
Activatable Peptides for Rapid and Simple Visualization of Protease Activity Secreted in Living Cells

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
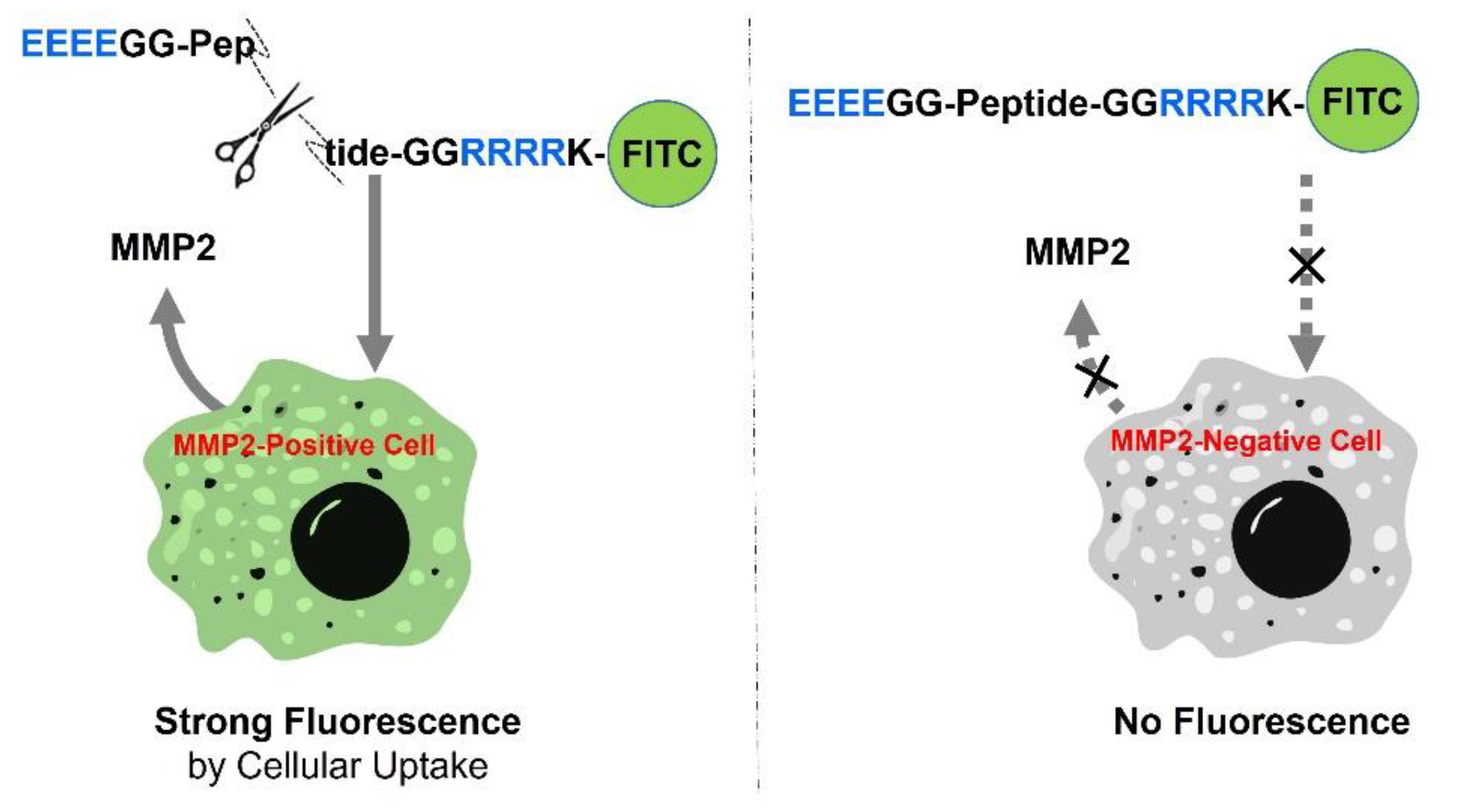
2.1. Principle of MMP-Activated Cellular Imaging
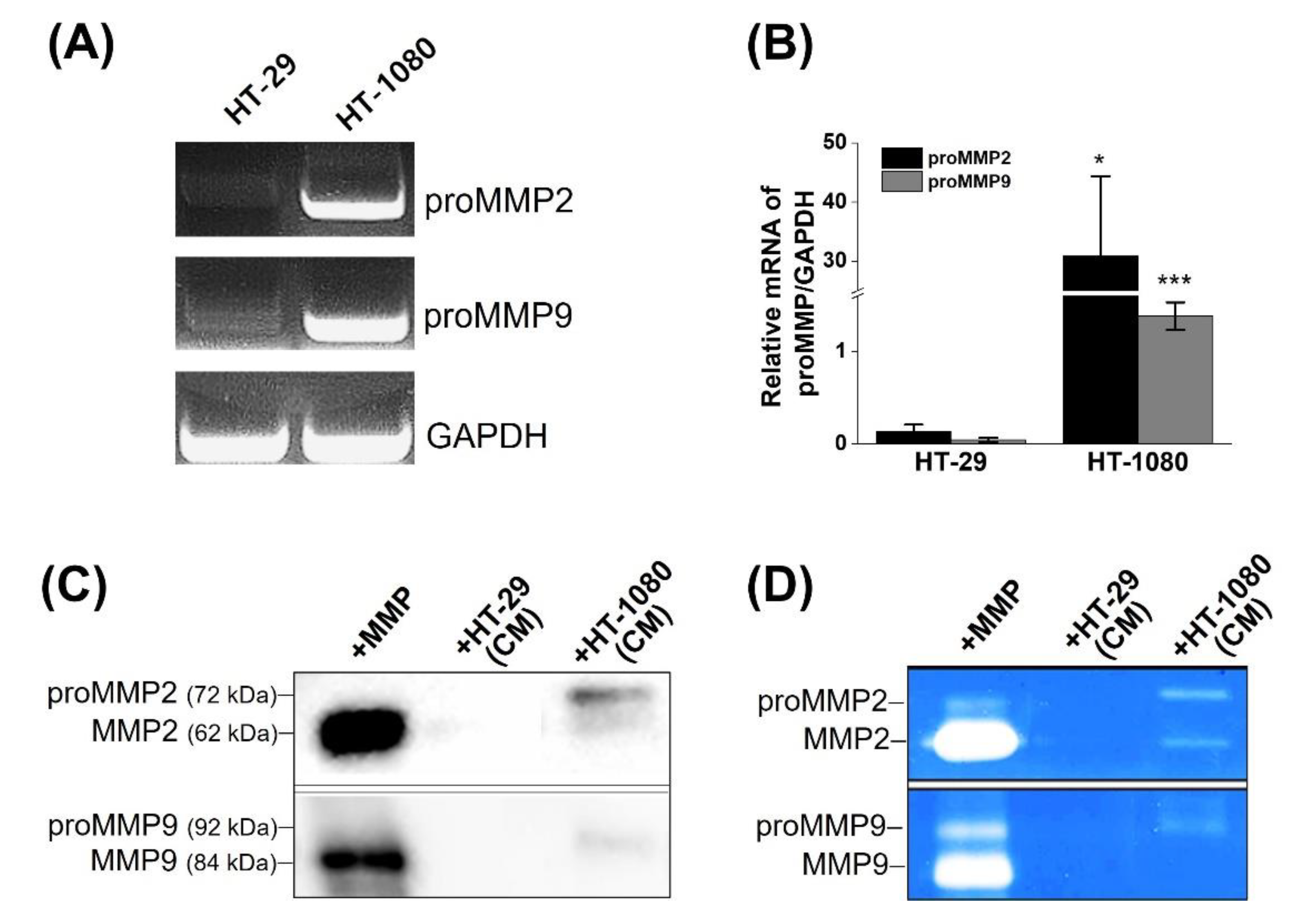
2.2. Determination of MMP2 Expression in Cells
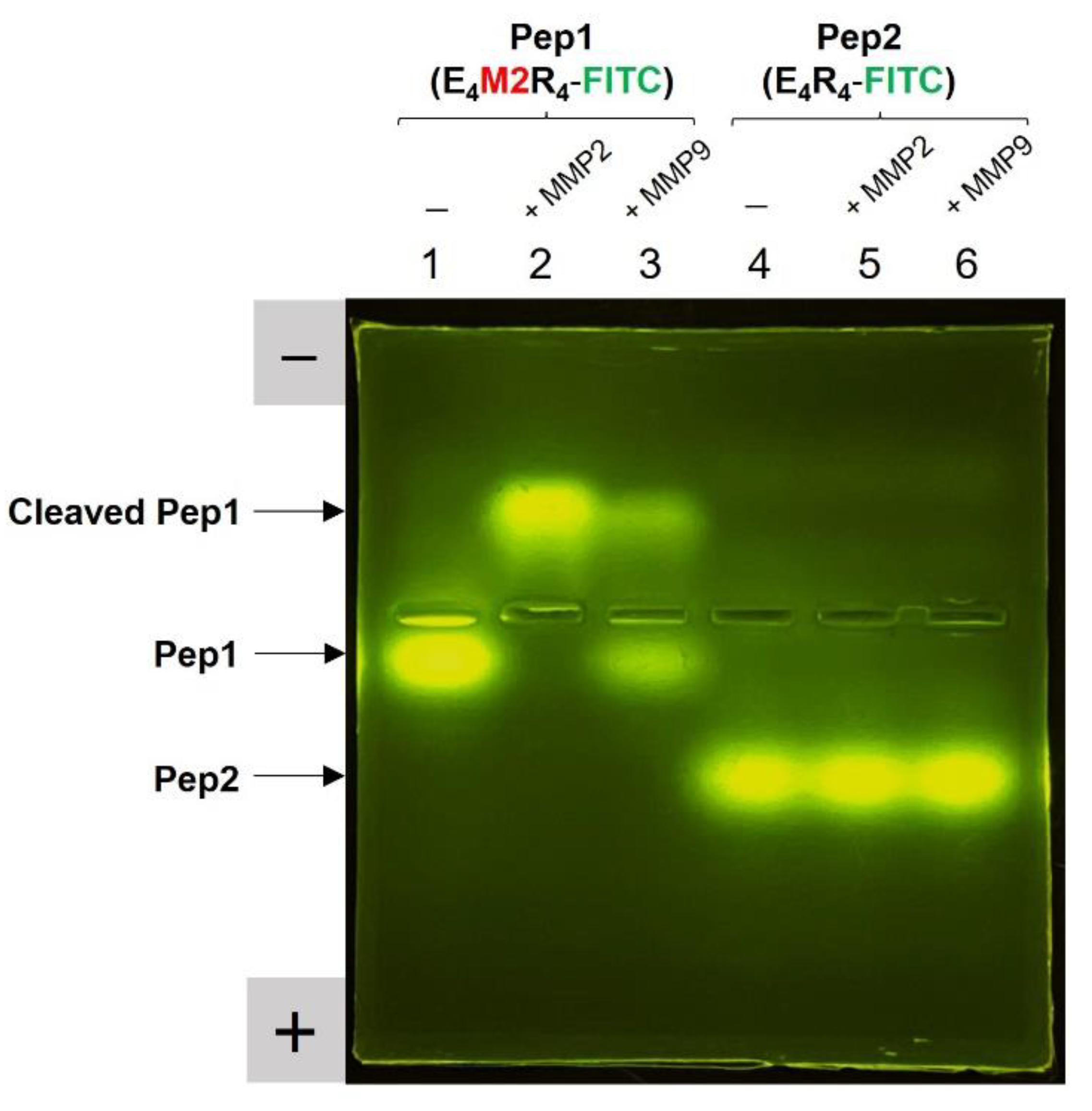
2.3. Peptide Substrate Specificity for MMP2
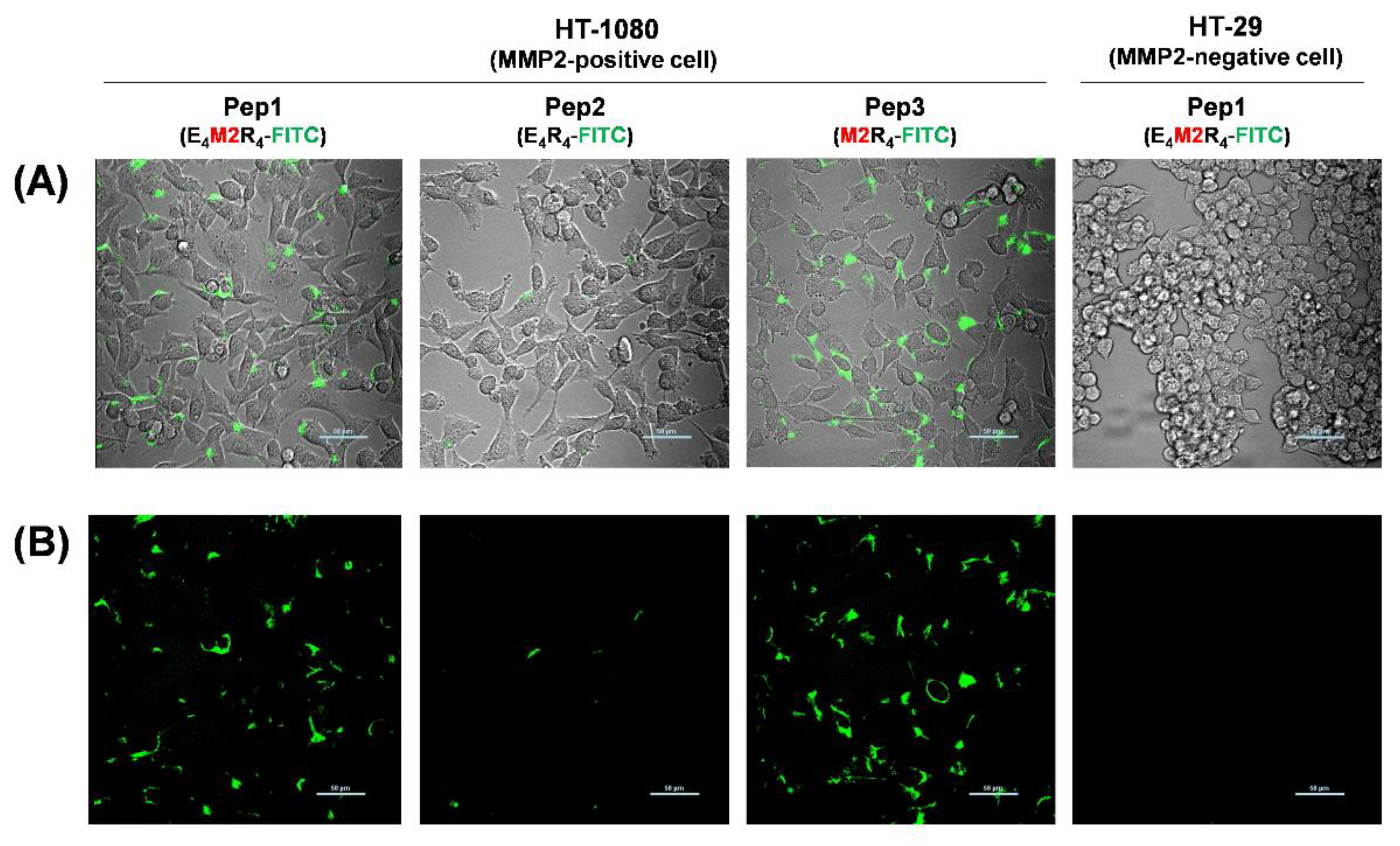
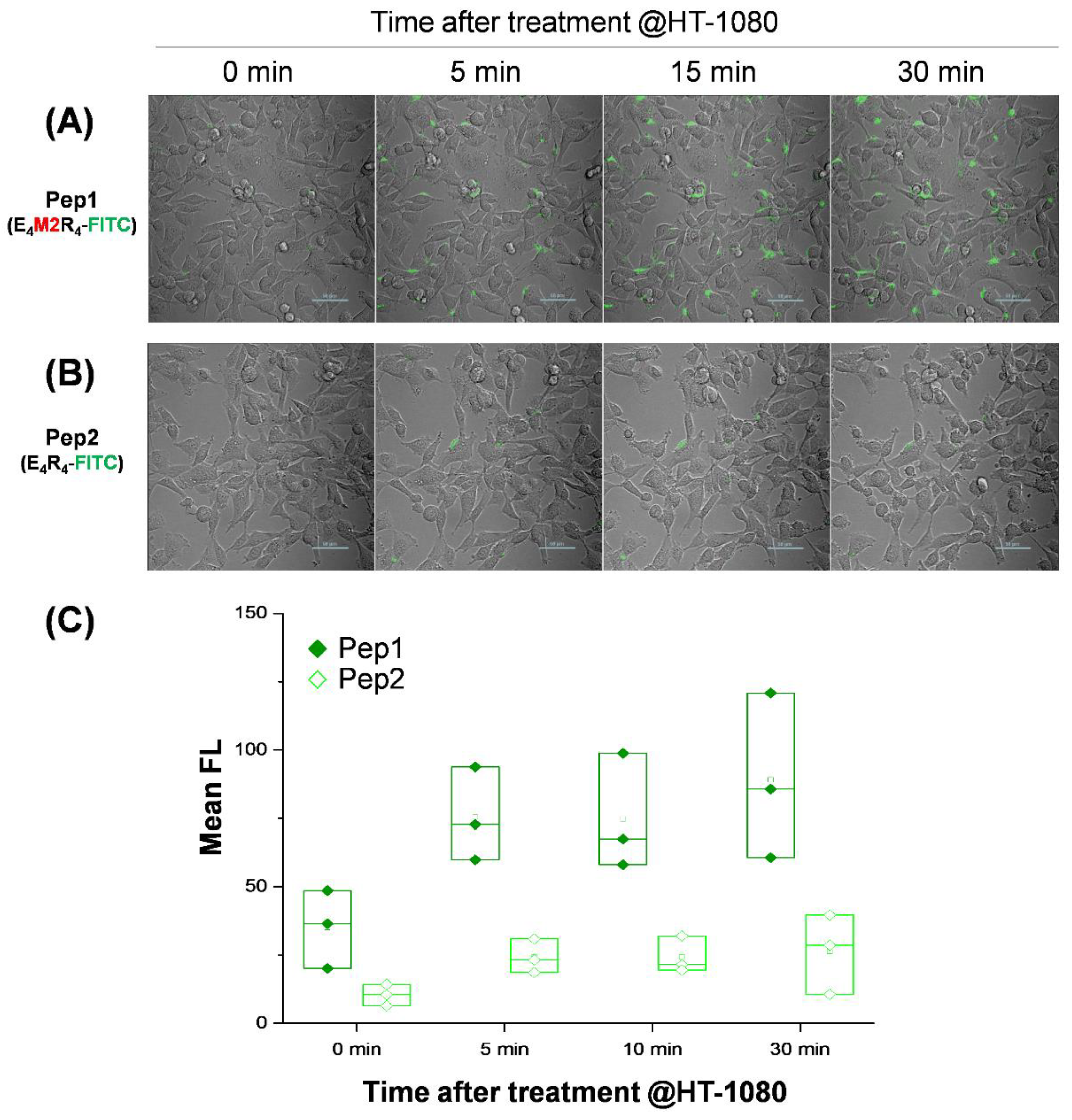
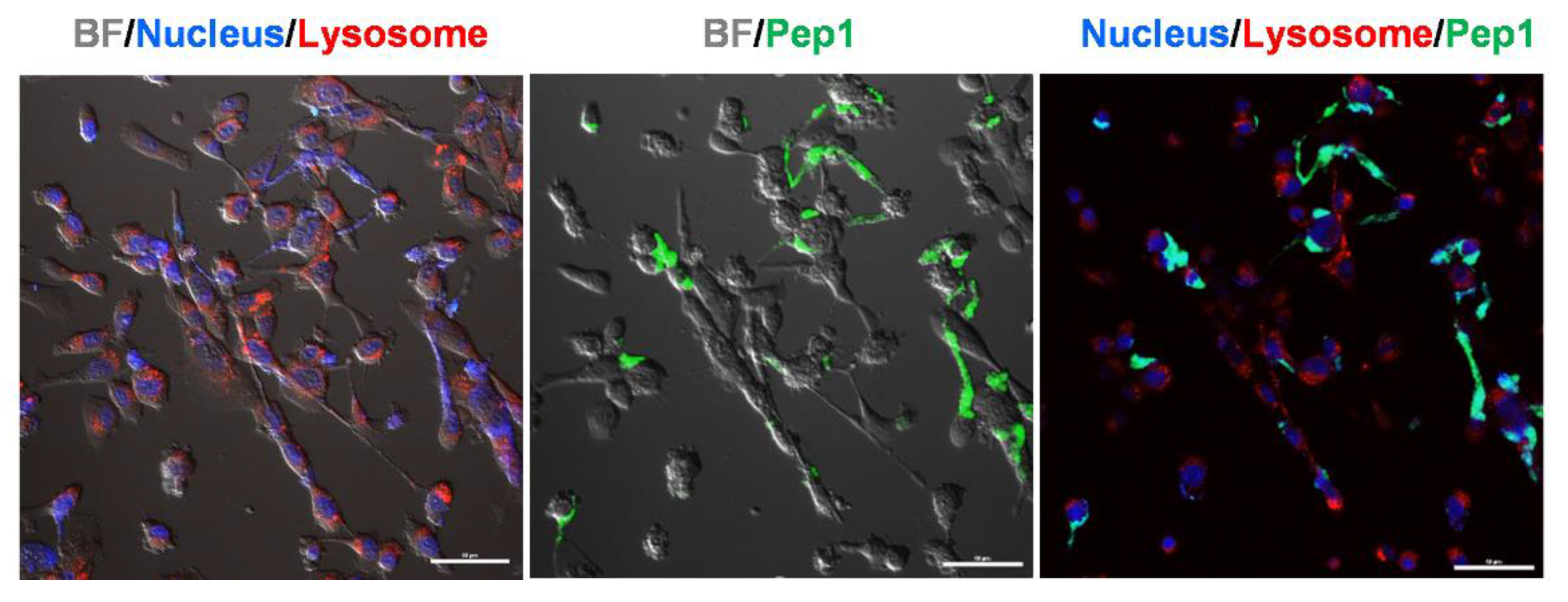
2.4. Live Cell Imaging in Response to MMP2 Activity






3. Materials and Methods
3.1. Materials
3.2. Cell Culture
3.3. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Real-Time PCR
3.4. Western Blot and Zymography
3.5. Fluorescent Gel Electrophoresis
3.6. Cell Imaging
3.7. Cell Viability Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopez-Otin, C.; Overall, C.M. Protease degradomics: A new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002, 3, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.S. Proteases: History, discovery, and roles in health and disease. J. Biol. Chem. 2019, 294, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Fingleton, B.; Rothenberg, M.L.; Matrisian, L.M. Matrix metalloproteinases: Biologic activity and clinical implications. J. Clin. Oncol. 2000, 18, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54. [Google Scholar] [CrossRef]
- Zucker, S.; Cao, J. Imaging metalloproteinase activity in vivo. Nat. Med. 2001, 7, 655–656. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Lin, J.; Lu, J.; Liu, B.F.; Zeng, S.; Luo, Q. Detection of MMP activity in living cells by a genetically encoded surface-displayed FRET sensor. Biochim. Biophys. Acta 2007, 1773, 400–407. [Google Scholar] [CrossRef][Green Version]
- Lee, H.; Kim, Y.P. Fluorescent and bioluminescent nanoprobes for in vitro and in vivo detection of matrix metalloproteinase activity. BMB Rep. 2015, 48, 313–318. [Google Scholar] [CrossRef]
- Lee, H.; Kim, S.J.; Shin, H.; Kim, Y.P. Collagen-iImmobilized extracellular FRET reporter for visualizing protease activity secreted by living cells. ACS Sens. 2020, 5, 655–664. [Google Scholar] [CrossRef]
- Lei, Z.; Jian, M.; Li, X.; Wei, J.; Meng, X.; Wang, Z. Biosensors and bioassays for determination of matrix metalloproteinases: State of the art and recent advances. J. Mater. Chem. B 2020, 8, 3261–3291. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; So, M.K.; Rao, J. Protease-modulated cellular uptake of quantum dots. Nano Lett. 2006, 6, 1988–1992. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yhee, J.Y.; Kim, S.A.; Koo, H.; Son, S.; Ryu, J.H.; Youn, I.C.; Choi, K.; Kwon, I.C.; Kim, K. Optical imaging of cancer-related proteases using near-infrared fluorescence matrix metalloproteinase-sensitive and cathepsin B-sensitive probes. Theranostics 2012, 2, 179–189. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, S.M.; Kim, G.B.; Kim, Y.P. Gold nanoparticle-based fluorescence quenching via metal coordination for assaying protease activity. Gold Bull. 2012, 45, 213–219. [Google Scholar] [CrossRef][Green Version]
- Zeng, W.H.; Wu, L.Y.; Sun, Y.D.; Wang, Y.Q.; Wang, J.F.; Ye, D.J. Ratiometric imaging of MMP-2 activity facilitates tumor detection using activatable near-infrared fluorescent semiconducting polymer nanoparticles. Small 2021, 17, 2101924. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, T.W.; Lo, P.C.; Wilson, B.C.; Zheng, G. “Zipper” molecular beacons: A generalized strategy to optimize the performance of activatable protease probes. Bioconjug. Chem. 2009, 20, 1836–1842. [Google Scholar] [CrossRef]
- Mulhall, H.J.; Hughes, M.P.; Kazmi, B.; Lewis, M.P.; Labeed, F.H. Epithelial cancer cells exhibit different electrical properties when cultured in 2D and 3D environments. Biochim. Biophys. Acta. 2013, 1830, 5136–5141. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in cancer: The relevance of drug-membrane interaction studies. Biochim. Biophys. Acta. 2016, 1858, 2231–2244. [Google Scholar] [CrossRef]
- Shi, D.L. Cancer cell surface negative charges: A bio-physical manifestation of the Warburg effect. Nano Life 2017, 7, 1771001. [Google Scholar] [CrossRef]
- Chen, B.D.; Le, W.J.; Wang, Y.L.; Li, Z.Q.; Wang, D.; Ren, L.; Lin, L.; Cui, S.B.; Hu, J.J.; Hu, Y.H.; et al. Targeting negative surface charges of cancer cells by multifunctional nanoprobes. Theranostics 2016, 6, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Young, C.C.; Vedadghavami, A.; Bajpayee, A.G. Bioelectricity for Drug Delivery: The promise of cationic therapeutics. Bioelectricity 2020, 2, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.W.; Monterrey, J.C.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Patterns of MMP-2 and MMP-9 expression in human cancer cell lines. Oncol. Rep. 2009, 21, 1323–1333. [Google Scholar] [PubMed]
- Roomi, M.W.; Kalinovsky, T.; Monterrey, J.; Rath, M.; Niedzwiecki, A. In vitro modulation of MMP-2 and MMP-9 in adult human sarcoma cell lines by cytokines, inducers and inhibitors. Int. J. Oncol. 2013, 43, 1787–1798. [Google Scholar] [CrossRef]
- Turk, B.E.; Huang, L.L.; Piro, E.T.; Cantley, L.C. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol. 2001, 19, 661–667. [Google Scholar] [CrossRef]
- Bosshard, H.R.; Marti, D.N.; Jelesarov, I. Protein stabilization by salt bridges: Concepts, experimental approaches and clarification of some misunderstandings. J. Mol. Recognit. 2004, 17, 1–16. [Google Scholar] [CrossRef]
- Allolio, C.; Magarkar, A.; Jurkiewicz, P.; Baxova, K.; Javanainen, M.; Mason, P.E.; Sachl, R.; Cebecauer, M.; Hof, M.; Horinek, D.; et al. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. USA 2018, 115, 11923–11928. [Google Scholar] [CrossRef]
- De Jong, H.; Bonger, K.M.; Löwik, D.W.P.M. Activatable cell-penetrating peptides: 15 years of research. RSC Chem. Biol. 2020, 1, 192–203. [Google Scholar] [CrossRef]
- Rasheed, S.; Nelson-Rees, W.A.; Toth, E.M.; Arnstein, P.; Gardner, M.B. Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer 1974, 33, 1027–1033. [Google Scholar] [CrossRef]
- Hutmacher, D.W.; Loessner, D.; Rizzi, S.; Kaplan, D.L.; Mooney, D.J.; Clements, J.A. Can tissue engineering concepts advance tumor biology research? Trends Biotechnol. 2010, 28, 125–133. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, T.H.; Jang, J.W.; Jang, Y.J.; Lee, K.H.; Lee, S.T. Analysis of matrix metalloproteinase mRNAs expressed in hepatocellular carcinoma cell lines. Mol. Cells 2001, 12, 32–40. [Google Scholar] [PubMed]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, G.B.; Lippitz, A.; Kim, Y.M.; Jung, D.; Unger, W.E.S.; Kim, Y.P.; Lee, T.G. Plasma-polymerized antifouling biochips for label-free measurement of protease activity in cell culture media. Sens. Actuators B Chem. 2019, 281, 527–534. [Google Scholar] [CrossRef]
- Nguyen, D.L.; Kim, H.; Kim, D.; Lee, J.O.; Gye, M.C.; Kim, Y.P. Detection of matrix metalloproteinase activity by bioluminescence via intein-mediated biotinylation of luciferase. Sensors 2018, 18, 875. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.P.; Lee, B.S.; Kim, E.; Choi, I.S.; Moon, D.W.; Lee, T.G.; Kim, H.S. Activity-based assay of matrix metalloproteinase on nonbiofouling surfaces using time-of flight secondary ion mass spectrometry. Anal. Chem. 2008, 80, 5094–5102. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.P.; Oh, Y.H.; Oh, E.; Ko, S.; Han, M.K.; Kim, H.S. Energy transfer-based multiplexed assay of proteases by using gold nanoparticle and quantum dot conjugates on a surface. Anal. Chem. 2008, 80, 4634–4641. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Kim, K.H.; Park, Y.H.; Ko, S.; Kim, Y.P. Colorimetric assay of matrix metalloproteinase activity based on metal-induced self-assembly of carboxy gold nanoparticles. Biosens. Bioelectron. 2013, 41, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.X.; Shi, N.Q.; Qi, X.R. Distinct transduction modes of arginine-rich cell-penetrating peptides for cargo delivery into tumor cells. Int. J. Pharm. 2011, 419, 200–208. [Google Scholar] [CrossRef]
- Zahid, M.; Robbins, P.D. Cell-type specific penetrating peptides: Therapeutic promises and challenges. Molecules 2015, 20, 13055–13070. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Lee, Y.; Best-Popescu, C.; Gao, L. High-speed compressed-sensing fluorescence lifetime imaging microscopy of live cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2004176118. [Google Scholar] [CrossRef]
- Choi, S. Hydrophoresis—A Microfluidic Principle for Directed Particle Migration in Flow. BioChip J. 2020, 14, 72–83. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.-B.; Lee, J.M.; Nguyen, D.L.; Lee, J.; Kim, Y.-P. Activatable Peptides for Rapid and Simple Visualization of Protease Activity Secreted in Living Cells. Int. J. Mol. Sci. 2022, 23, 1605. https://doi.org/10.3390/ijms23031605
Kim G-B, Lee JM, Nguyen DL, Lee J, Kim Y-P. Activatable Peptides for Rapid and Simple Visualization of Protease Activity Secreted in Living Cells. International Journal of Molecular Sciences. 2022; 23(3):1605. https://doi.org/10.3390/ijms23031605
Chicago/Turabian StyleKim, Gae-Baik, Jeong Min Lee, Duc Long Nguyen, Joonseok Lee, and Young-Pil Kim. 2022. "Activatable Peptides for Rapid and Simple Visualization of Protease Activity Secreted in Living Cells" International Journal of Molecular Sciences 23, no. 3: 1605. https://doi.org/10.3390/ijms23031605
APA StyleKim, G.-B., Lee, J. M., Nguyen, D. L., Lee, J., & Kim, Y.-P. (2022). Activatable Peptides for Rapid and Simple Visualization of Protease Activity Secreted in Living Cells. International Journal of Molecular Sciences, 23(3), 1605. https://doi.org/10.3390/ijms23031605