
Molecular Insights into Salinity Responsiveness in Contrasting Genotypes of Rice at the Seedling Stage

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
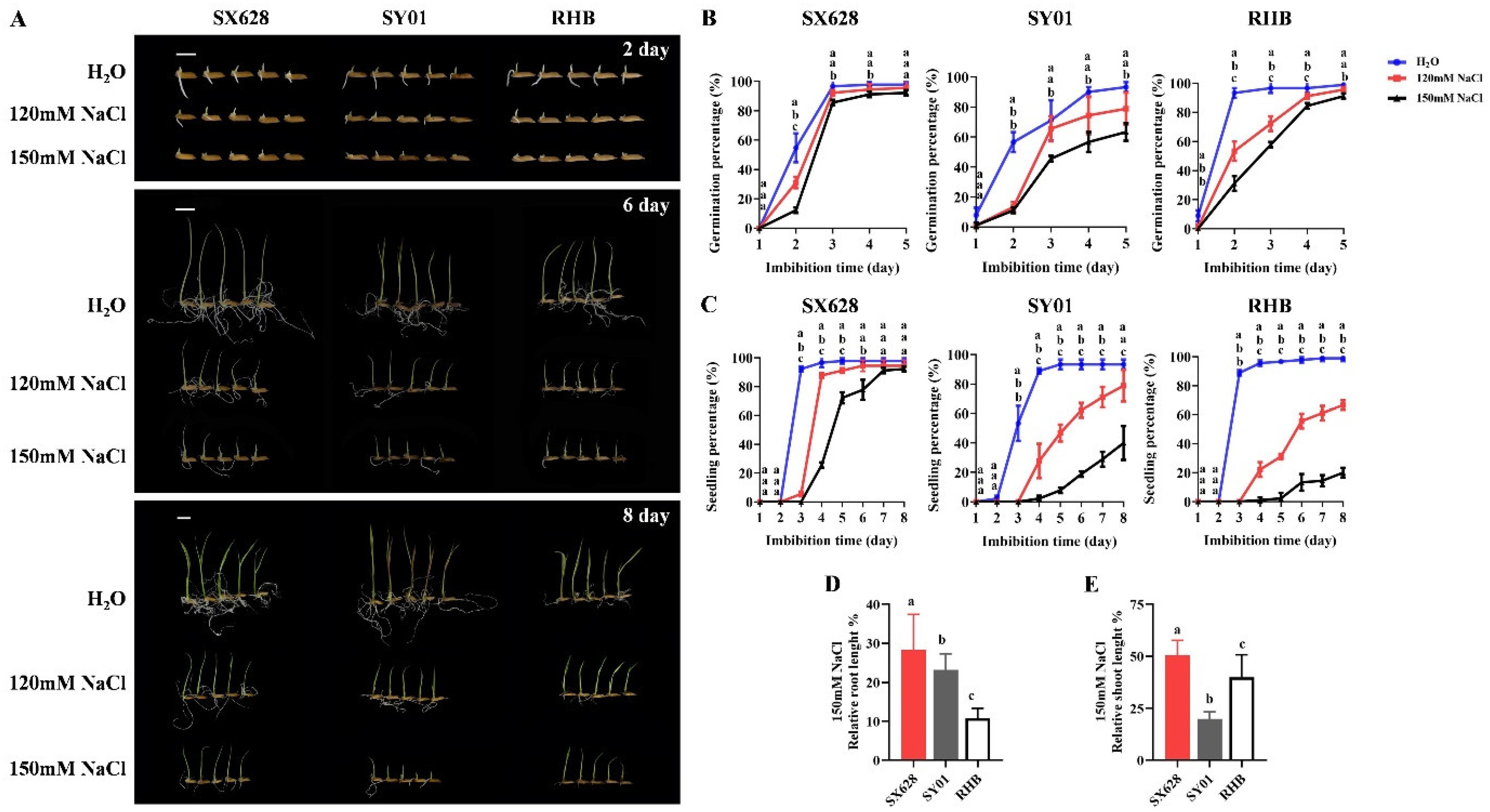
2.1. Phenotypic Characterization of Resistant and Sensitive Rice Cultivars in Response to Salinity Stress at Seedling Stage
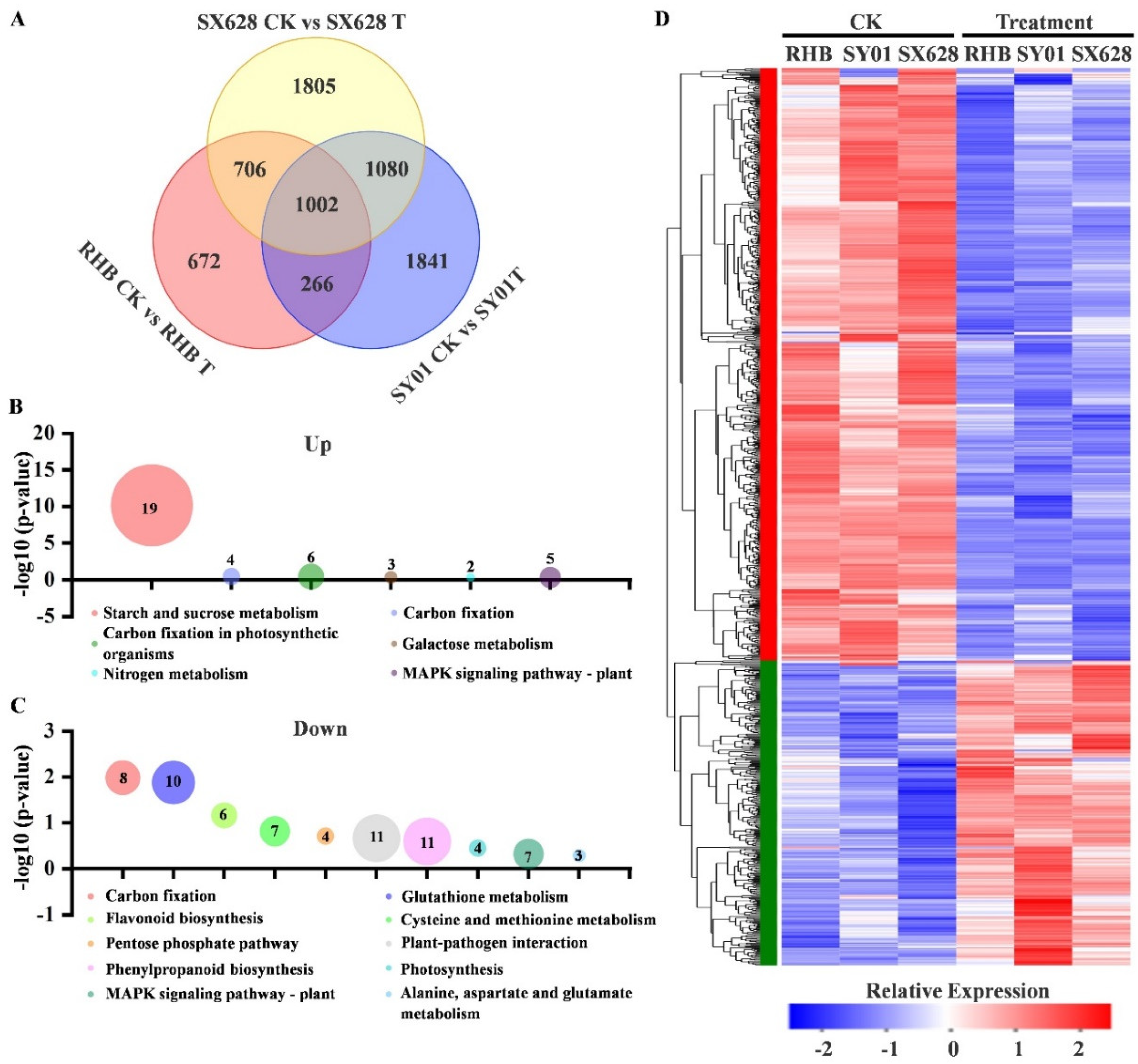
2.2. Transcriptome Analysis of Response to Salinity in Contrasting Genotypes Revealed That Several Metabolic Pathways Were Altered in Highly Salt-Tolerant and Salt-Sensitive Plants
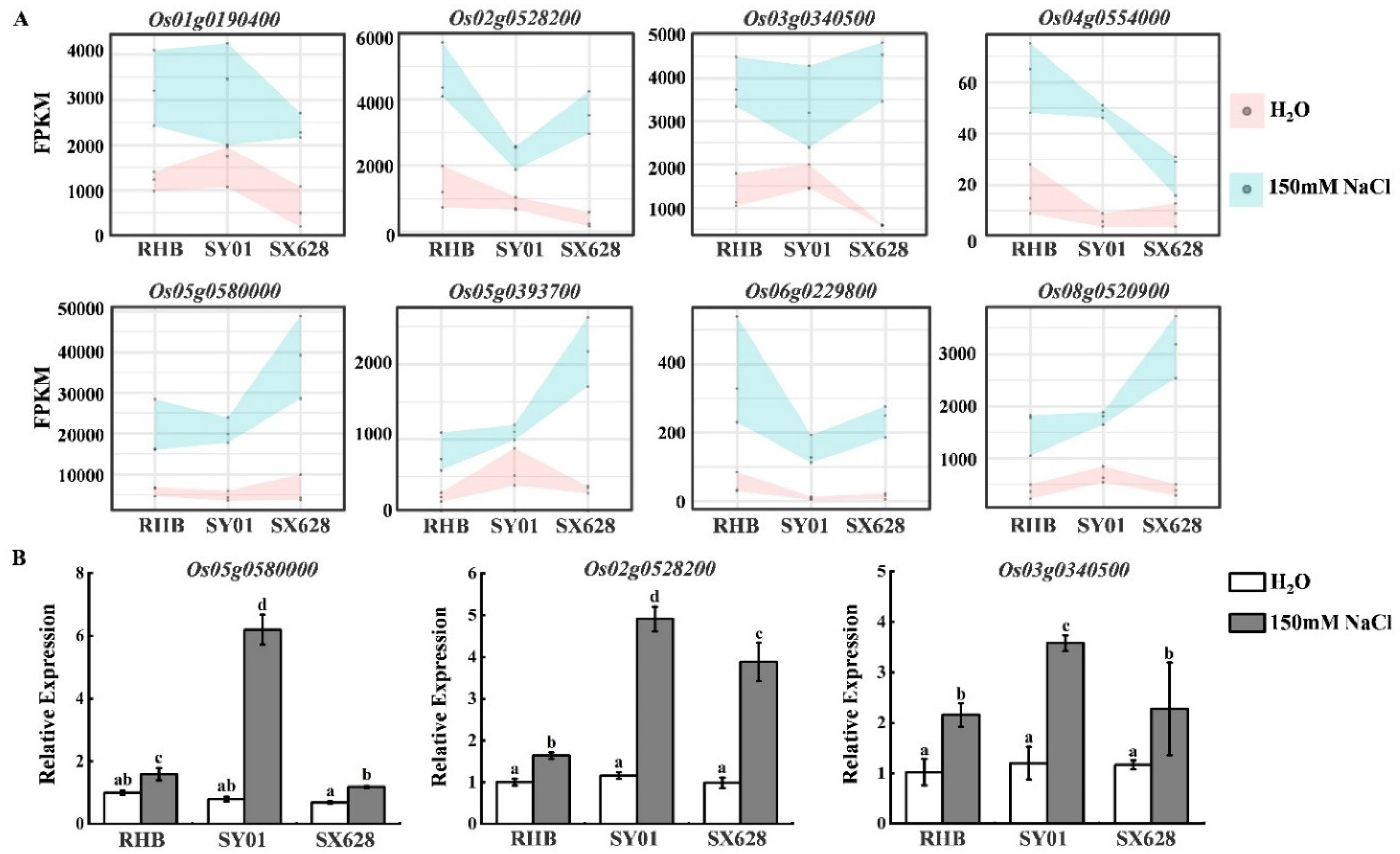
2.3. Activation of Genes in Rice after Salinity Treatment at Seedling Stage Is Associated with Starch and Sucrose Metabolism Pathways
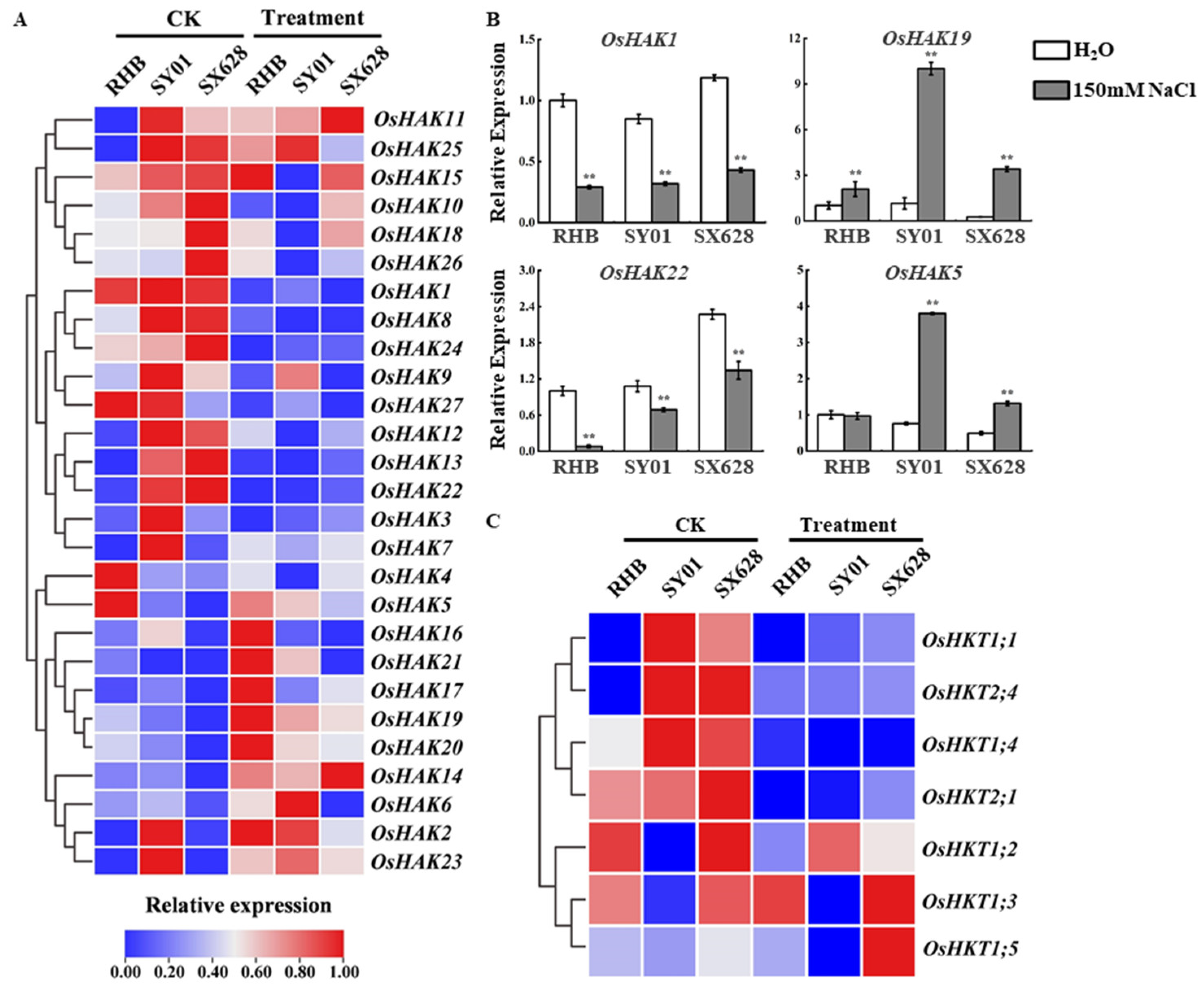
2.4. Dynamic Expression of HAKs and HKTs among Contrasting Rice Cultivars and Salinity Treatment at Seedling Stage
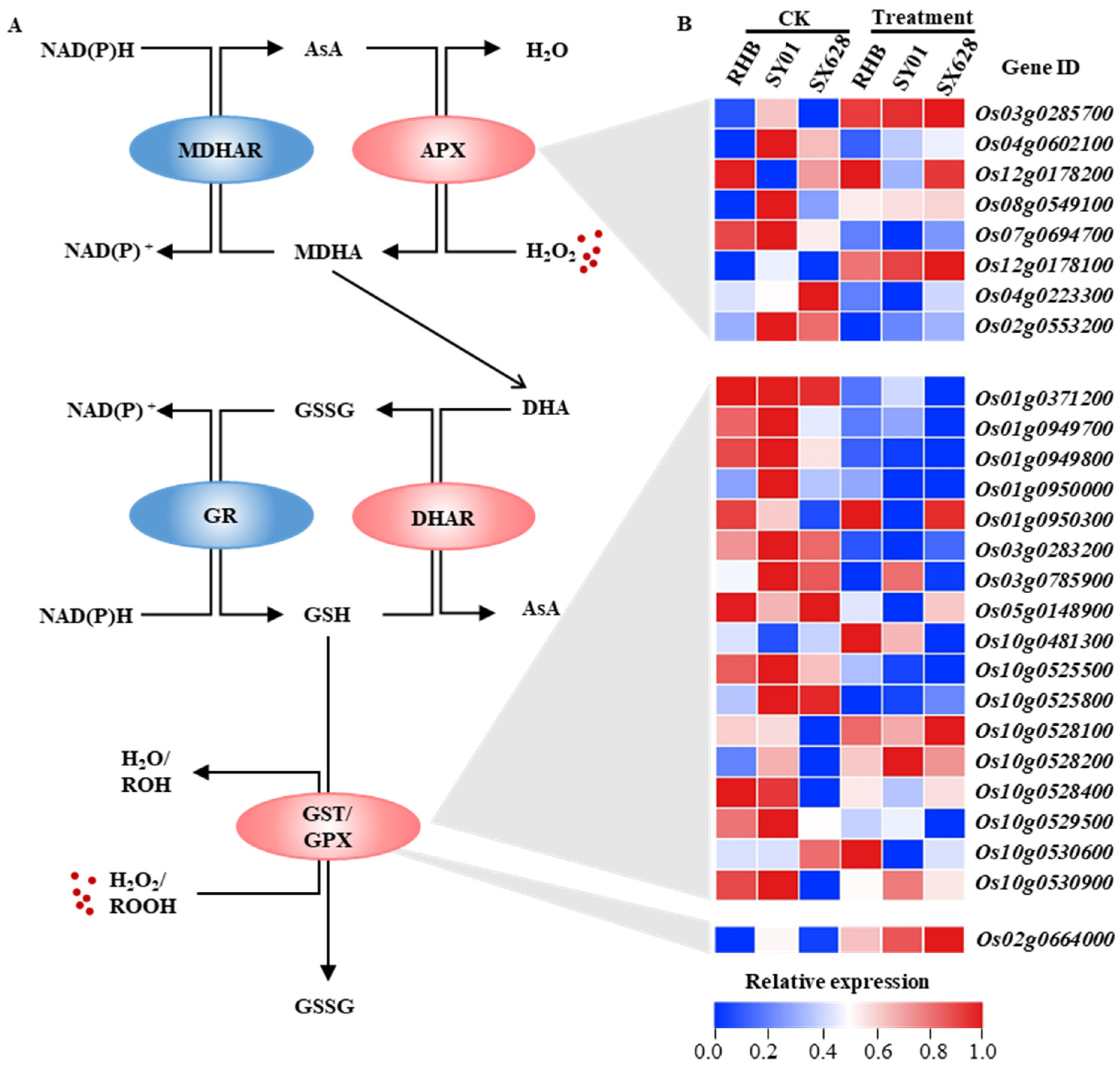
2.5. Several Genes Related to the AsA–GSH Pathway Displayed Significant Changes under Salinity Treatment in Contrasting Genotypes
3. Discussion
4. Materials and Methods
4.1. Evaluation of Seed Germination
4.2. Sample Preparation and RNA Sequencing
4.3. Transcriptome Data Analysis
4.4. Expression Analysis by Quantitative Reverse-Transcription PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reddy, I.N.B.L.; Kim, B.K.; Yoon, I.S.; Kim, K.H.; Kwon, T.R. Salt tolerance in rice: Focus on mechanisms and approaches. Rice Sci. 2017, 24, 123–144. [Google Scholar] [CrossRef]
- Zeng, L.; Poss, J.A.; Wilson, C.; Draz, A.S.E.; Gregorio, G.B.; Grieve, C.M. Evaluation of salt tolerance in rice genotypes by physiological characters. Euphytica 2003, 129, 281–292. [Google Scholar] [CrossRef]
- Flowers, T.J. Improving crop salt tolerance. J. Exp. Bot. 2004, 55, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Ganie, S.A.; Molla, K.A.; Henry, R.J.; Bhat, K.V.; Mondal, T.K. Advances in understanding salt tolerance in rice. Theor. Appl. Genet. 2019, 132, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, B.; Huang, B. Mechanism of salinity tolerance in plants: Physiological, biochemical, and molecular characterization. Int. J. Genom. 2014, 2014, 701596. [Google Scholar] [CrossRef]
- Qiu, Q.S.; Guo, Y.; Quintero, F.J.; Pardo, J.M.; Schumaker, K.S.; Zhu, J.K. Regulation of vacuolar Na+/H+ exchange in Arabidopsis thaliana by the salt-overly-sensitive (SOS) pathway. J. Biol. Chem. 2004, 279, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Chen, Z.Z.; Zhou, X.F.; Yin, H.B.; Li, X.; Xin, X.F.; Hong, X.H.; Zhu, J.K.; Gong, Z.Z. Overexpression of SOS (Salt Overly Sensitive) genes increases salt tolerance in transgenic Arabidopsis. Mol. Plant 2009, 2, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Almeida, D.M.; Oliveira, M.M.; Saibo, N.J. Regulation of Na+ and K+ homeostasis in plants: Towards improved salt stress tolerance in crop plants. Genet. Mol. Biol. 2017, 40, 326–345. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S. Role of sodium and hydrogen (Na+/H+) antiporters in salt tolerance of plants: Present and future challenges. Afr. J. Biotechnol. 2011, 10, 13693–13704. [Google Scholar]
- Assaha, D.V.; Ueda, A.; Saneoka, H.; Al-Yahyai, R.; Yaish, M.W. The role of Na+ and K+ transporters in salt stress adaptation in glycophytes. Front. Physiol. 2017, 8, 509. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.; Gilliham, M.; Hrmova, M. Plant high-affinity potassium (HKT) transporters involved in salinity tolerance: Structural insights to probe differences in ion selectivity. Int. J. Mol. Sci. 2013, 14, 7660–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachtman, D.P.; Schroeder, J.I. Structure and transport mechanism of a high-affinity potassium uptake transporter from higher plants. Nature 1994, 370, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Byrt, C.S.; Xu, B.; Krishnan, M.; Lightfoot, D.J.; Athman, A.; Jacobs, A.K.; Watson-Haigh, N.S.; Plett, D.; Tester, M.; Gilliham, M. The Na+ transporter, Ta HKT 1; 5-D, limits shoot Na+ accumulation in bread wheat. Plant J. 2014, 80, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Salt-induced tissue-specific cytosine methylation downregulates expression of HKT genes in contrasting wheat (Triticum aestivum L.) genotypes. DNA Cell Biol. 2017, 36, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Uozumi, N.; Kim, E.J.; Rubio, F.; Yamaguchi, T.; Muto, S.; Tsuboi, A.; Bakker, E.P.; Nakamura, T.; Schroeder, J.I. The Arabidopsis HKT1 gene homolog mediates inward Na+ currents in Xenopus laevis oocytes and Na+ uptake in Saccharomyces cerevisiae. Plant Physiol. 2000, 122, 1249–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rus, A.; Baxter, I.; Muthukumar, B.; Gustin, J.; Lahner, B.; Yakubova, E.; Salt, D.E. Natural variants of At HKT1 enhance Na+ accumulation in two wild populations of Arabidopsis. PLoS Genet. 2006, 2, e210. [Google Scholar] [CrossRef] [Green Version]
- Shkolnik-Inbar, D.; Adler, G.; Bar-Zvi, D. ABI4 downregulates expression of the sodium transporter HKT1; 1 in Arabidopsis roots and affects salt tolerance. Plant J. 2013, 73, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guan, C.; Wang, P.; Ma, Q.; Bao, A.K.; Zhang, J.L.; Wang, S.M. The Effect of AtHKT1; 1 or AtSOS1 mutation on the expressions of Na+ or K+ transporter genes and ion homeostasis in Arabidopsis thaliana under salt stress. Int. J. Mol. Sci. 2019, 20, 1085. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Spielmeyer, W.; Lagudah, E.S.; Munns, R. Comparative mapping of HKT genes in wheat, barley, and rice, key determinants of Na+ transport, and salt tolerance. J. Exp. Bot. 2008, 59, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Lan, W.Z.; Wang, W.; Wang, S.M.; Li, L.G.; Buchanan, B.B.; Lin, H.X.; Gao, J.P.; Luan, S. A rice high-affinity potassium transporter (HKT) conceals a calcium-permeable cation channel. Proc. Natl. Acad. Sci. USA 2010, 107, 7089–7094. [Google Scholar] [CrossRef] [Green Version]
- Shohan, M.U.S.; Sinha, S.; Nabila, F.H.; Dastidar, S.G.; Seraj, Z.I. HKT1; 5 transporter gene expression and association of amino acid substitutions with salt tolerance across rice genotypes. Front. Plant Sci. 2019, 10, 1420. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Yoshida, K.; Nakayama, H.; Yamada, K.; Oiki, S.; Shinmyo, A. Two types of HKT transporters with different properties of Na+ and K+ transport in Oryza sativa. Plant J. 2001, 27, 129–138. [Google Scholar] [CrossRef]
- Chen, T.; Shabala, S.; Niu, Y.; Chen, Z.H.; Shabala, L.; Meinke, H.; Venkataraman, G.; Pareek, A.; Xu, J.; Zhou, M. Molecular mechanisms of salinity tolerance in rice. Crop J. 2021, 9, 506–520. [Google Scholar] [CrossRef]
- Almeida, P.; Katschnig, D.; De Boer, A.H. HKT transporters—State of the art. Int. J. Mol. Sci. 2013, 14, 20359–20385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, T.; Costa, A.; Kim, T.H.; Han, M.J.; Horie, R.; Leung, H.Y.; Miyao, A.; Hirochika, H.; An, G.; Schroeder, J.I. Rice OsHKT2; 1 transporter mediates large Na+ influx component into K+-starved roots for growth. EMBO J. 2007, 26, 3003–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, F.; Horie, T. A conserved primary salt tolerance mechanism mediated by HKT transporters: A mechanism for sodium exclusion and maintenance of high K+/Na+ ratio in leaves during salinity stress. Plant Cell Environ. 2010, 33, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jing, W.; Xiao, L.; Jin, Y.; Shen, L.; Zhang, W. The rice high-affinity potassium transporter1; 1 is involved in salt tolerance and regulated by an MYB-type transcription factor. Plant Physiol. 2015, 168, 1076–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, R.; Bhattacharjee, A.; Jain, M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci. Rep. 2016, 6, 23719. [Google Scholar] [CrossRef] [Green Version]
- Cartagena, J.A.; Yao, Y.; Mitsuya, S.; Tsuge, T. Comparative transcriptome analysis of root types in salt tolerant and sensitive rice varieties in response to salinity stress. Physiol. Plant. 2021, 173, 1629–1642. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Liu, J.; Zhang, L.; Luo, J.; Dong, H.; Ma, Y.; Zhao, X.; Chen, B.; Sui, N.; Zhou, Z.; et al. Effects of soil salinity on sucrose metabolism in cotton leaves. PLoS ONE 2016, 11, e0156241. [Google Scholar] [CrossRef]
- Thalmann, M.; Santelia, D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017, 214, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Kempa, S.; Krasensky, J.; Dal Santo, S.; Kopka, J.; Jonak, C. A central role of abscisic acid in stress-regulated carbohydrate metabolism. PLoS ONE 2008, 3, e3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.J.; Chen, J.Y.; Wang, S.J. Molecular regulation of starch accumulation in rice seedling leaves in response to salt stress. Acta Physiol. Plant. 2008, 30, 135–142. [Google Scholar] [CrossRef]
- Wang, X.C.; Chang, L.L.; Wang, B.C.; Wang, D.; Li, P.H.; Wang, L.M.; Yi, X.P.; Huang, Q.X.; Peng, M.; Guo, A.P. Comparative proteomics of Thellungiella halophila leaves from plants subjected to salinity reveals the importance of chloroplastic starch and soluble sugars in halophyte salt tolerance. Mol. Cell. Proteom. 2013, 12, 2174–2195. [Google Scholar] [CrossRef] [Green Version]
- Mathan, J.; Singh, A.; Ranjan, A. Sucrose transport in response to drought and salt stress involves ABA-mediated induction of OsSWEET13 and OsSWEET15 in rice. Physiol. Plant. 2021, 171, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, R.; La Camera, S.; Atanassova, R.; Dédaldéchamp, F.; Allario, T.; Pourtau, N.; Bonnemain, J.L.; Laloi, M.; Coutos-Thévenot, P.; Maurousset, L.; et al. Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 2013, 4, 272. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.Y.; Zhang, S.; Hu, Y.B.; Wu, F.C.; Hu, Q.D.; Chen, G.; Cai, J.; Wu, T.; Moran, N.; Yu, L.; et al. The role of a potassium transporter OsHAK5 in potassium acquisition and transport from roots to shoots in rice at low potassium supply levels. Plant Physiol. 2014, 166, 945–959. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.Y.; Feng, H.M.; Zhang, S.; Xiao, H.J.; Hu, Q.D.; Chen, G.; Xuan, W.; Moran, N.; Murphy, A.; Yu, L.; et al. The potassium transporter OsHAK5 alters rice architecture via ATP-dependent transmembrane auxin fluxes. Plant Commun. 2020, 1, 100052. [Google Scholar] [CrossRef]
- Chen, J.H.; Jiang, H.W.; Hsieh, E.J.; Chen, H.Y.; Chien, C.T.; Hsieh, H.L.; Lin, T.P. Drought and salt stress tolerance of an Arabidopsis glutathione S-transferase U17 knockout mutant are attributed to the combined effect of glutathione and abscisic acid. Plant Physiol. 2012, 158, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Sahoo, A.; Devendran, R.; Jain, M. Over-expression of a rice tau class glutathione s-transferase gene improves tolerance to salinity and oxidative stresses in Arabidopsis. PLoS ONE 2014, 9, e92900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yang, B.; He, Y.; Zhan, C.; Cheng, Y.; Zhang, J.; Zhang, H.; Cheng, J.; Wang, Z. A quantitative trait locus, qSE3, promotes seed germination and seedling establishment under salinity stress in rice. Plant J. 2019, 97, 1089–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Xu, T.; Liu, Y.; Chen, T.; Zhang, Q.; Li, W.; Zhou, H.; Zhang, Y.; Zhang, Z. Molecular Insights into Salinity Responsiveness in Contrasting Genotypes of Rice at the Seedling Stage. Int. J. Mol. Sci. 2022, 23, 1624. https://doi.org/10.3390/ijms23031624
Zhang J, Xu T, Liu Y, Chen T, Zhang Q, Li W, Zhou H, Zhang Y, Zhang Z. Molecular Insights into Salinity Responsiveness in Contrasting Genotypes of Rice at the Seedling Stage. International Journal of Molecular Sciences. 2022; 23(3):1624. https://doi.org/10.3390/ijms23031624
Chicago/Turabian StyleZhang, Jingjing, Tingting Xu, Yiran Liu, Tong Chen, Qiuxin Zhang, Weiyan Li, Hongkai Zhou, Yuexiong Zhang, and Zemin Zhang. 2022. "Molecular Insights into Salinity Responsiveness in Contrasting Genotypes of Rice at the Seedling Stage" International Journal of Molecular Sciences 23, no. 3: 1624. https://doi.org/10.3390/ijms23031624
APA StyleZhang, J., Xu, T., Liu, Y., Chen, T., Zhang, Q., Li, W., Zhou, H., Zhang, Y., & Zhang, Z. (2022). Molecular Insights into Salinity Responsiveness in Contrasting Genotypes of Rice at the Seedling Stage. International Journal of Molecular Sciences, 23(3), 1624. https://doi.org/10.3390/ijms23031624