
Transcriptomics and Functional Analysis of Copper Stress Response in the Sulfate-Reducing Bacterium Desulfovibrio alaskensis G20

 and
and 
Abstract
:1. Introduction
2. Results
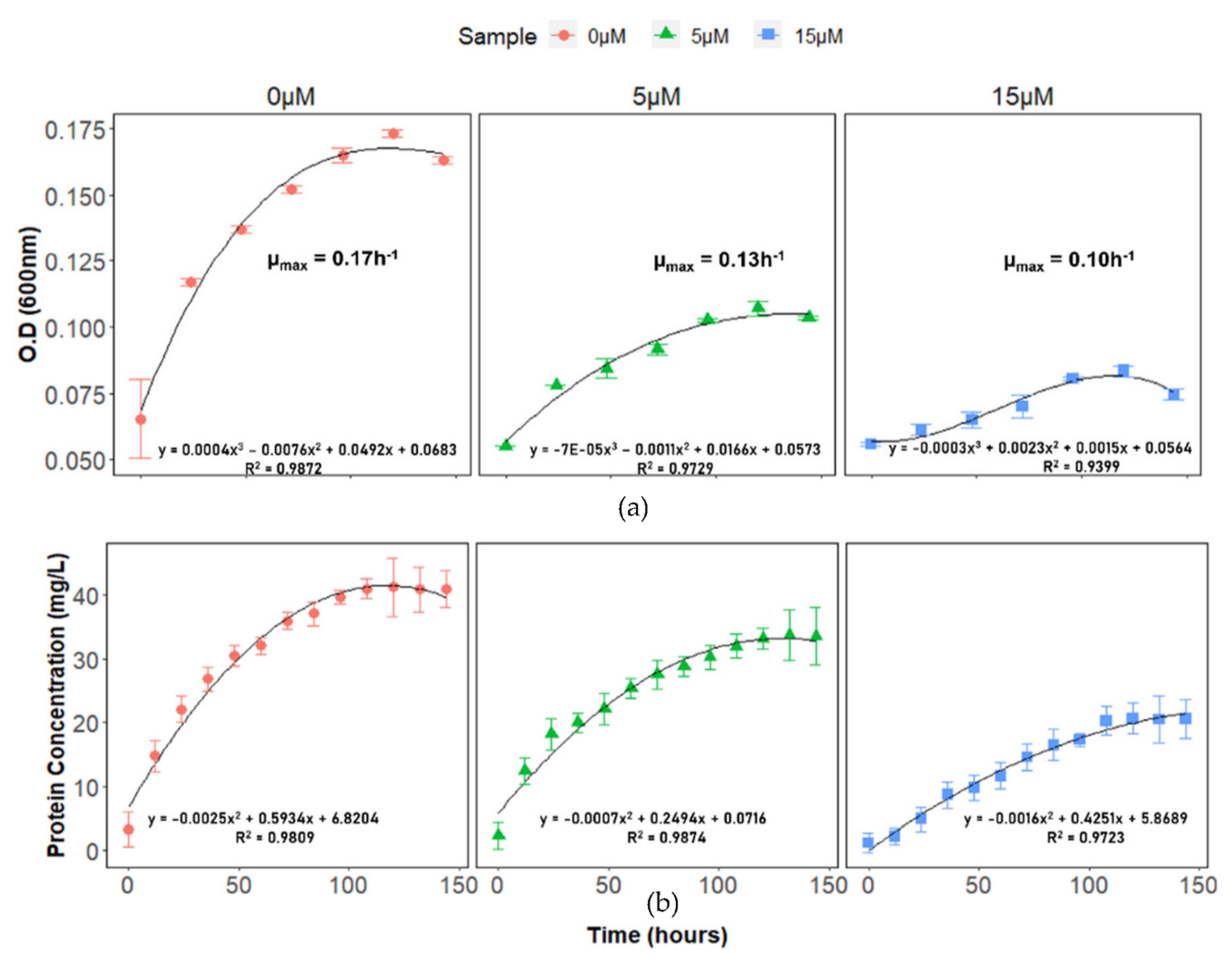
2.1. DA-G20 Growth under Cu(II) Stress
2.2. Differential Gene Expression Analysis during Cu(II) Stress
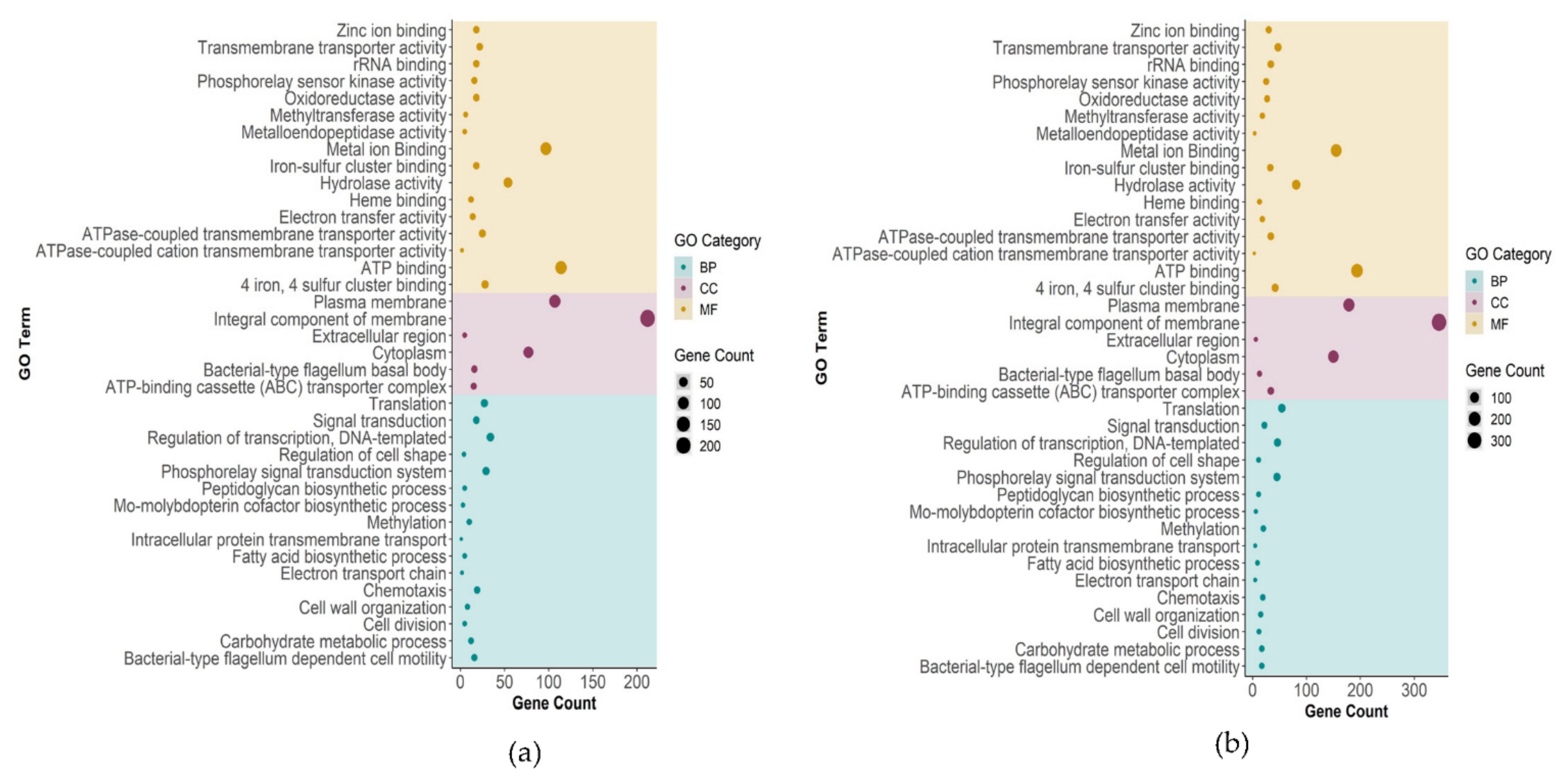
2.3. Gene Ontology Analyses of DEGs
2.3.1. Biological Process, Molecular Function, and Cellular Component Enrichment Analyses
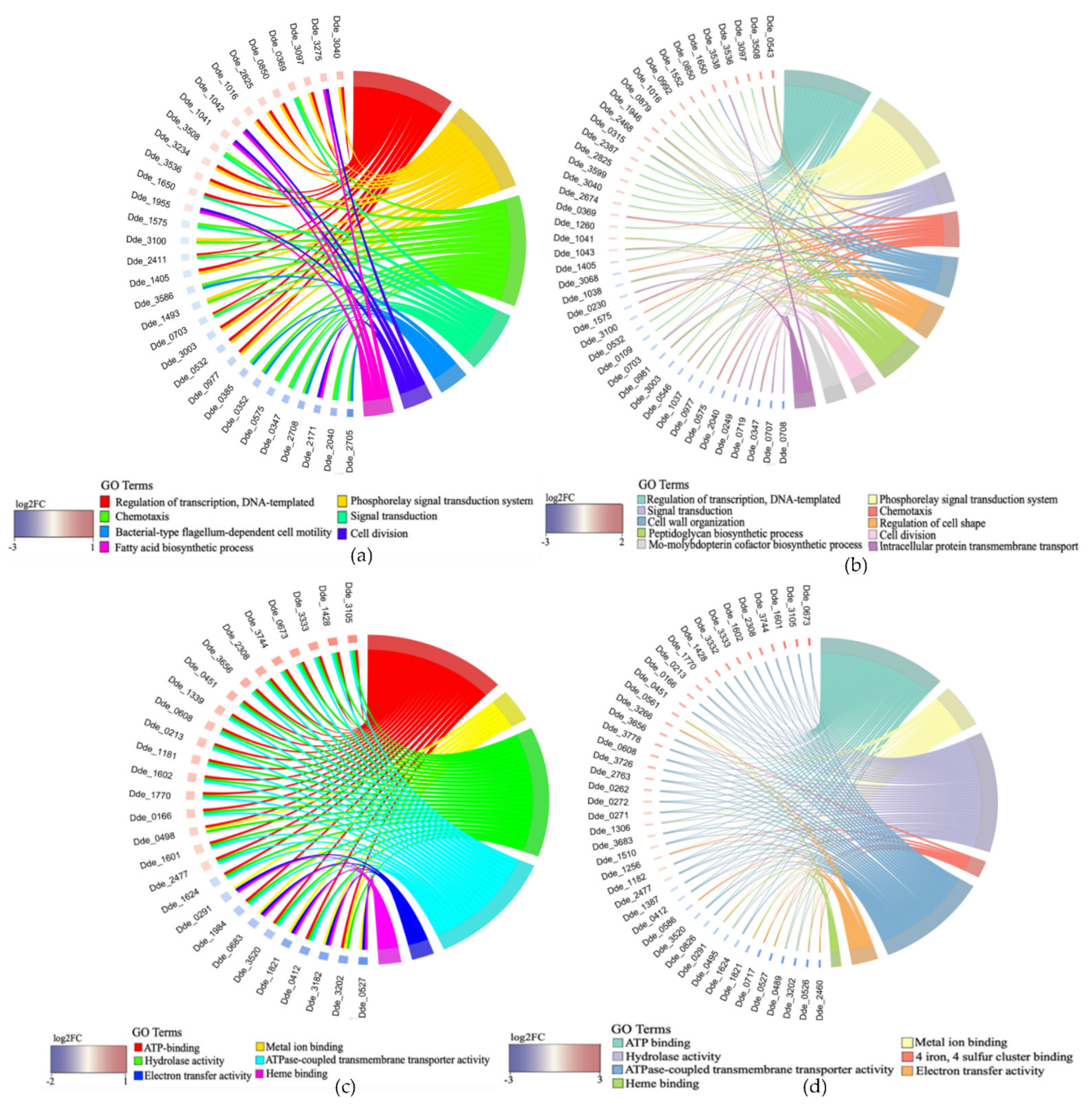
2.3.2. Association between Genes and Enriched GO Terms
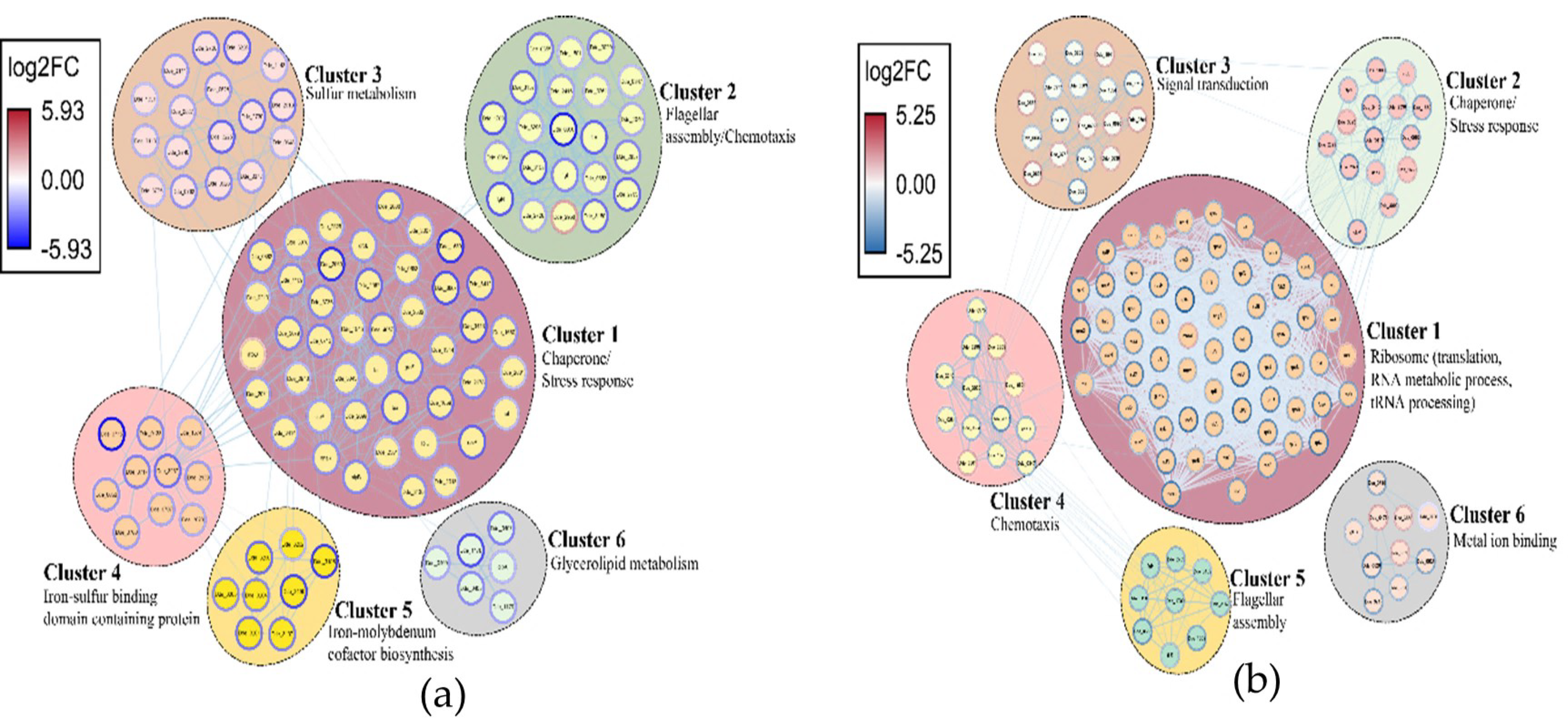
2.3.3. Network Analysis of the DEGs and Identification of Clusters
2.3.4. Metabolomic Profile of Control and Test Samples
3. Discussion
3.1. Downregulation of Translation Machinery
3.2. Modulation in Transporter Related Activity
3.3. Regulation of Oxidative Stress Response
3.4. Impact on Chemotaxis and Signal Transduction System
3.5. Some Atypical Transcriptional Changes Induced by Cu(II) Ions
3.5.1. Significant Upregulation of ApbE Family Lipoprotein
3.5.2. Differential Expression of DNA Repair Genes
3.5.3. Modulation of Methylation Related Activity
3.5.4. Regulation of Genes Associated with Cell Division and Cell Wall Organization
3.5.5. Role of Ascorbic Acid and Alkaline Phosphatase
4. Materials and Methods
4.1. Bacterial Strain and Growth Conditions
4.2. Copper Toxicity Experimental Setup
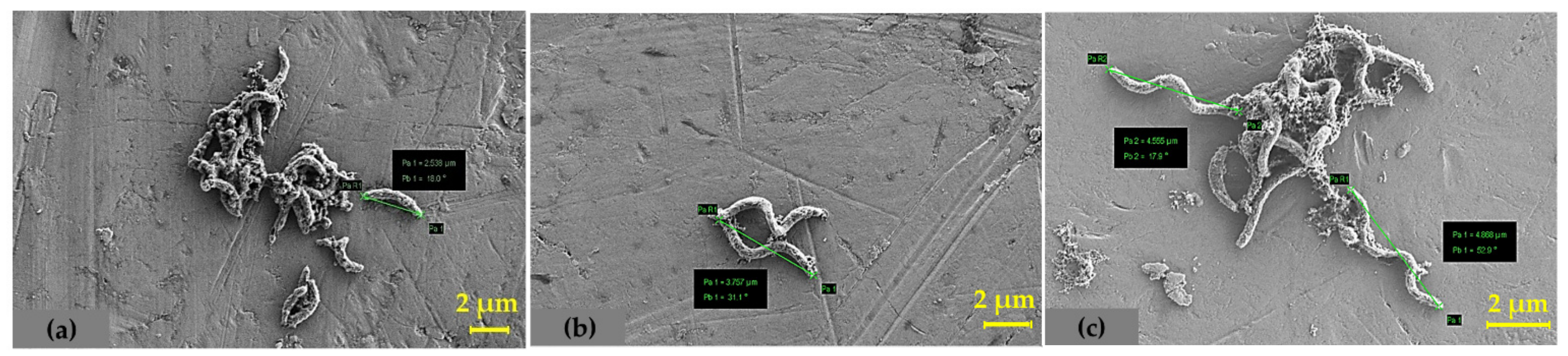
4.3. Determination of Total Cell Protein and SEM Analysis
4.4. RNA Isolation
4.5. Complementary DNA (cDNA) Library Preparation and Sequencing
4.6. QC of Raw RNA Sequencing Reads and Data Analysis
4.7. RT-qPCR Validation
4.8. Network Analysis Using Cytoscape
4.9. Bioinformatics Analysis
4.10. Targeted Metabolomics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Giachino, A.; Waldron, K.J. Copper tolerance in bacteria requires the activation of multiple accessory pathways. Mol. Microbiol. 2020, 114, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Salah, I.; Parkin, I.P.; Allan, E. Copper as an antimicrobial agent: Recent advances. RSC Adv. 2021, 11, 18179–18186. [Google Scholar] [CrossRef]
- Oursel, B.; Garnier, C.; Durrieu, G.; Mounier, S.; Omanović, D.; Lucas, Y. Dynamics and fates of trace metals chronically input in a Mediterranean coastal zone impacted by a large urban area. Mar. Pollut. Bull. 2013, 69, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Sani Rajesh, K.; Peyton Brent, M.; Brown Laura, T. Copper-Induced Inhibition of Growth of Desulfovibrio desulfuricans G20: Assessment of Its Toxicity and Correlation with Those of Zinc and Lead. Appl. Environ. Microbiol. 2001, 67, 4765–4772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. Exp. Suppl. 2012, 101, 133–164. [Google Scholar] [CrossRef] [Green Version]
- Vargas, I.T.; Fischer, D.A.; Alsina, M.A.; Pavissich, J.P.; Pastén, P.A.; Pizarro, G.E. Copper Corrosion and Biocorrosion Events in Premise Plumbing. Materials 2017, 10, 1036. [Google Scholar] [CrossRef]
- Taylor, A.A.; Tsuji, J.S.; Garry, M.R.; McArdle, M.E.; Goodfellow, W.L.; Adams, W.J.; Menzie, C.A. Critical Review of Exposure and Effects: Implications for Setting Regulatory Health Criteria for Ingested Copper. Environ. Manag. 2020, 65, 131–159. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Kuila, A. Bioremediation of heavy metals by microbial process. Environ. Technol. Innov. 2019, 14, 100369. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Occurrence of Copper Proteins through the Three Domains of Life: A Bioinformatic Approach. J. Proteome Res. 2008, 7, 209–216. [Google Scholar] [CrossRef]
- Arguello, J.; Raimunda, D.; Padilla-Benavides, T. Mechanisms of copper homeostasis in bacteria. Front. Cell. Infect. Microbiol. 2013, 3, 73. [Google Scholar] [CrossRef] [Green Version]
- Rensing, C.; Grass, G. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev. 2003, 27, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Bhamidimarri, S.P.; Young, T.R.; Shanmugam, M.; Soderholm, S.; Baslé, A.; Bumann, D.; van den Berg, B. Acquisition of ionic copper by the bacterial outer membrane protein OprC through a novel binding site. PLoS Biol. 2021, 19, e3001446. [Google Scholar] [CrossRef] [PubMed]
- Barwinska-Sendra, A.; Waldron, K.J. The Role of Intermetal Competition and Mis-Metalation in Metal Toxicity. Adv. Microb. Physiol. 2017, 70, 315–379. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.L.; Grass, G.; Rensing, C. Copper toxicity and the origin of bacterial resistance—New insights and applications. Metallomics 2011, 3, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Raimunda, D.; González-Guerrero, M.; Leeber, B.W., 3rd; Argüello, J.M. The transport mechanism of bacterial Cu+-ATPases: Distinct efflux rates adapted to different function. Biometals 2011, 24, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Fan, W. Bioremediation of heavy metal-contaminated soils by sulfate-reducing bacteria. Ann. N. Y. Acad. Sci. 2008, 1140, 446–454. [Google Scholar] [CrossRef]
- Hussain, A.; Hasan, A.; Javid, A.; Qazi, J.I. Exploited application of sulfate-reducing bacteria for concomitant treatment of metallic and non-metallic wastes: A mini review. 3 Biotech 2016, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Sani, R.K.; Geesey, G.G.; Peyton, B.M. Assessment of lead toxicity to Desulfovibrio desulfuricans G20: Influence of components of lactate C medium. Adv. Environ. Res. 2001, 5, 269–276. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Thakur, P.; Saxena, P.; Rauniyar, S.; Gopalakrishnan, V.; Singh, R.N.; Gadhamshetty, V.; Gnimpieba, E.Z.; Jasthi, B.K.; Sani, R.K. Gene Sets and Mechanisms of Sulfate-Reducing Bacteria Biofilm Formation and Quorum Sensing with Impact on Corrosion. Front. Microbiol. 2021, 12, 3120. [Google Scholar] [CrossRef]
- Ayangbenro, A.S.; Olanrewaju, O.S.; Babalola, O.O. Sulfate-Reducing Bacteria as an Effective Tool for Sustainable Acid Mine Bioremediation. Front. Microbiol. 2018, 9, 1986. [Google Scholar] [CrossRef]
- Miran, W.; Jang, J.; Nawaz, M.; Shahzad, A.; Jeong, S.E.; Jeon, C.O.; Lee, D.S. Mixed sulfate-reducing bacteria-enriched microbial fuel cells for the treatment of wastewater containing copper. Chemosphere 2017, 189, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Jalali, K.; Baldwin, S.A. The role of sulphate reducing bacteria in copper removal from aqueous sulphate solutions. Water Res. 2000, 34, 797–806. [Google Scholar] [CrossRef]
- Andrei, A.; Öztürk, Y.; Khalfaoui-Hassani, B.; Rauch, J.; Marckmann, D.; Trasnea, P.-I.; Daldal, F.; Koch, H.-G. Cu Homeostasis in Bacteria: The Ins and Outs. Membranes 2020, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Monds, R.D.; Lee, T.K.; Colavin, A.; Ursell, T.; Quan, S.; Cooper, T.F.; Huang, K.C. Systematic perturbation of cytoskeletal function reveals a linear scaling relationship between cell geometry and fitness. Cell Rep. 2014, 9, 1528–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiler-Samerotte, K.A.; Li, S.; Lazaris, C.; Taylor, A.; Ziv, N.; Ramjeawan, C.; Paaby, A.B.; Siegal, M.L. Extent and context dependence of pleiotropy revealed by high-throughput single-cell phenotyping. PLoS Biol. 2020, 18, e3000836. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Petit-Härtlein, I.; Rome, K.; de Rosny, E.; Molton, F.; Duboc, C.; Gueguen, E.; Rodrigue, A.; Covès, J. Biophysical and physiological characterization of ZraP from Escherichia coli, the periplasmic accessory protein of the atypical ZraSR two-component system. Biochem. J. 2015, 472, 205–216. [Google Scholar] [CrossRef]
- Adhikari, S.; Curtis, P.D. DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev. 2016, 40, 575–591. [Google Scholar] [CrossRef]
- Barton, L.L.; Goulhen, F.; Bruschi, M.; Woodards, N.A.; Plunkett, R.M.; Rietmeijer, F.J.M. The bacterial metallome: Composition and stability with specific reference to the anaerobic bacterium Desulfovibrio desulfuricans. BioMetals 2007, 20, 291–302. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Nikolskaya, A.N.; Koonin, E.V. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 2001, 203, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, N. Lipid Flippases for Bacterial Peptidoglycan Biosynthesis. Lipid Insights 2015, 8 (Suppl. S1), 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakaran, P.; Ashraf, M.A.; Aqma, W.S. Microbial stress response to heavy metals in the environment. RSC Adv. 2016, 6, 109862–109877. [Google Scholar] [CrossRef]
- Pal, A.; Bhattacharjee, S.; Saha, J.; Sarkar, M.; Mandal, P. Bacterial survival strategies and responses under heavy metal stress: A comprehensive overview. Crit. Rev. Microbiol. 2021, 48, 1–29. [Google Scholar] [CrossRef]
- Plugge, C.; Zhang, W.; Scholten, J.; Stams, A. Metabolic Flexibility of Sulfate-Reducing Bacteria. Front. Microbiol. 2011, 2, 81. [Google Scholar] [CrossRef] [Green Version]
- Joudeh, N.; Saragliadis, A.; Schulz, C.; Voigt, A.; Almaas, E.; Linke, D. Transcriptomic Response Analysis of Escherichia coli to Palladium Stress. Front. Microbiol. 2021, 12, 2840. [Google Scholar] [CrossRef]
- Hu, P.; Brodie Eoin, L.; Suzuki, Y.; McAdams Harley, H.; Andersen Gary, L. Whole-Genome Transcriptional Analysis of Heavy Metal Stresses in Caulobacter crescentus. J. Bacteriol. 2005, 187, 8437–8449. [Google Scholar] [CrossRef] [Green Version]
- Volpicella, M.; Leoni, C.; Manzari, C.; Chiara, M.; Picardi, E.; Piancone, E.; Italiano, F.; D’Erchia, A.; Trotta, M.; Horner, D.S.; et al. Transcriptomic analysis of nickel exposure in Sphingobium sp. ba1 cells using RNA-seq. Sci. Rep. 2017, 7, 8262. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, C.J.; Brown, N.L.; Constantinidou, C.; Patel, M.D.; Hobman, J.L. The expression profile of Escherichia coli K-12 in response to minimal, optimal and excess copper concentrations. Microbiology 2005, 151 Pt 4, 1187–1198. [Google Scholar] [CrossRef] [Green Version]
- Pontes, M.H.; Sevostyanova, A.; Groisman, E.A. When Too Much ATP Is Bad for Protein Synthesis. J. Mol. Biol. 2015, 427, 2586–2594. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Crowley, D.E. Global gene expression responses to cadmium toxicity in Escherichia coli. J. Bacteriol. 2005, 187, 3259–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gault, M.; Effantin, G.; Rodrigue, A. Ni exposure impacts the pool of free Fe and modifies DNA supercoiling via metal-induced oxidative stress in Escherichia coli K-12. Free Radic. Biol. Med. 2016, 97, 351–361. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, J.S.; Shaw, A.M. Differential gene regulation in the Ag nanoparticle and Ag(+)-induced silver stress response in Escherichia coli: A full transcriptomic profile. Nanotoxicology 2014, 8 (Suppl. S1), 177–184. [Google Scholar] [CrossRef] [PubMed]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Piepenbreier, H.; Fritz, G.; Gebhard, S. Transporters as information processors in bacterial signalling pathways. Mol. Microbiol. 2017, 104, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Porcheron, G.; Garenaux, A.; Proulx, J.; Sabri, M.; Dozois, C. Iron, copper, zinc, and manganese transport and regulation in pathogenic Enterobacteria: Correlations between strains, site of infection and the relative importance of the different metal transport systems for virulence. Front. Cell. Infect. Microbiol. 2013, 3, 90. [Google Scholar] [CrossRef] [Green Version]
- El-Awady, R.; Saleh, E.; Hashim, A.; Soliman, N.; Dallah, A.; Elrasheed, A.; Elakraa, G. The Role of Eukaryotic and Prokaryotic ABC Transporter Family in Failure of Chemotherapy. Front. Pharmacol. 2017, 7, 535. [Google Scholar] [CrossRef] [Green Version]
- Herrou, J.; Willett, J.W.; Czyż, D.M.; Babnigg, G.; Kim, Y.; Crosson, S. Conserved ABC Transport System Regulated by the General Stress Response Pathways of Alpha- and Gammaproteobacteria. J. Bacteriol. 2017, 199, e00746-16. [Google Scholar] [CrossRef] [Green Version]
- Ayangbenro, A.S.; Babalola, O.O. A New Strategy for Heavy Metal Polluted Environments: A Review of Microbial Biosorbents. Int. J. Environ. Res. Public Health 2017, 14, 94. [Google Scholar] [CrossRef]
- Biemans-Oldehinkel, E.; Mahmood, N.A.B.N.; Poolman, B. A sensor for intracellular ionic strength. Proc. Natl. Acad. Sci. USA 2006, 103, 10624–10629. [Google Scholar] [CrossRef] [Green Version]
- Fantino, J.R.; Py, B.; Fontecave, M.; Barras, F. A genetic analysis of the response of Escherichia coli to cobalt stress. Environ. Microbiol. 2010, 12, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, S.P.; Summers, A.O. Correction to: Transcriptional responses of Escherichia coli during recovery from inorganic or organic mercury exposure. BMC Genom. 2018, 19, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Jacobsen, F.E.; Giedroc, D.P. Coordination chemistry of bacterial metal transport and sensing. Chem. Rev. 2009, 109, 4644–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nies, D.H. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol. Rev. 2003, 27, 313–339. [Google Scholar] [CrossRef]
- Chien, C.C.; Huang, C.H.; Lin, Y.W. Characterization of a heavy metal translocating P-type ATPase gene from an environmental heavy metal resistance Enterobacter sp. isolate. Appl. Biochem. Biotechnol. 2013, 169, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Valencia, E.Y.; Braz, V.S.; Guzzo, C.; Marques, M.V. Two RND proteins involved in heavy metal efflux in Caulobacter crescentus belong to separate clusters within proteobacteria. BMC Microbiol. 2013, 13, 79. [Google Scholar] [CrossRef] [Green Version]
- Chandrangsu, P.; Rensing, C.; Helmann, J.D. Metal homeostasis and resistance in bacteria. Nat. Rev. Microbiol. 2017, 15, 338–350. [Google Scholar] [CrossRef]
- Lewinson, O.; Lee, A.T.; Rees, D.C. A P-type ATPase importer that discriminates between essential and toxic transition metals. Proc. Natl. Acad. Sci. USA 2009, 106, 4677. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, R.D.; Hentges, D.J.; Campbell, B.J.; Barrett, J.T. Factors related to the oxygen tolerance of anaerobic bacteria. Appl. Environ. Microbiol. 1978, 36, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.R.; Selby, T.; Coleman, J.P.; Smith, C.J. Oxidative stress response in an anaerobe, Bacteroides fragilis: A role for catalase in protection against hydrogen peroxide. J. Bacteriol. 1996, 178, 6895–6903. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, R.A.; Holmes, W.R.; Fontaine, C.P.; Maret, W. Cytosolic zinc buffering and muffling: Their role in intracellular zinc homeostasis. Metallomics 2010, 2, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Krishna Deepak, R.N.V.; Chandrakar, B.; Sankararamakrishnan, R. Comparison of metal-binding strength between methionine and cysteine residues: Implications for the design of metal-binding motifs in proteins. Biophys. Chem. 2017, 224, 32–39. [Google Scholar] [CrossRef]
- Lee, L.J.; Barrett, J.A.; Poole, R.K. Genome-wide transcriptional response of chemostat-cultured Escherichia coli to zinc. J. Bacteriol. 2005, 187, 1124–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoostal, M.J.; Bidart-Bouzat, M.G.; Bouzat, J.L. Local adaptation of microbial communities to heavy metal stress in polluted sediments of Lake Erie. FEMS Microbiol. Ecol. 2008, 65, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Uda, N.R.; Upert, G.; Angelici, G.; Nicolet, S.; Schmidt, T.; Schwede, T.; Creus, M. Zinc-selective inhibition of the promiscuous bacterial amide-hydrolase DapE: Implications of metal heterogeneity for evolution and antibiotic drug design. Metallomics 2014, 6, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Jaworska, H.; Lemanowicz, J. Heavy metal contents and enzymatic activity in soils exposed to the impact of road traffic. Sci. Rep. 2019, 9, 19981. [Google Scholar] [CrossRef]
- Wadhams, G.H.; Armitage, J.P. Making sense of it all: Bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 2004, 5, 1024–1037. [Google Scholar] [CrossRef]
- Singh, K.; Senadheera, D.B.; Cvitkovitch, D.G. An intimate link: Two-component signal transduction systems and metal transport systems in bacteria. Future Microbiol. 2014, 9, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Berg, H.C. The Rotary Motor of Bacterial Flagella. Annu. Rev. Biochem. 2003, 72, 19–54. [Google Scholar] [CrossRef]
- Yung, M.C.; Ma, J.; Salemi, M.R.; Phinney, B.S.; Bowman, G.R.; Jiao, Y. Shotgun Proteomic Analysis Unveils Survival and Detoxification Strategies by Caulobacter crescentus during Exposure to Uranium, Chromium, and Cadmium. J. Proteome Res. 2014, 13, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Chourey, K.; Thompson, M.R.; Morrell-Falvey, J.; Verberkmoes, N.C.; Brown, S.D.; Shah, M.; Zhou, J.; Doktycz, M.; Hettich, R.L.; Thompson, D.K. Global molecular and morphological effects of 24-hour chromium(VI) exposure on Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 2006, 72, 6331–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, J.; Eriksen, M.; Kallipolitis, B.; Valentin-Hansen, P. Down-regulation of outer membrane proteins by noncoding RNAs: Unraveling the cAMP-CRP- and sigmaE-dependent CyaR-ompX regulatory case. J. Mol. Biol. 2008, 383, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular Mechanisms of Two-Component Signal Transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, B.J.; Downs, D.M. The apbE gene encodes a lipoprotein involved in thiamine synthesis in Salmonella typhimurium. J. Bacteriol. 1998, 180, 885–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, B.J.; Downs, D.M. A periplasmic location is essential for the role of the ApbE lipoprotein in thiamine synthesis in Salmonella typhimurium. J. Bacteriol. 1999, 181, 7285–7290. [Google Scholar] [CrossRef] [Green Version]
- Skovran, E.; Downs, D.M. Lack of the ApbC or ApbE protein results in a defect in Fe-S cluster metabolism in Salmonella enterica serovar Typhimurium. J. Bacteriol. 2003, 185, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Bertsova, Y.V.; Fadeeva, M.S.; Kostyrko, V.A.; Serebryakova, M.V.; Baykov, A.A.; Bogachev, A.V. Alternative pyrimidine biosynthesis protein ApbE is a flavin transferase catalyzing covalent attachment of FMN to a threonine residue in bacterial flavoproteins. J. Biol. Chem. 2013, 288, 14276–14286. [Google Scholar] [CrossRef] [Green Version]
- Helbig, K.; Grosse, C.; Nies, D.H. Cadmium toxicity in glutathione mutants of Escherichia coli. J. Bacteriol. 2008, 190, 5439–5454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, S.; Hao, X.; Su, J.Q.; Xue, X.; Yan, Y.; Zhu, Y.G.; Ye, J. Transcriptomic Analysis Reveals Adaptive Responses of an Enterobacteriaceae Strain LSJC7 to Arsenic Exposure. Front. Microbiol. 2016, 7, 636. [Google Scholar] [CrossRef] [Green Version]
- Górecka, K.M.; Krepl, M.; Szlachcic, A.; Poznański, J.; Šponer, J.; Nowotny, M. RuvC uses dynamic probing of the Holliday junction to achieve sequence specificity and efficient resolution. Nat. Commun. 2019, 10, 4102. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, C.W.; Segall, A.M. DNA repair, a novel antibacterial target: Holliday junction-trapping peptides induce DNA damage and chromosome segregation defects. Mol. Microbiol. 2006, 59, 1129–1148. [Google Scholar] [CrossRef] [PubMed]
- Casadesús, J.; Low, D. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. MMBR 2006, 70, 830–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The Epigenomic Landscape of Prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendall, M.L.; Luong, K.; Wetmore, K.M.; Blow, M.; Korlach, J.; Deutschbauer, A.; Malmstrom, R.R. Exploring the Roles of DNA Methylation in the Metal-Reducing Bacterium Shewanella oneidensis MR-1. J. Bacteriol. 2013, 195, 4966–4974. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 15553–15557. [Google Scholar] [CrossRef] [Green Version]
- Erickson Harold, P.; Anderson David, E.; Osawa, M. FtsZ in Bacterial Cytokinesis: Cytoskeleton and Force Generator All in One. Microbiol. Mol. Biol. Rev. 2010, 74, 504–528. [Google Scholar] [CrossRef] [Green Version]
- Chien, A.C.; Hill, N.S.; Levin, P.A. Cell size control in bacteria. Curr. Biol. 2012, 22, R340–R349. [Google Scholar] [CrossRef] [Green Version]
- Palacios, P.; Vicente, M.; Sánchez, M. Dependency of Escherichia coli cell-division size, and independency of nucleoid segregation on the mode and level of ftsZ expression. Mol. Microbiol. 1996, 20, 1093–1098. [Google Scholar] [CrossRef]
- Bach, J.N.; Albrecht, N.; Bramkamp, M. Imaging DivIVA dynamics using photo-convertible and activatable fluorophores in Bacillus subtilis. Front. Microbiol. 2014, 5, 59. [Google Scholar] [CrossRef]
- Wu, L.; Wang, Y.; Zhou, S.; Zhu, Y.; Chen, X. Enzyme-induced Cu2+/Cu+ conversion as the electrochemical signal for sensitive detection of ethyl carbamate. Anal. Chim. Acta 2021, 1151, 338256. [Google Scholar] [CrossRef] [PubMed]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 23 December 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.; Petrzelkova, K.; Yeoman, C.J.; Vlckova, K.; Mrázek, J.; Koppova, I.; Carbonero, F.; Ulanov, A.; Modry, D.; Todd, A.; et al. Gut microbiome composition and metabolomic profiles of wild western lowland gorillas (Gorilla gorilla gorilla) reflect host ecology. Mol. Ecol. 2015, 24, 2551–2565. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Protein Name | log2FC | Standard Error |
|---|---|---|---|
| Dde_2535 * | ApbE family lipoprotein | 3.95 | ±0.37 |
| Dde_2958 * | Flagellar basal body rod protein | 3.12 | ±0.31 |
| Dde_3047 * | Protein serine/threonine phosphatase | 3.08 | ±1.19 |
| Dde_1981 * | Uncharacterized protein | 3.05 | ±0.93 |
| Dde_0899 * | Uncharacterized protein | 2.98 | ±0.48 |
| Dde_1205 * | Uncharacterized protein | 2.96 | ±0.44 |
| Dde_2895 * | Teichoic-acid-transporting ATPase | 2.85 | ±0.96 |
| Dde_1929 * | Uncharacterized protein | 2.82 | ±0.49 |
| Dde_2799 * | Phage regulatory protein, Rha family | 2.81 | ±0.63 |
| Dde_0900 * | RNA polymerase sigma factor, sigma-70 family | 2.77 | ±0.54 |
| Dde_0111 # | Zinc resistance-associated protein | −8.56 | ±0.30 |
| Dde_4025 # | Uncharacterized protein | −7.07 | ±0.64 |
| Dde_2170 # | UPF0235 protein Dde_2170 | −6.48 | ±0.39 |
| Dde_2819 # | Uncharacterized protein | −6.11 | ±0.27 |
| Dde_3737 # | Putative GAF sensor protein | −5.98 | ±0.77 |
| Dde_2991 # | Transcription termination/antitermination protein, NusG | −5.97 | ±0.36 |
| Dde_3226 # | Phage shock protein A, PspA | −5.68 | ±0.27 |
| Dde_1010 # | Uncharacterized protein | −5.52 | ±0.22 |
| Dde_0221 # | Response regulator receiver protein | −5.51 | ±0.41 |
| Dde_0715 # | Uncharacterized protein | −5.46 | ±0.35 |
| Dde_3047 ** | Protein serine/threonine phosphatase | 3.05 | ±1.03 |
| Dde_2958 ** | Flagellar basal body rod protein | 2.48 | ±0.36 |
| Dde_0959 ** | AIG2 family protein | 2.04 | ±0.60 |
| Dde_3729 ** | ABC transporter related protein | 2.01 | ±0.55 |
| Dde_4035 ** | Uncharacterized protein | 1.87 | ±0.60 |
| Dde_1264 ** | PAS modulated sigma54 specific transcriptional regulator, Fis family | 1.82 | ±0.67 |
| Dde_3378 ** | Uncharacterized protein | 1.80 | ±0.61 |
| Dde_0930 ** | Uncharacterized protein | 1.70 | ±0.53 |
| Dde_3061 ** | M18 family aminopeptidase | 1.68 | ±0.69 |
| Dde_0925 ** | Uncharacterized protein | 1.67 | ±0.62 |
| Dde_0715 ## | Uncharacterized protein | −5.93 | ±0.32 |
| Dde_2170 ## | UPF0235 protein Dde_2170 | −5.72 | ±0.37 |
| Dde_4025 ## | Uncharacterized protein | −5.58 | ±0.63 |
| Dde_0356 ## | Flagellar basal body rod protein FlgB | −5.34 | ±0.35 |
| Dde_0221 ## | Response regulator receiver protein | −5.17 | ±0.42 |
| Dde_0563 ## | Uncharacterized protein | −5.10 | ±0.37 |
| Dde_1010 ## | Uncharacterized protein | −4.88 | ±0.23 |
| Dde_2560 ## | Thioredoxin peroxidase | −4.84 | ±0.29 |
| Dde_1689 ## | OmpA/MotB domain protein | −4.78 | ±0.25 |
| Dde_0283 ## | Uncharacterized protein | −4.64 | ±0.26 |
| Enriched GO Terms in SP1 (0 vs. 5 µM Cu) | ||||
|---|---|---|---|---|
| Gene Ontology (GO) Term | GO ID | Total Gene Count | −Log10 (p-Value) | z-Score * |
| Regulation of transcription, DNA-templated (BP) | GO:0006355 | 34 | 16.5 | 0 |
| Phosphorelay signal transduction system (BP) | GO:0000160 | 29 | 2.60 | −1.29 |
| Translation (BP) | GO:0006412 | 27 | 48.14 | −4.81 |
| Chemotaxis (BP) | GO:0006935 | 19 | 2.74 | −2.98 |
| Signal transduction (BP) | GO:0007165 | 18 | 2.73 | −1.88 |
| ATP binding (MF) | GO:0005524 | 114 | 16.55 | 2.06 |
| Metal ion binding (MF) | GO:0046872 | 97 | 2.97 | −2.94 |
| Hydrolase activity (MF) | GO:0016787 | 54 | 1.96 | 0 |
| 4 iron, 4 sulfur cluster binding (MF) | GO:0051539 | 28 | 2.32 | −0.75 |
| ATPase-coupled transmembrane transporter activity (MF) | GO:0042626 | 25 | 1.64 | 3.40 |
| Integral component of membrane (CC) | GO:0016021 | 212 | 3.38 | −0.41 |
| Plasma membrane (CC) | GO:0005886 | 99 | 1.94 | −0.90 |
| Cytoplasm (CC) | GO:0005737 | 76 | 1.77 | −2.52 |
| ATP-binding cassette (ABC) transporter complex (CC) | GO:0043190 | 15 | 1.58 | 2.32 |
| Bacterial-type flagellum basal body (CC) | GO:0009425 | 9 | 5.05 | −3.00 |
| Enriched GO terms in SP2 (0 vs. 15 µM Cu) | ||||
| Translation (BP) | GO:0006412 | 54 | 51.75 | −7.07 |
| Regulation of transcription, DNA-templated (BP) | GO:0006355 | 45 | 3.59 | 1.5 |
| Phosphorelay signal transduction system (BP) | GO:0000160 | 45 | 2.30 | 0.15 |
| Signal transduction (BP) | GO:0007165 | 22 | 5.76 | −0.85 |
| Methylation (BP) | GO:0032259 | 20 | 4.26 | 0 |
| ATP binding (MF) | GO:0005524 | 194 | 72.25 | 1.29 |
| Metal ion binding (MF) | GO:0046872 | 152 | 3.30 | −1.13 |
| Hydrolase activity (MF) | GO:0016787 | 81 | 4.37 | 1.22 |
| Transmembrane transporter activity (MF) | GO:0022857 | 47 | 3.20 | 4.23 |
| 4 iron, 4 sulfur cluster binding (MF) | GO:0051539 | 42 | 2.33 | 0.3 |
| Integral component of membrane (CC) | GO:0016021 | 346 | 17.47 | 4.3 |
| Plasma membrane (CC) | GO:0005886 | 160 | 1.66 | 1.42 |
| Cytoplasm (CC) | GO:0005737 | 149 | 1.52 | −5.65 |
| ATP-binding cassette (ABC) transporter complex (CC) | GO:0043190 | 34 | 1.73 | 4.11 |
| Bacterial-type flagellum basal body (CC) | GO:0009425 | 6 | 2.76 | −0.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripathi, A.K.; Saxena, P.; Thakur, P.; Rauniyar, S.; Samanta, D.; Gopalakrishnan, V.; Singh, R.N.; Sani, R.K. Transcriptomics and Functional Analysis of Copper Stress Response in the Sulfate-Reducing Bacterium Desulfovibrio alaskensis G20. Int. J. Mol. Sci. 2022, 23, 1396. https://doi.org/10.3390/ijms23031396
Tripathi AK, Saxena P, Thakur P, Rauniyar S, Samanta D, Gopalakrishnan V, Singh RN, Sani RK. Transcriptomics and Functional Analysis of Copper Stress Response in the Sulfate-Reducing Bacterium Desulfovibrio alaskensis G20. International Journal of Molecular Sciences. 2022; 23(3):1396. https://doi.org/10.3390/ijms23031396
Chicago/Turabian StyleTripathi, Abhilash Kumar, Priya Saxena, Payal Thakur, Shailabh Rauniyar, Dipayan Samanta, Vinoj Gopalakrishnan, Ram Nageena Singh, and Rajesh Kumar Sani. 2022. "Transcriptomics and Functional Analysis of Copper Stress Response in the Sulfate-Reducing Bacterium Desulfovibrio alaskensis G20" International Journal of Molecular Sciences 23, no. 3: 1396. https://doi.org/10.3390/ijms23031396
APA StyleTripathi, A. K., Saxena, P., Thakur, P., Rauniyar, S., Samanta, D., Gopalakrishnan, V., Singh, R. N., & Sani, R. K. (2022). Transcriptomics and Functional Analysis of Copper Stress Response in the Sulfate-Reducing Bacterium Desulfovibrio alaskensis G20. International Journal of Molecular Sciences, 23(3), 1396. https://doi.org/10.3390/ijms23031396