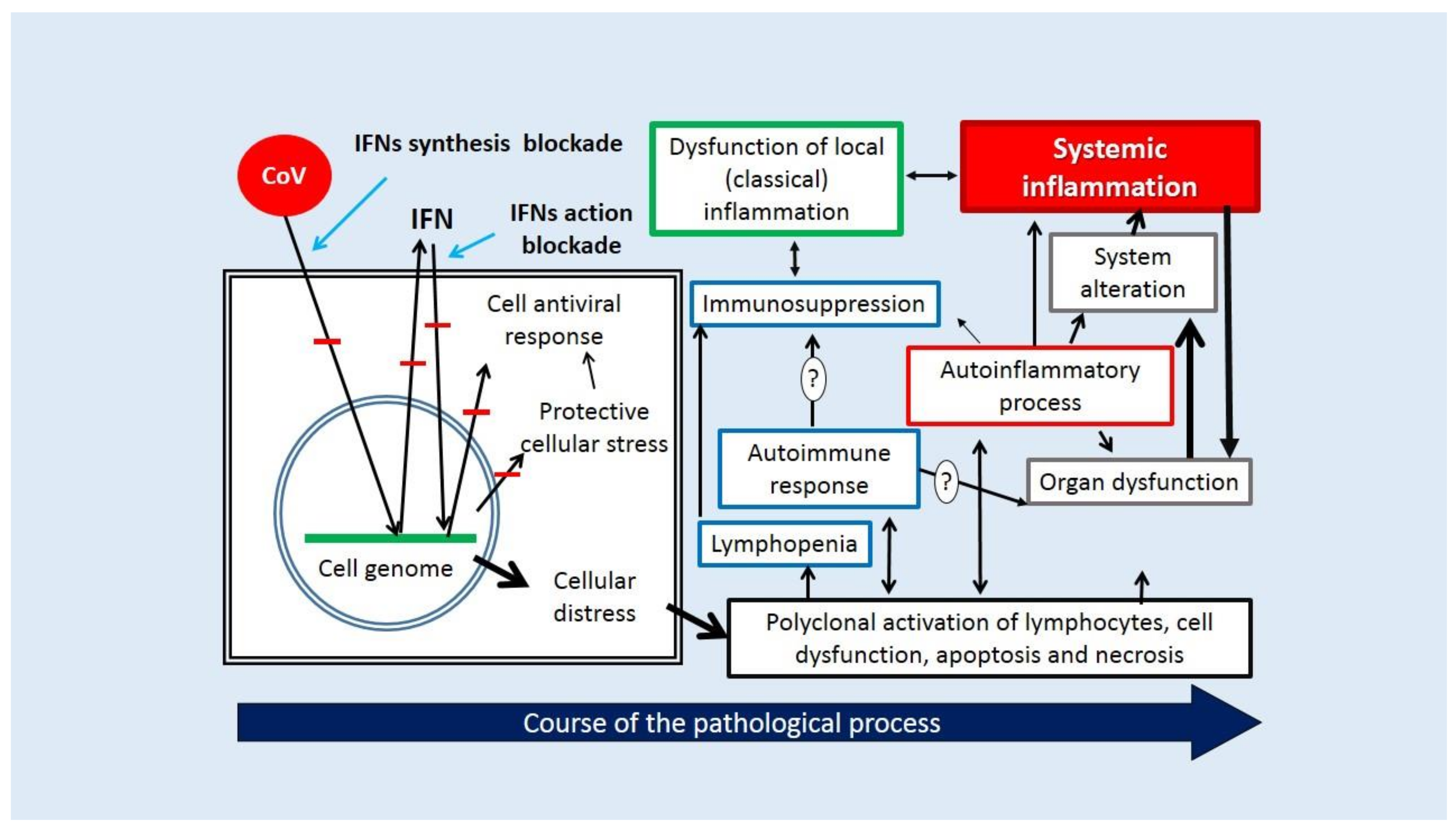{kind=link}
| Receptor [Ref] | Expression on Cells | Receptor Functions |
|---|---|---|
| ACE2 [83,84] | Epithelial cells, macrophages, platelets, endothelial cells, smooth muscle cells, many other cells. | Main receptor for SARS-CoV-1 and SARS-CoV-2; neutralization of Ang II; formation of anti-inflammatory 1-7 Ang. |
| Chondroitin sulfate [55,79] | Most of the cells. | Component of the cell glycocalyx; coreceptor for SARS-CoV-2 and several other viruses. |
| Neuropilin 1 (NRP1, CD304) [85,86,87] | Nerve cells of the brain and nasal cavity, endothelial cells. | Coreceptor for binding SARS-CoV-2; coreceptor for many growth factors. The receptor function of NRP-1 depends on the cleavage of S-protein by furin. |
| AXL [88] | Expression of AXL > ACE2 in many tissues, and in the lungs and bronchi. | A putative alternative receptor for SARS-CoV-2; binds several growth factors and phosphatidylserine. |
| CD147 (Basigin) [89,90] | It is widely expressed in human tissues, highly expressed on cells of the immune system. | The coreceptor and activator of ACE2 expression; receptor for many other viruses; polyfunctional membrane chaperone that binds and is activated by cyclophelin A and B. |
| GRP78 (BiP, HSPA5) [91,92,93,94,95] | On different cells. | Coreceptor for SARS-CoV-2; an inducible HSP of the HSP70 family; a cellular stress factor; a gateway for many viruses. |
| SR-B1 [35] | Proliferating cells, hepatocytes, macrophages, adrenal cells. | Coreceptor for SARS-CoV-2; the main receptor for the hepatitis C virus, the HDL receptor; SR. |
| SR-H2 (Stabilin-2) [96] | Macrophages, endothelial cells. | SR; a lectin; could potentially be involved in the uptake of SARS-CoV-2 by macrophages. |
| CD209L (L-SIGN) [97,98,99] | Endothelial cells, alveolar epithelium. | An independent receptor and a cofactor for ACE2 in SARS-CoV-2 infection; cell adhesion factor. |
| CD209 (DC-SIGN) [97,98,100] | Macrophages (including alveolar), dendritic cells. | SR and alternative receptor for SARS-CoV-2; Binding of various PAMPs, including SARS-CoV-1/2 and other viruses, cell adhesion. |
| SR-E2 (Dectin-1, CD369) [100] | Macrophages, monocytes, dendritic cells, neutrophils, eosinophils. | SR, binds PAMP (β-1,3 and β-1,6 glycans). Can recognize glycans on the SARS-CoV-2 S protein and integrate with TLR2 and TLR4. |
| SR-E3 (CD206, Mannose receptor 1) [100] | Macrophages, monocytes, dendritic cells. | SR, can bind mannose-rich microbial glycans, SARS-CoV-2 S1 protein and integrate with TLR2 and TLR4. |
| DPP4, (CD26) [101,102] | It is widely expressed on the surface of many cell types, including the respiratory epithelium. | The main receptor for MERS-CoV, a possible alternative receptor for SARS-CoV-2; a multifunctional receptor. |
| ANPE [63,103]. | Various epithelial cells, neutrophils. | A putative alternative receptor and ACE2 coreceptor for SARS-CoV-2; a well-known receptor for the entry of many CoVs. |
| ENPEP (CD249) [63,104]. | The expression pattern is similar to that of ACE2. | A putative alternative receptor and ACE2 coreceptor for SARS-CoV-2. Participates in the regulation of vascular tone. |
| ASGr1 [105] | Hepatocytes. | Receptor for many viruses, including hepatitis B virus; a possible alternative receptor for SARS-CoV-2. |
| KIM-1/TIM-1 [106] | Epithelium of the lungs and kidneys. | Probably an alternative ACE2 receptor for SARS-CoV-2. |
| Proteins | Functions |
|---|---|
| Nsp1 | Suppresses the host protein synthesis (including IFN-I and RIG-I) through association with ribosomes [155,156]. Inhibits IRF3 phosphorylation and IFN production; suppresses ISG induction due to Tyk2 and STAT2 depletion and inhibition of STAT1 phosphorylation [40,157]. Changes the structure and function of the cytoskeleton [158]. |
| Nsp2 | Can participate in the binding of nucleic acids and the regulation of intracellular signaling pathways [159]. Participates in the processes of viral replication [160]. Proteins that interact with nsp2 are involved in several biological processes, such as endosomal transport and translation [161]. Changes the synthesis and modification of lipids [158]. |
| Nsp3 | Papain-like protease (PLpro) and deubiquitinase. Participates in the proteolysis of 1a/1ab polyproteins [10,162]. Cleaves IRF3 [163]. By removing ubiquitin in the structures of signaling pathways, they disrupt the development of antiviral cellular stress, including translocation of IRF3 into the cell nucleus and inhibition of RIG-I, STING, TRAF3, TBK1, IRF3 [164]. Nsp3 SARS-CoV-1 c promotes inactivation of conjugated ubiquitin-like molecules such as interferon, which is involved in different pathways of the innate antiviral immune response regulation [165,166]. However, the deubiquitinase and IFN-blocking functions of nsp3 SARS-CoV-2 seem to be significantly lower than that of nsp3 SARS-CoV-1 [150]. Participates in the RTC formation [167]. |
| Nsp4 | An ER-localized transmembrane protein (as nsp3 and nsp6), is considered to be involved in the assembly of virus-induced cytoplasmic double-membrane vesicles [168]. Participates in the RTC formation [167]. |
| Nsp5 | 3-chymotrypsin-like “main” protease (3CLpro) is involved in the proteolysis of 1a/1ab polyproteins [10]. Inhibits the translocation of IRF3 into the cell nucleus and promotes the degradation of IRF3 [169]. It regulates (by proteolysis) the development of pro-inflammatory stress in different directions: activating the formation of NLRP12 by inflammasomes and inhibiting kinase - TGF-beta activated kinase 1 (MAP3K7 - mitogen-activated protein kinase 7) binding protein 1 (TAB1) [163]. Participates in the RTC formation [167]. |
| Nsp6 | Inhibits the phosphorylation of IRF3, STAT1, and STAT2 [110]. Potentially contributes to SARS-CoV-2 infection by impairing the ability of autophagosomes to deliver viral components to lysosomes for degradation [170,171]. Probably, along with orf7a, can interact with sigma receptors, which are involved in lipid remodeling and the ER stress response [161]. Participates in the RTC formation [167]. |
| Nsp7 | During viral RNA replication, the nsp12 cofactor forms a primase complex with nsp8 [167,172]. Participates in the RTC formation [167,173]. |
| Nsp8 | The nsp12 cofactor in viral RNA replication [162]. Participates in the formation of the RTC [167,173]. Forms a primase complex with nsp7, which plays a decisive role in the regulation of the activity of RNA-dependent RNA polymerase (RdRP) nsp12. [174,175]. Suppresses the transport of membrane proteins in cells infected with SARS-CoV-2, disrupts IFN secretion [176]. |
| Nsp9 | RNA-binding protein, interacting with nsp12, is one of the key factors of RTC [173,175]. Suppresses the transport of membrane proteins in cells infected with SARS-CoV-2, disrupts the secretion of IFN and some other cytokines [176]. Interferes with the activation of IRF3 and the induction of IFN synthesis by inhibiting TBK1 [20,138]. |
| Nsp10 | The cofactor of nsp14 and nsp16 forms functional complexes with them upon methylation of viral RNA [154,172]. Acting on NKRF (NF-κB-repressive factor), can regulate the levels of IL-8, NKRF and form a unique immune signature in COVID-19 [177]. Participates in the RTC formation [167]. |
| Nsp11 | Includes only 13 amino acid residues [178]. The role of the nsp11 protein in cells infected with CoV has not yet been studied [179]. |
| Nsp12 | RNA-dependent RNA polymerase is a key enzyme mediating the synthesis of all viral RNA molecules [162]. A key component of RTC [167,173,175]. |
| Nsp13 | Helicase, 5′-triphosphatase. Inhibits the phosphorylation of TBK1, which leads to a decrease in IRF3 activation, inhibits the phosphorylation of STAT1 and STAT2 [40]. Participates in viral replication [170]. Changes the structure and function of the cytoskeleton [158]. Participates in the RTC formation [167,173,175]. |
| Nsp14 | 3′-5 ′exoribonuclease and N-7-methyltransferase. Inhibits nuclear translocation of IRF3 [150]. Participates in the RTC formation [167,175]. As an exonuclease, it provides the ability to correct errors in the RNA synthesis complex, which allows SARS-CoV-2 to maintain its large genome [180]. |
| Nsp15 | NendoU, a uridylate-specific endoribonuclease. Inhibits nuclear translocation of IRF3 [150]. Participates in the RTC formation [167,175]. Meanwhile, nsp15-deficient CoVs are viable and can replicate [181], but with some delay and errors [182]. Another possible mechanism of nsp15 action is an evasion of viral double-stranded RNA recognition by host sensors in macrophages, including MDA5, PKR, and OAS/RNaseL [182]. |
| Nsp16 | 2-Oʹ-methyltransferase, which blocks the recognition of viral RNA by PRR [183,184] by forming the morpho-functional complexes with nsp10 [174]. Participates in the RTC formation [167,175]. |
| Orf3a | Disrupts the IFN signaling pathways by inhibiting STAT1 phosphorylation [40,177]. Can regulate the effects of IL-6 action by inhibiting STAT3 and STAT5 [177]. Promotes lysosomal degradation of the α-chain of the IFN-I receptor (IFNAR1) and induces ER stress (shown in SARS-CoV-1) [185,186]. Forms transmembrane ionic potassium-specific channels that facilitate the release of the virus from the cell [187]. It is assumed that viral ion channels can also promote membrane fusion and regulate viral replication and/or the packaging of genomic RNA into viral particles [187]. Activates NF-kB, NLRP3 inflammasomes, promoting cytokine storm [188,189]. Blocks the fusion of autophagosomes with lysosomes, contributing to the survival of the virus [136]. |
| Orf3b | Interferes with nuclear translocation of IRF3 [190]. Can cause a specific antibody response as N and orf8 proteins [191]. Orf3b is not expressed in many strains of SARS-CoV-2 [150], however, SARS-CoV-2 orf3b can suppress IFN-I induction more efficiently than its orthologue SARS-CoV-1 [190]. |
| Orf6 | Inhibits IRF3 (via action on TBK1) phosphorylation [177]. Binds to KPNA2 (karyopherin subunit alpha 2) and the Nup98-Rae1 complex, which inhibits nuclear translocation of IRF3 and STAT1 [40,150,192]. Causes changes in signaling pathways that include the activation of apoptosis through caspase-3-mediated, ER stress, and JNK-dependent pathways (JNK-c-Jun N-terminal kinases) [158]. Inhibition of STAT1 causes compensatory hyperactivation of STAT3 in cells infected with SARS-CoV-2, which promotes overproduction and activation of plasminogen activator inhibitor-1 (PAI-1) and can lead to coagulopathy [193]. |
| Orf7a | Inhibits the STAT2 phosphorylation [40]. According to the results of in silico calculations, can interact with sigma receptors, as nsp6 [194]. Has structural homology to ICAM-1, binds to Mac-1 and the integrin receptor LFA-1 on leukocytes. The orf7a expression leads to apoptosis, blocking the cell cycle, activation of NF-kB, and mitogen-activated protein kinase (MAPK) [195]. Binds to CD14 (TLR4 cofactor), promoting the production of proinflammatory cytokines [196]. Promotes the release of SARS-CoV-2 from the cell membrane by inhibiting the membrane protein BST-2 [197]. |
| Orf7b | Inhibits STAT1 and STAT2 phosphorylation [40]. |
| Orf8 | Interacts with a variety of host proteins and blocks the class 1 major histocompatibility complex (MHC-I) protein in the ER lumen [198,199]. Inhibits IFN signaling pathways and activation of ISG promoters [200]. Orf8, being secreted into the extracellular space can activate the IL-17 signaling pathway by interacting with the host’s IL17RA [201]. Forms cation channels upon assembly in the lipid bilayer, like orf3a and E-protein [202]. |
| Orf9b | Blocks the signaling pathways from TNF receptors by acting on TRAF3 and TRAF6 (TNF receptor-associated factor 3 and 6), disrupts IFN-I synthesis, and induces ATG5-mediated autophagy in host cells [189]. Blocks TOM70, a key adapter that transmits an antiviral signal from the mitochondrial RLR/MAVS pathway to TBK1/IRF3 to induce an IFN response [203,204]. Interacts with RIG-I, MDA-5, MAVS, STING, and TBK1, prevents phosphorylation and nuclear translocation of IRF3, NF-κB activation, and inhibits TRIF (TLR adapter) [205,206]. |
| S | Plays a key role in the process of receptor recognition and cell membrane fusion [207]. The structure of this protein allows it to interact with many receptors in host cells (Table 1). |
| E | Promotes the assembly and release of the virus, has the properties of a viroporin membrane channel, which can contribute to damage to the epithelial barrier, the pathogenesis, and the severity of COVID-19 [208]. The function of the ion channel is associated with the activation of inflammasomes and the development of hyperinflammation in the lungs [209,210]. These effects of E-protein are capable of initiating a cytokine storm and the development of an experimental analog of ARDS [211]. |
| M | The dominant structural protein can bind to other structural proteins such as S and E and determines the shape of the viral envelope [210]. Participates in the packaging of the genome in the viral particle, structures the viral particle [172]. Inhibits STAT1 phosphorylation [40]. M interacts with RIG-I, MAVS, and TBK1, thus preventing the formation of a multiprotein complex containing RIG-I, MAVS, TRAF3, and TBK1, and subsequently preventing phosphorylation, nuclear translocation, and activation of IRF3 [212]. |
| N | Binds viral RNA and protects the viral genome, participates in the assembly of the genomic RNA of the virus [172]. Modifies the signaling pathway of the cytokine TGF-β, blocking the apoptosis of infected host cells, but can also induce apoptosis by activating the mitochondrial pathway [158]. Promotes hyperinflammation by activating NLRP3 inflammasome [213]. It interferes with RIG-I activation, interferes with the association between TBK1 and IRF3, and prevents nuclear translocation of IRF3 [214]. Inhibits the formation of G3BP1-dependent stress granules during the development of the cell’s antiviral response and blocks the host mRNA [215]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716. https://doi.org/10.3390/ijms23031716
Gusev E, Sarapultsev A, Solomatina L, Chereshnev V. SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. International Journal of Molecular Sciences. 2022; 23(3):1716. https://doi.org/10.3390/ijms23031716
Chicago/Turabian StyleGusev, Evgenii, Alexey Sarapultsev, Liliya Solomatina, and Valeriy Chereshnev. 2022. "SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19" International Journal of Molecular Sciences 23, no. 3: 1716. https://doi.org/10.3390/ijms23031716
APA StyleGusev, E., Sarapultsev, A., Solomatina, L., & Chereshnev, V. (2022). SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. International Journal of Molecular Sciences, 23(3), 1716. https://doi.org/10.3390/ijms23031716