
Nonsteroidal Anti-Inflammatory Drugs as PPARγ Agonists Can Induce PRODH/POX-Dependent Apoptosis in Breast Cancer Cells: New Alternative Pathway in NSAID-Induced Apoptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
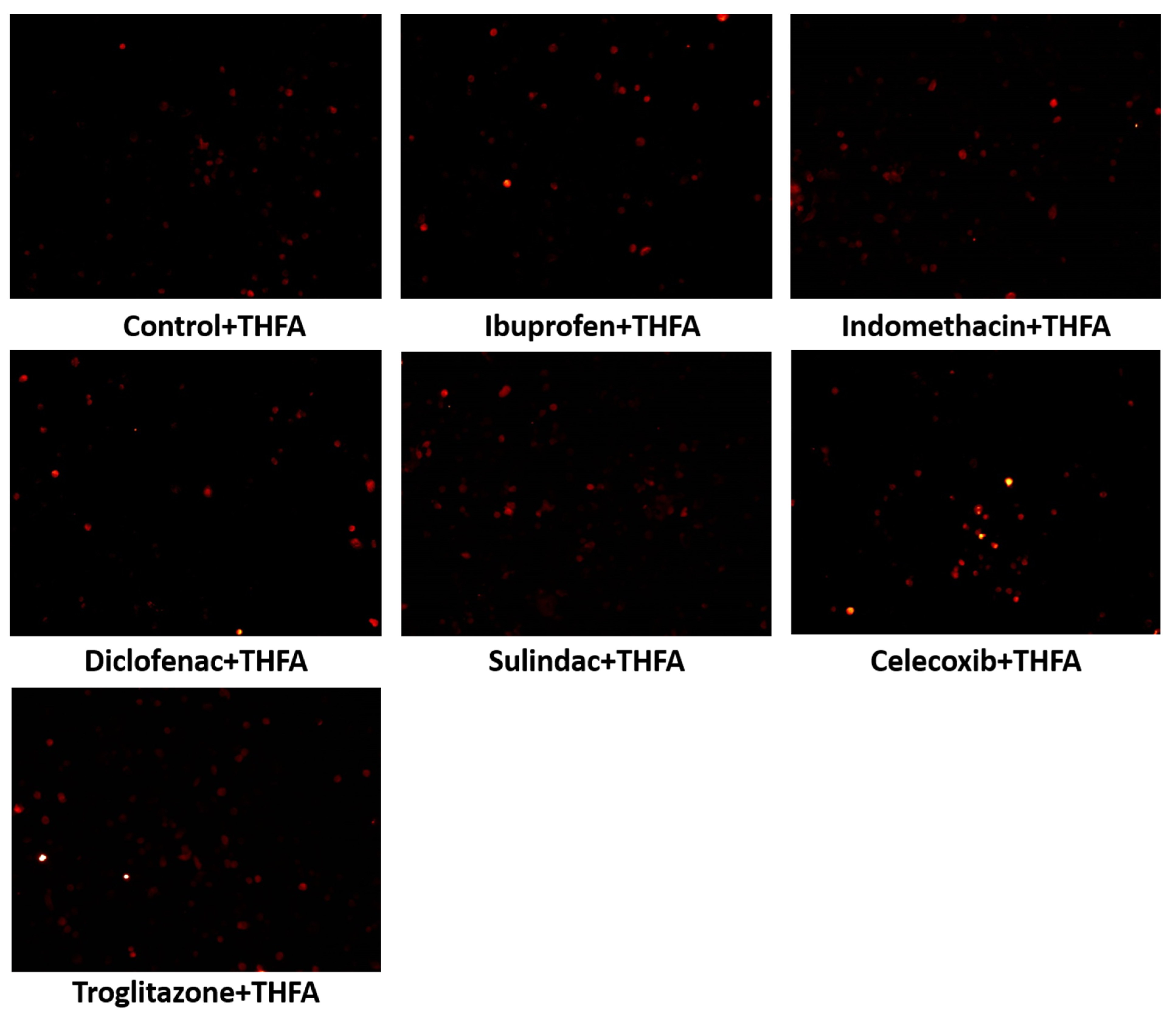
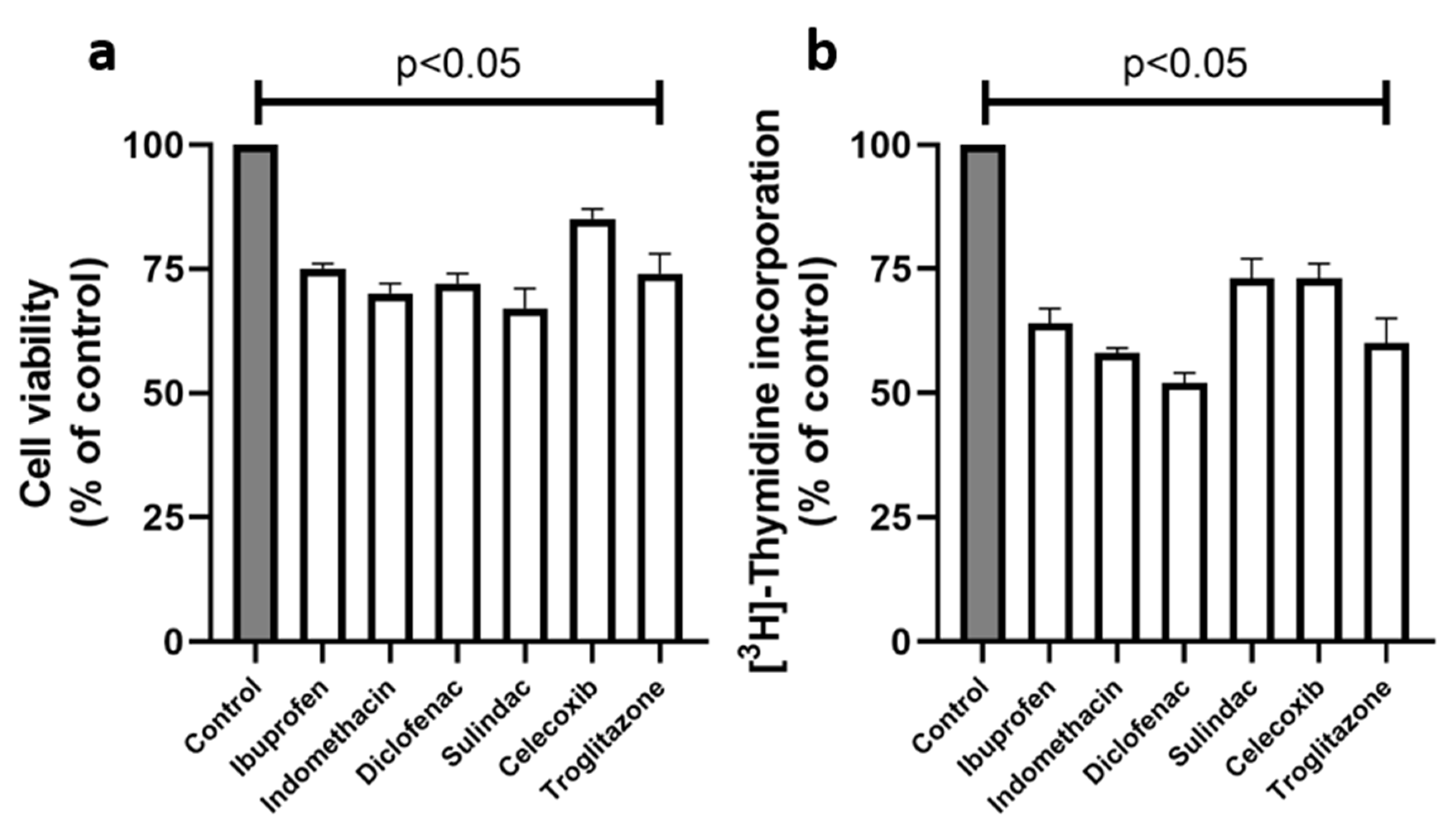
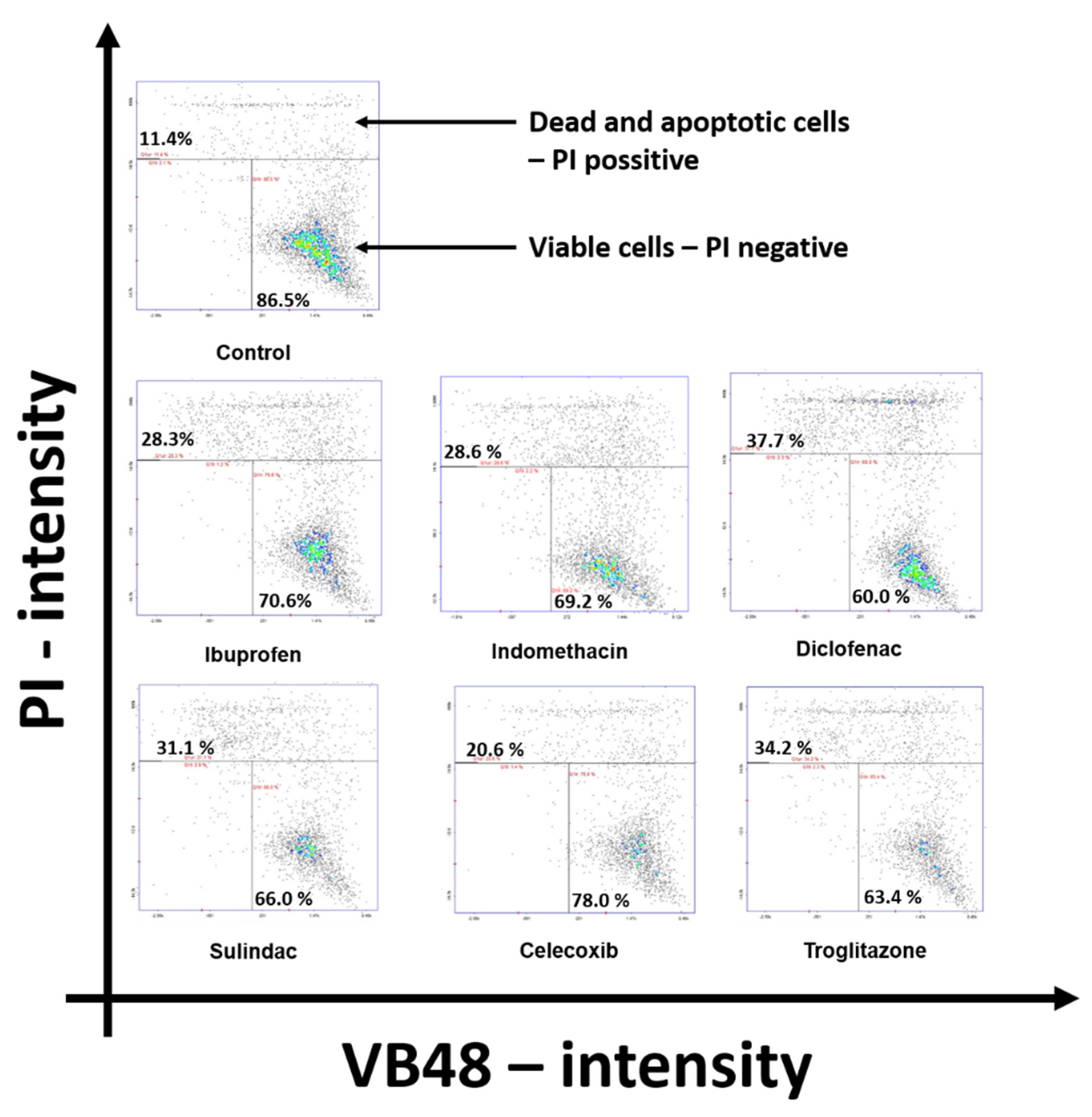
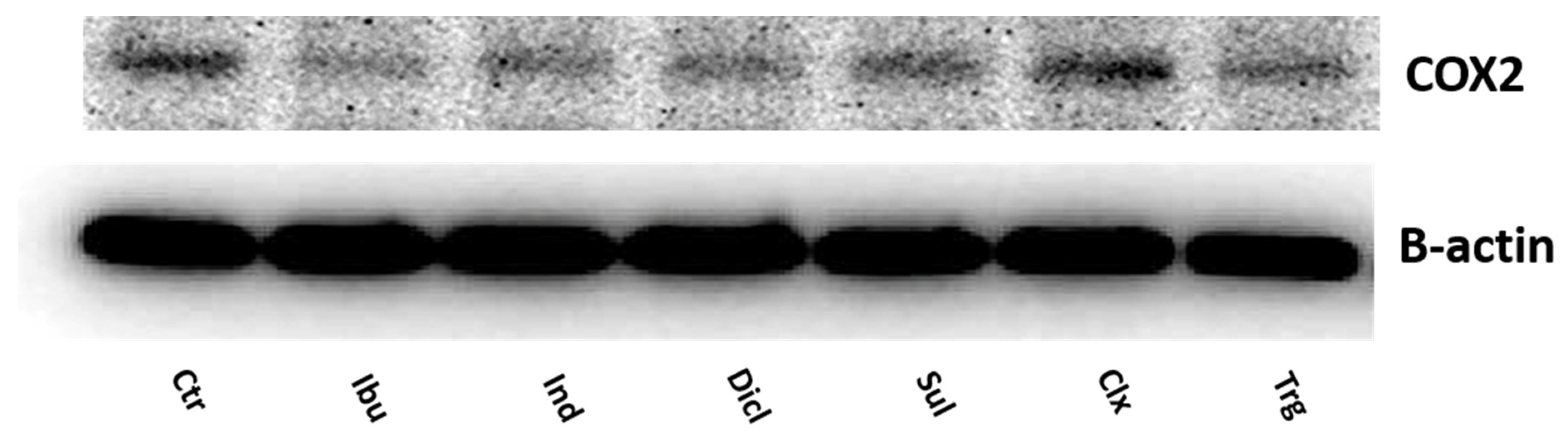
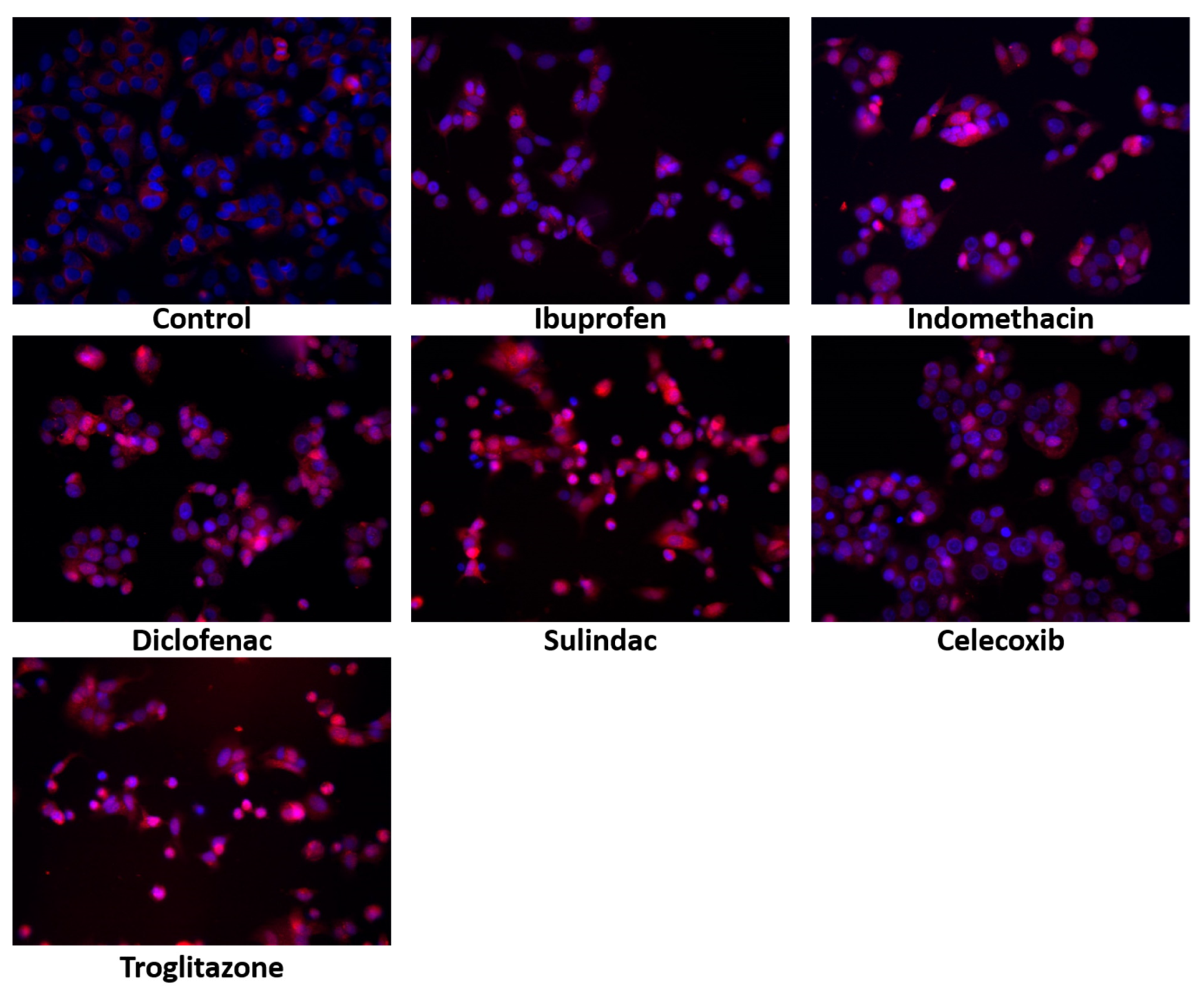
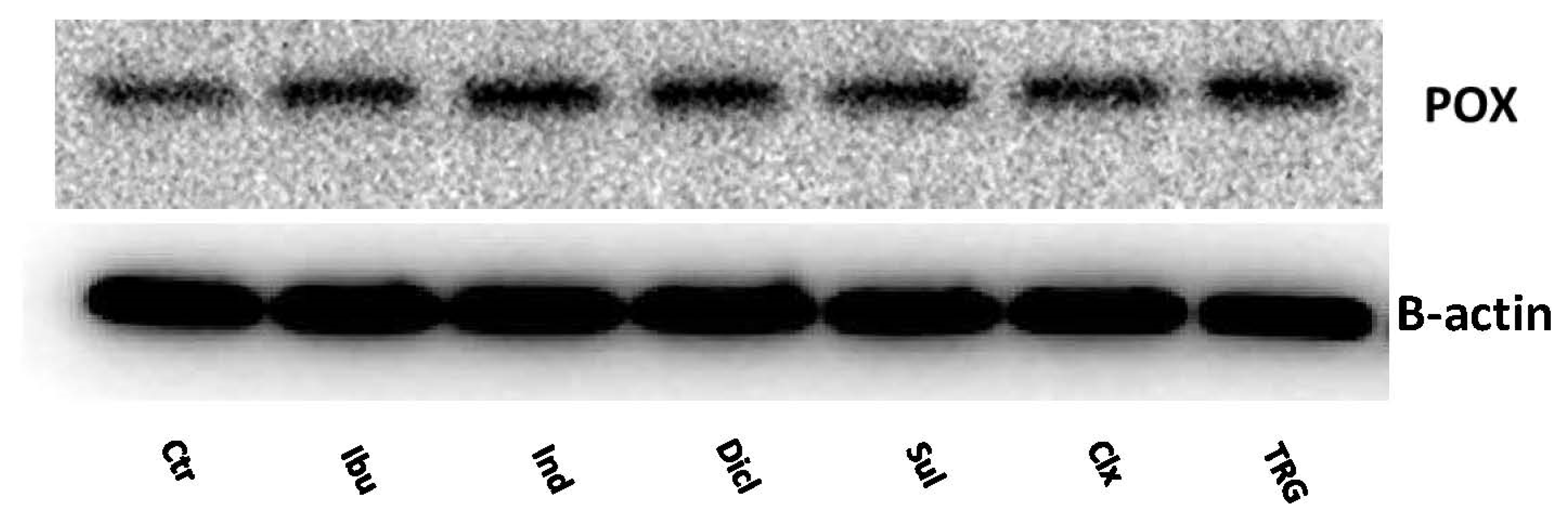
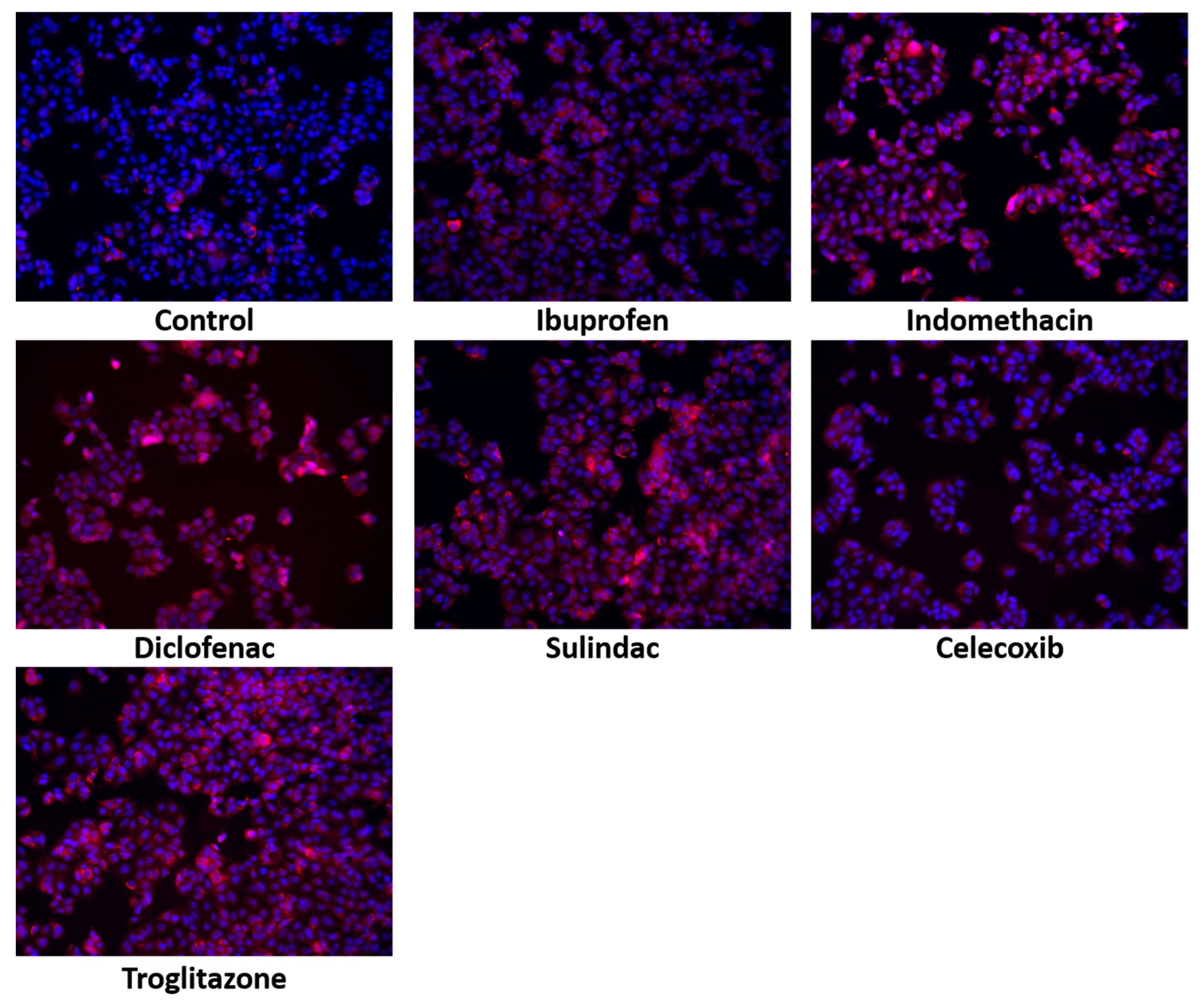
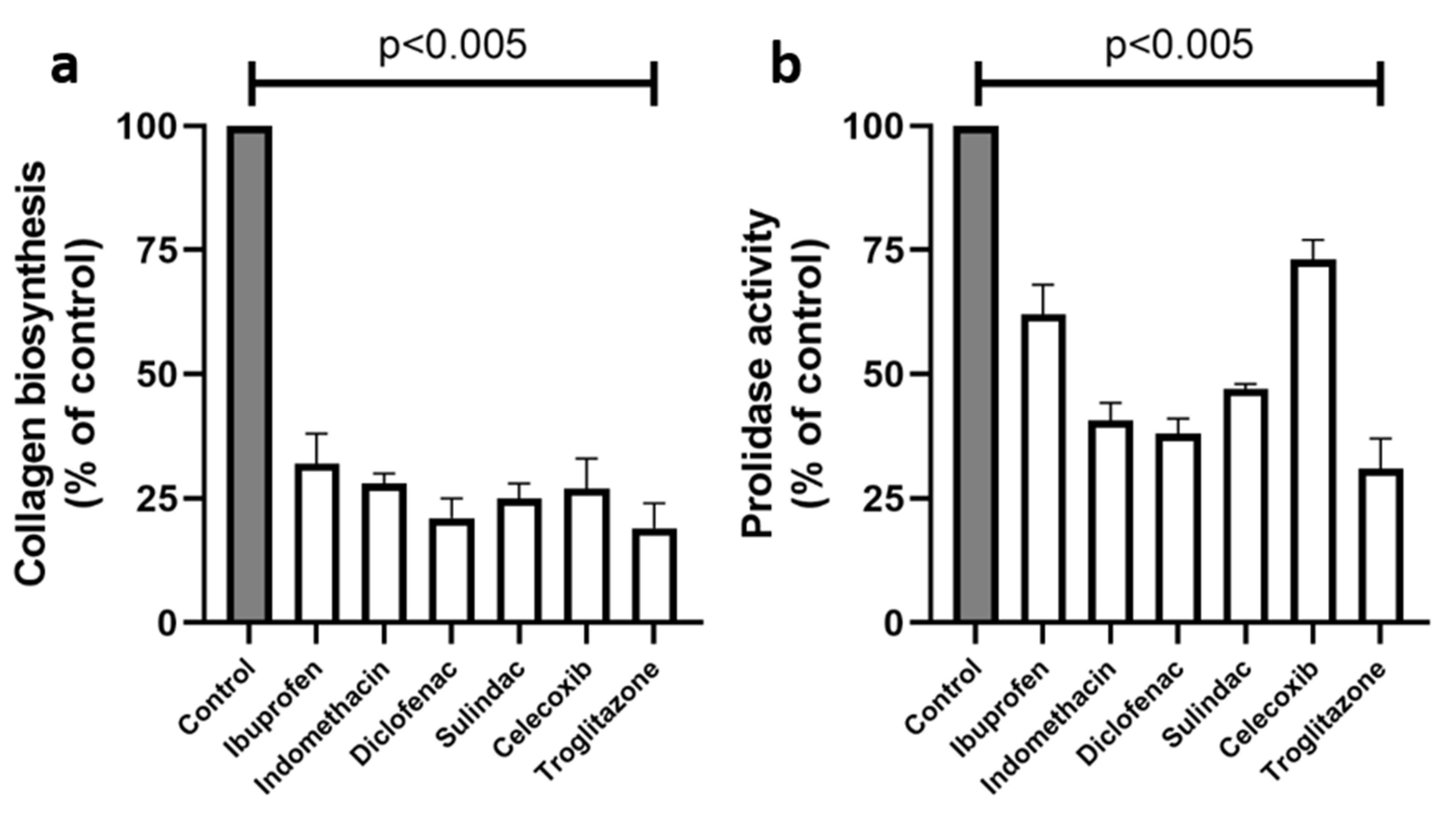
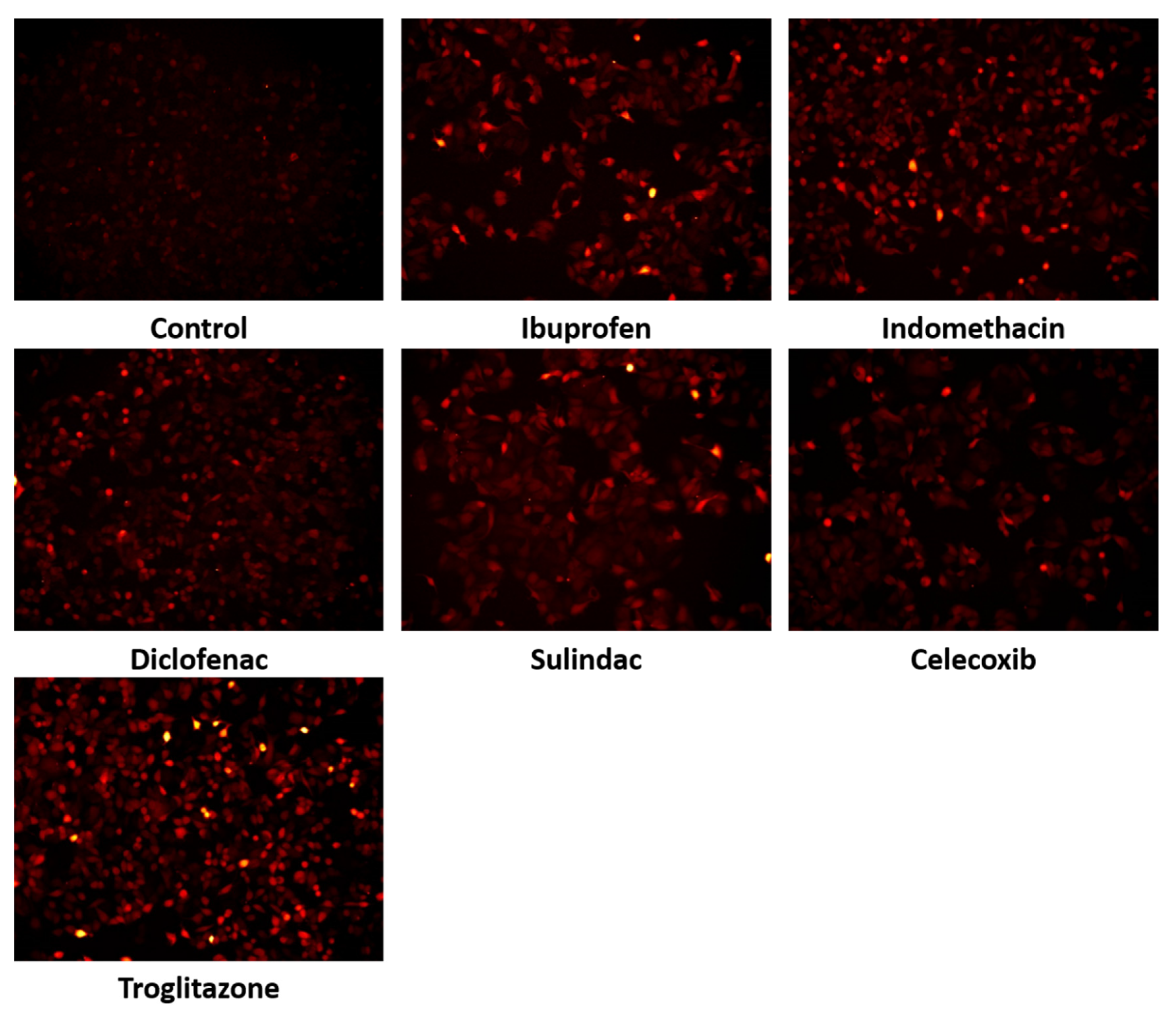
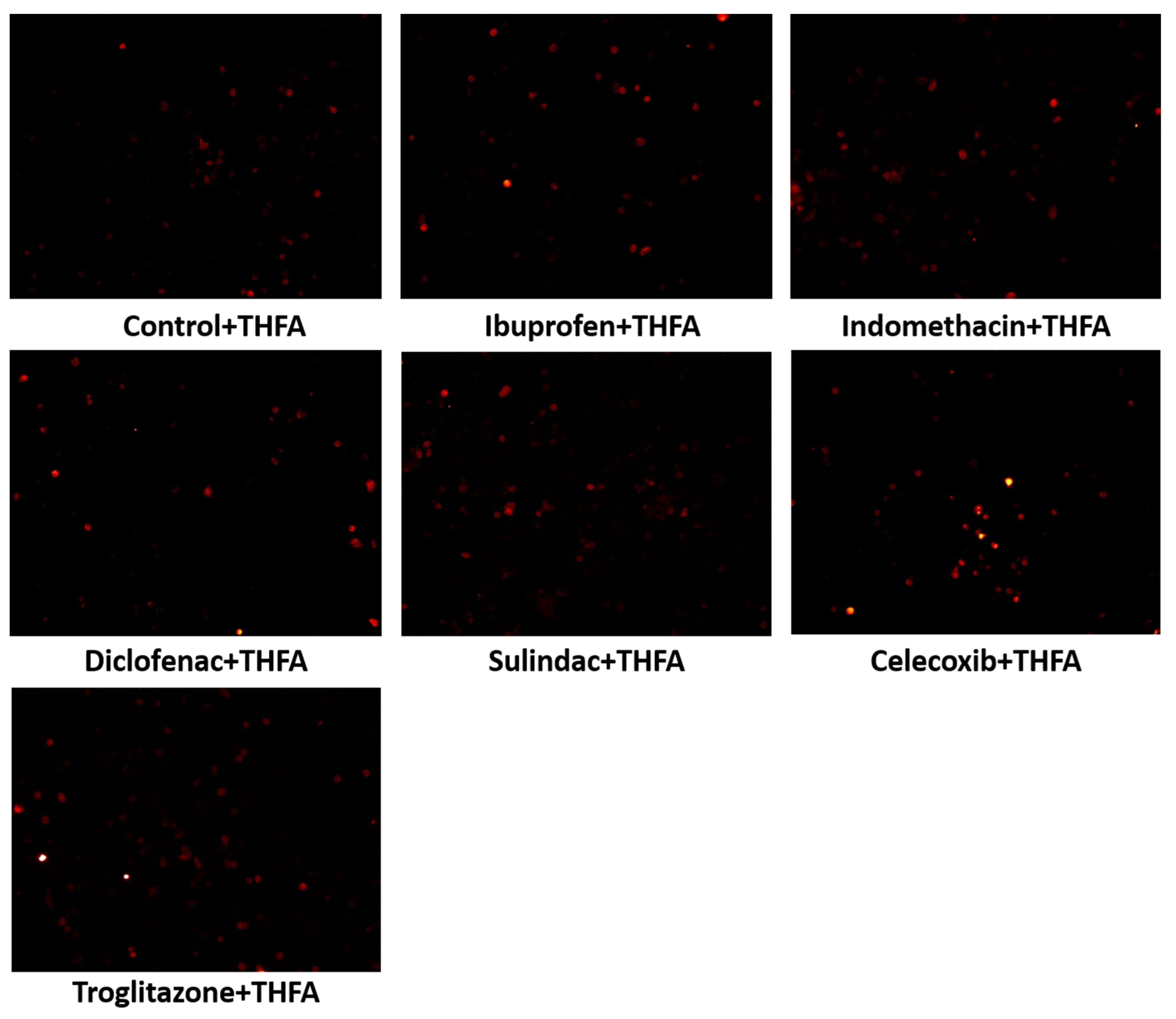
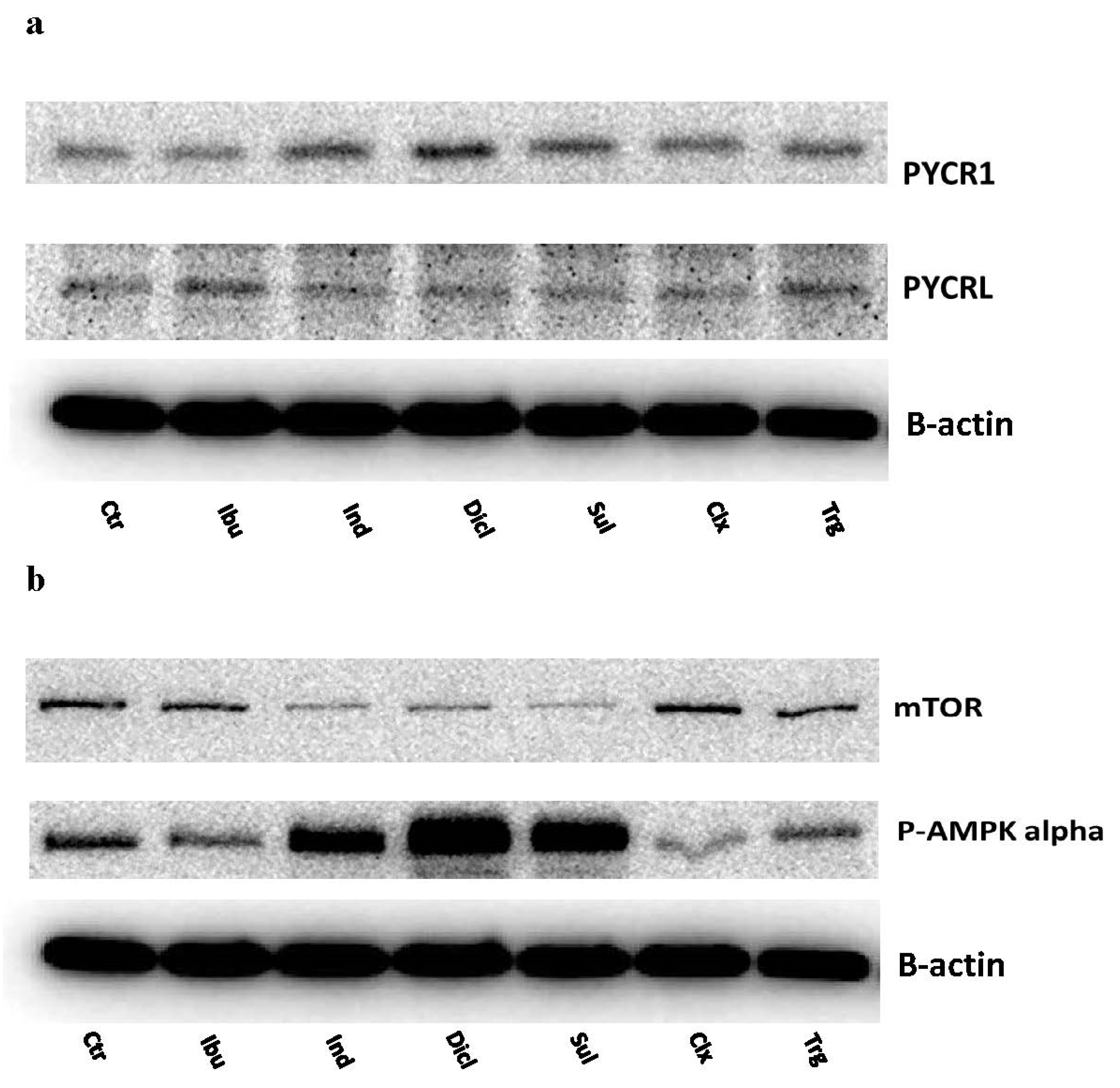
2. Results
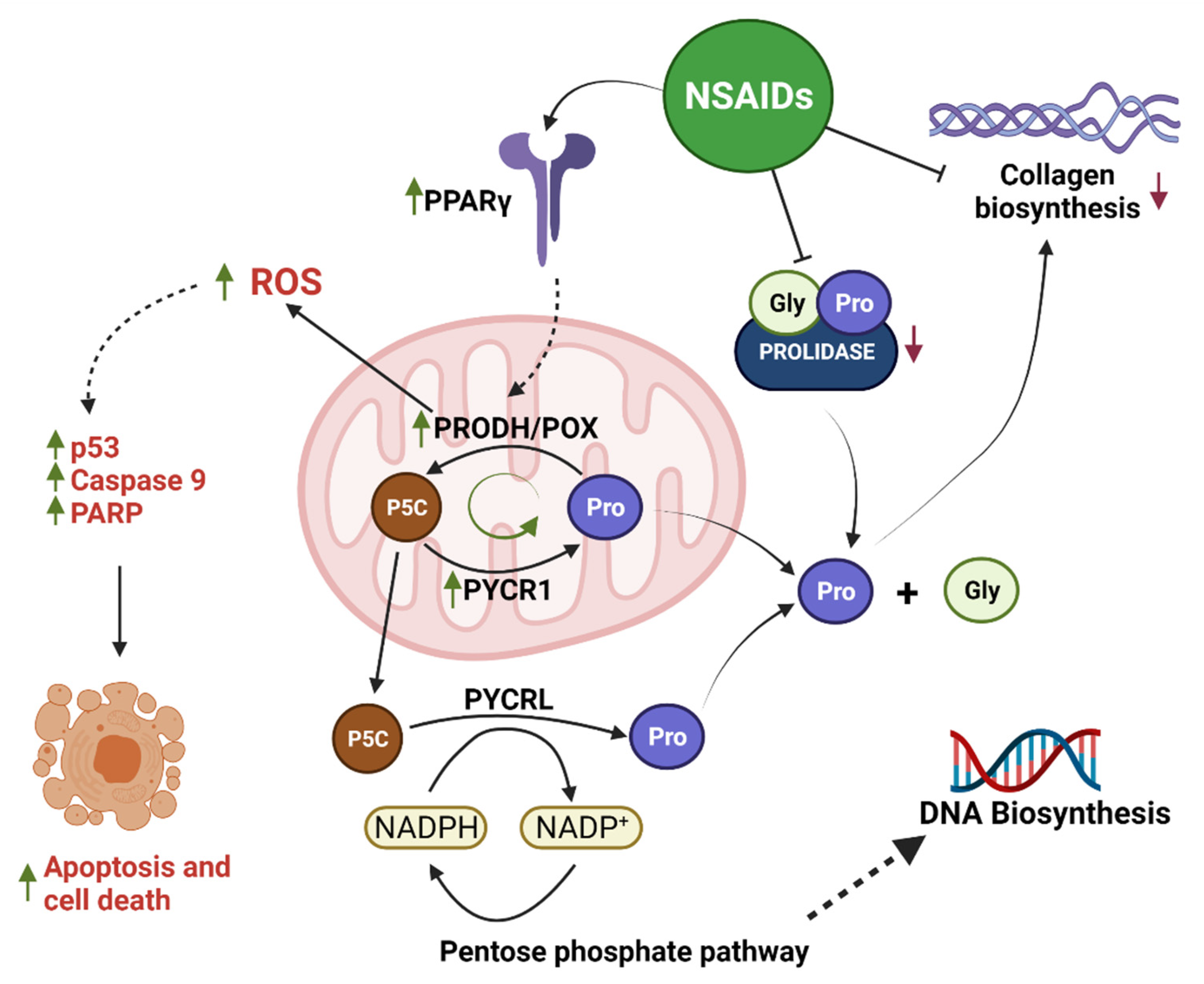
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Cell Culture
4.2.2. Cell Viability Test MTT
4.2.3. DNA Biosynthesis
4.2.4. Collagen Biosynthesis
4.2.5. SDS PAGE and Western Blotting
4.2.6. ROS Formation
4.2.7. Cytometric Assay for Apoptosis
4.2.8. Immunofluorescent Analysis
4.2.9. Prolidase Activity
4.2.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| COX | cyclooxygenase |
| EGFR | epidermal growth factor receptor |
| MAPK | mitogen-activated protein kinase |
| mTOR | mechanistic target of rapamycin |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| P5C–∆1 | pyrroline-5-carboxylate |
| P5CDH | P5C dehydrogenase |
| PARP | poly (ADP-ribose) polymerase |
| PGE2 | prostaglandin E2 |
| PPAR | peroxisome proliferator-activated receptor |
| PRODH/POX | proline dehydrogenase/proline oxidase |
| PYCR1 | mitochondrial P5C reductase |
| PYCRL | cytosolic P5C reductase |
| ROS | reactive oxygen species |
References
- Chan, A.T. Long-term Use of Aspirin and Nonsteroidal Anti-inflammatory Drugs and Risk of Colorectal Cancer. JAMA 2005, 294, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Shebl, F.M.; Hsing, A.W.; Park, Y.; Hollenbeck, A.R.; Chu, L.W.; Meyer, T.E.; Koshiol, J. Non-Steroidal Anti-Inflammatory Drugs Use Is Associated with Reduced Risk of Inflammation-Associated Cancers: NIH-AARP Study. PLoS ONE 2014, 9, e114633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Wilson, M.; Elwin, C.-E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Knox, S.S. From ‘omics’ to complex disease: A systems biology approach to gene-environment interactions in cancer. Cancer Cell Int. 2010, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Moon, H.-G.; Hancha, W.; Yom, C.K.; Kim, W.H.; Kim, J.H.; Noh, D.-Y. COX2 overexpression is a prognostic marker for Stage III breast cancer. Breast Cancer Res. Treat. 2011, 132, 51–59. [Google Scholar] [CrossRef]
- Menter, D.G.; DuBois, R.N. Prostaglandins in Cancer Cell Adhesion, Migration, and Invasion. Int. J. Cell Biol. 2012, 2012, 723419. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [Green Version]
- Eibl, G.; Bruemmer, D.; Okada, Y.; Duffy, J.P.; Law, R.E.; Reber, H.A.; Hines, O.J. PGE2 is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2003, 306, 887–897. [Google Scholar] [CrossRef]
- Matos, P.; Jordan, P. Beyond COX-inhibition: ‘Side-Effects’ of ibuprofen on neoplastic development and progression. Curr. Pharm. Des. 2015, 21, 2978–2982. [Google Scholar] [CrossRef]
- Patel, M.I.; Subbaramaiah, K.; Du, B.; Chang, M.; Yang, P.; Newman, R.A.; Cordon-Cardo, C.; Thaler, H.T.; Dannenberg, A.J. Celecoxib Inhibits Prostate Cancer Growth: Evidence of a Cyclooxygenase-2-Independent Mechanism. Clin. Cancer Res. 2005, 11, 1999–2007. [Google Scholar] [CrossRef] [Green Version]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention—Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Rigas, B.; Shiff, S. Is inhibition of cyclooxygenase required for the chemopreventive effect of NSAIDs in colon cancer? A model reconciling the current contradiction. Med. Hypotheses 2000, 54, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs Inhibit Tumorigenesis, but How? Clin. Cancer Res. 2013, 20, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. COX-Independent Mechanisms of Cancer Chemoprevention by Anti-Inflammatory Drugs. Front. Oncol. 2013, 3, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akrami, H.; Moradi, B.; Farahani, D.B.; Mehdizadeh, K. Ibuprofen reduces cell proliferation through inhibiting Wnt/β catenin signaling pathway in gastric cancer stem cells. Cell Biol. Int. 2018, 42, 949–958. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Yoshitani, S.; Takashima, S.; Okumura, A.; Murakami, A.; Hosokawa, M. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001, 61, 2424–2428. [Google Scholar]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Okazaki, S.; Shioi, R.; Noguchi-Yachide, T.; Ishikawa, M.; Makishima, M.; Hashimoto, Y.; Yamaguchi, T. Structure-activity relationship studies of non-carboxylic acid peroxisome proliferator-activated receptor α/δ (PPARα/δ) dual agonists. Bioorg. Med. Chem. 2016, 24, 5455–5461. [Google Scholar] [CrossRef]
- Blitek, A.; Szymanska, M. Peroxisome proliferator-activated receptor (PPAR) isoforms are differentially expressed in peri-implantation porcine conceptuses. Theriogenology 2017, 101, 53–61. [Google Scholar] [CrossRef]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef]
- Heudobler, D.; Rechenmacher, M.; Lüke, F.; Vogelhuber, M.; Pukrop, T.; Herr, W.; Ghibelli, L.; Gerner, C.; Reichle, A. Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue. Int. J. Mol. Sci. 2018, 19, 3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, B.; Samadi, N.; Baradaran, B.; Shafiei-Irannejad, V.; Zarghami, N. Peroxisome Proliferator-Activated Receptor Ligands and Their Role in Chronic Myeloid Leukemia: Therapeutic Strategies. Chem. Biol. Drug Des. 2016, 88, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef]
- Kitamura, S.; Miyazaki, Y.; Shinomura, Y.; Kondo, S.; Kanayama, S.; Matsuzawa, Y. Peroxisome proliferator-activated receptor gamma induces growth arrest and differentiation markers of human colon cancer cells. JPN J. Cancer Res. 1999, 90, 75–80. [Google Scholar] [CrossRef]
- Nazim, U.M.; Moon, J.-H.; Lee, Y.-J.; Seol, J.-W.; Park, S.-Y. PPARγ activation by troglitazone enhances human lung cancer cells to TRAIL-induced apoptosis via autophagy flux. Oncotarget 2017, 8, 26819–26831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewoniewska, S.; Oscilowska, I.; Huynh, T.Y.L.; Prokop, I.; Baszanowska, W.; Bielawska, K.; Palka, J. Troglitazone-Induced PRODH/POX-Dependent Apoptosis Occurs in the Absence of Estradiol or ERβ in ER-Negative Breast Cancer Cells. J. Clin. Med. 2021, 10, 4641. [Google Scholar] [CrossRef]
- Hirase, N.; Yanase, T.; Mu, Y.-M.; Muta, K.; Umemura, T.; Takayanagi, R.; Nawata, H. Thiazolidinedione Induces Apoptosis and Monocytic Differentiation in the Promyelocytic Leukemia Cell Line HL60. Oncology 1999, 57, 17–26. [Google Scholar] [CrossRef]
- Pandhare, J.; Cooper, S.K.; Phang, J.M. Proline oxidase, a proapoptotic gene, is induced by troglitazone: Evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 2044–2052. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 2012, 8, 1407–1409. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Liu, W.; Hancock, C.; Christian, K.J. The Proline Regulatory Axis and Cancer. Front. Oncol. 2012, 2, 60. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Pandhare, J.; Liu, Y. The Metabolism of Proline as Microenvironmental Stress Substrate. J. Nutr. 2008, 138, 2008S–2015S. [Google Scholar] [CrossRef] [PubMed]
- Donald, S.P.; Sun, X.Y.; A Hu, C.; Yu, J.; Mei, J.M.; Valle, D.; Phang, J.M. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001, 61, 1810–1815. [Google Scholar] [PubMed]
- Hu, C.-A.A.; Donald, S.P.; Yu, J.; Lin, W.-W.; Liu, Z.; Steel, G.; Obie, C.; Valle, D.; Phang, J.M. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol. Cell. Biochem. 2006, 295, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Hu, C.-A.; Phang, J.M. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: The role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 2006, 25, 5640–5647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, J.J.; Fendt, S.-M.; Becker, D.F. The Proline Cycle As a Potential Cancer Therapy Target. Biochemistry 2018, 57, 3433–3444. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Borchert, G.L.; Donald, S.P.; Surażynski, A.; Hu, C.-A.; Weydert, C.J.; Oberley, L.W.; Phang, J.M. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis 2005, 26, 1335–1342. [Google Scholar] [CrossRef]
- Dannenberg, A.J.; Lippman, S.M.; Mann, J.R.; Subbaramaiah, K.; Dubois, R.N. Cyclooxygenase-2 and Epidermal Growth Factor Receptor: Pharmacologic Targets for Chemoprevention. J. Clin. Oncol. 2005, 23, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Borchert, G.L.; Surazynski, A.; Phang, J.M. Proline oxidase, a p53-induced gene, targets COX-2/PGE2 signaling to induce apoptosis and inhibit tumor growth in colorectal cancers. Oncogene 2008, 27, 6729–6737. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Liu, W.; Zabirnyk, O.; Wang, H.; Shiao, Y.-H.; Nickerson, M.L.; Khalil, S.; Anderson, L.M.; Perantoni, A.O.; Phang, J.M. miR-23b* targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 2010, 29, 4914–4924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, S.A.; Kochevar, G.J. Identification of a p53-response element in the promoter of the proline oxidase gene. Biochem. Biophys. Res. Commun. 2008, 369, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.; Beebe-Donk, J.; Doss, H.; Doss, D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: A critical review of non-selective COX-2 blockade (Review). Oncol. Rep. 2005, 13, 559–583. [Google Scholar] [CrossRef] [PubMed]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-Inflammatory Drugs as Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef] [Green Version]
- Grösch, S.; Tegeder, I.; Niederberger, E.; Bräutigam, L.; Geisslinger, G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J. 2001, 15, 1–12. [Google Scholar] [CrossRef]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996, 52, 237–245. [Google Scholar] [CrossRef]
- Misiura, M.; Ościłowska, I.; Bielawska, K.; Pałka, J.; Miltyk, W. PRODH/POX-Dependent Celecoxib-Induced Apoptosis in MCF-7 Breast Cancer. Pharmaceuticals 2021, 14, 874. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Celińska-Janowicz, K.; Zaręba, I.; Klupczyńska, A.; Kokot, Z.; Nowaszewska, B.K.; Reszeć, J.; Borys, J.; Miltyk, W. Proline-Dependent Induction of Apoptosis in Oral Squamous Cell Carcinoma (OSCC)—The Effect of Celecoxib. Cancers 2020, 12, 136. [Google Scholar] [CrossRef] [Green Version]
- Puhl, A.C.; Milton, F.A.; Cvoro, A.; Sieglaff, D.H.; Campos, J.L.D.O.; Bernardes, A.; Filgueira, C.; Lindemann, J.A.L.; Deng, T.; Neves, F.A.R.; et al. Mechanisms of Peroxisome Proliferator Activated Receptor γ Regulation by Non-steroidal Anti-inflammatory Drugs. Nucl. Recept. Signal. 2015, 13, e004. [Google Scholar] [CrossRef] [Green Version]
- Kononczuk, J.; Czyzewska, U.; Moczydlowska, J.; Surażyński, A.; Palka, J.; Miltyk, W. Proline Oxidase (POX) as A Target for Cancer Therapy. Curr. Drug Targets 2015, 16, 1464–1469. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. PEPD is a pivotal regulator of p53 tumor suppressor. Nat. Commun. 2017, 8, 2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conaghan, P.G. A turbulent decade for NSAIDs: Update on current concepts of classification, epidemiology, comparative efficacy, and toxicity. Rheumatol. Int. 2011, 32, 1491–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Peterkofsky, B.; Chojkier, M.; Bateman, J. Determination of Collagen Synthesis in Tissue and Cell Culture Systems. In Immunochemistry of the Extracellular Matrix, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 19–48. ISBN 9781351073424. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Myara, I.; Charpentier, C.; Lemonnier, A. Optimal conditions for prolidase assay by proline colorimetric determination: Application to iminodipeptiduria. Clin. Chim. Acta 1982, 125, 193–205. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazberuk, A.; Chalecka, M.; Palka, J.; Surazynski, A. Nonsteroidal Anti-Inflammatory Drugs as PPARγ Agonists Can Induce PRODH/POX-Dependent Apoptosis in Breast Cancer Cells: New Alternative Pathway in NSAID-Induced Apoptosis. Int. J. Mol. Sci. 2022, 23, 1510. https://doi.org/10.3390/ijms23031510
Kazberuk A, Chalecka M, Palka J, Surazynski A. Nonsteroidal Anti-Inflammatory Drugs as PPARγ Agonists Can Induce PRODH/POX-Dependent Apoptosis in Breast Cancer Cells: New Alternative Pathway in NSAID-Induced Apoptosis. International Journal of Molecular Sciences. 2022; 23(3):1510. https://doi.org/10.3390/ijms23031510
Chicago/Turabian StyleKazberuk, Adam, Magda Chalecka, Jerzy Palka, and Arkadiusz Surazynski. 2022. "Nonsteroidal Anti-Inflammatory Drugs as PPARγ Agonists Can Induce PRODH/POX-Dependent Apoptosis in Breast Cancer Cells: New Alternative Pathway in NSAID-Induced Apoptosis" International Journal of Molecular Sciences 23, no. 3: 1510. https://doi.org/10.3390/ijms23031510
APA StyleKazberuk, A., Chalecka, M., Palka, J., & Surazynski, A. (2022). Nonsteroidal Anti-Inflammatory Drugs as PPARγ Agonists Can Induce PRODH/POX-Dependent Apoptosis in Breast Cancer Cells: New Alternative Pathway in NSAID-Induced Apoptosis. International Journal of Molecular Sciences, 23(3), 1510. https://doi.org/10.3390/ijms23031510