
Selective Boosting of CCR7-Acting Chemokines; Short Peptides Boost Chemokines with Short Basic Tails, Longer Peptides Boost Chemokines with Long Basic Tails

 , , , , , , and
, , , , , , and 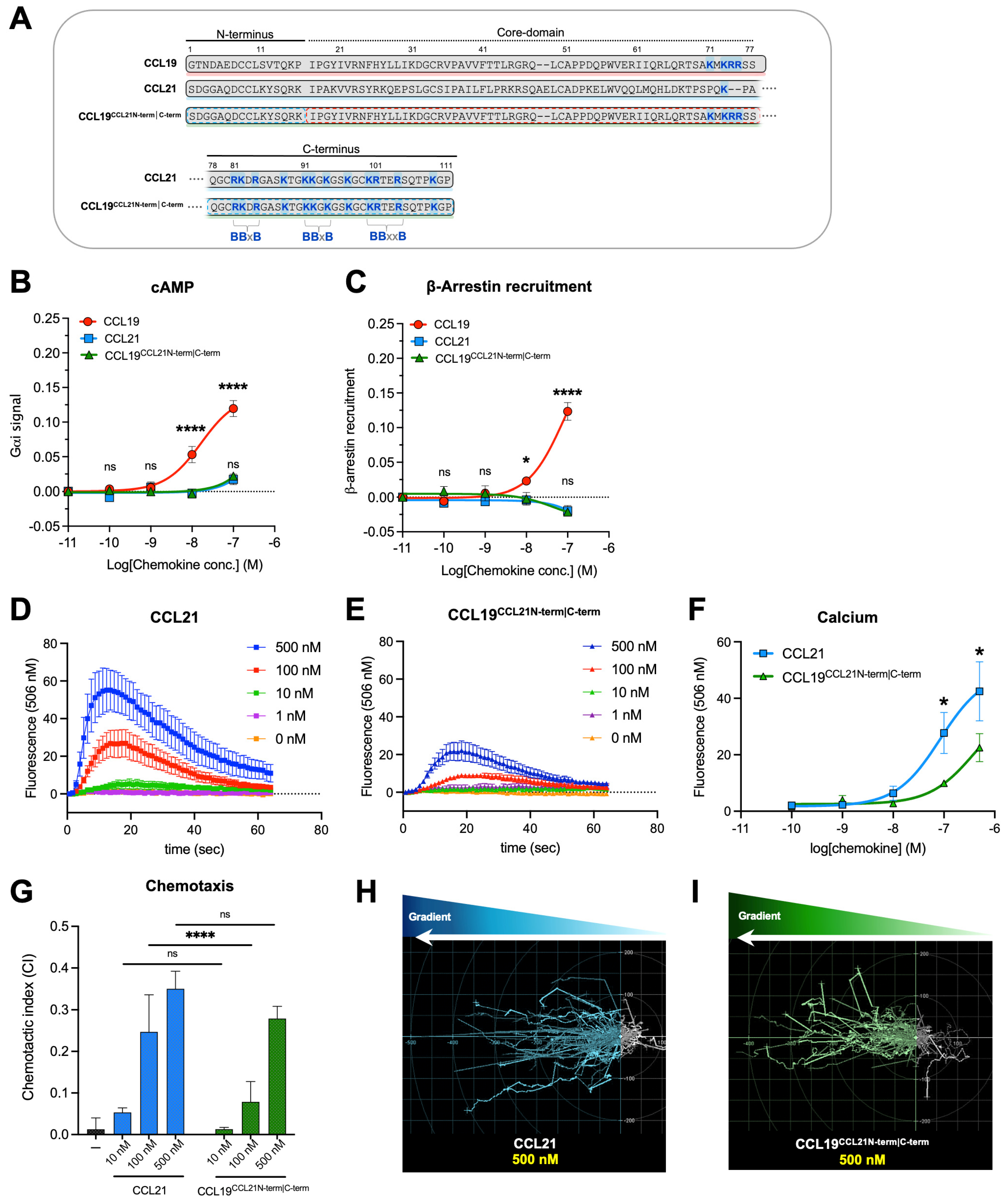{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The CCL21 N- and C-Terminus Cooperatively Reduce Potency
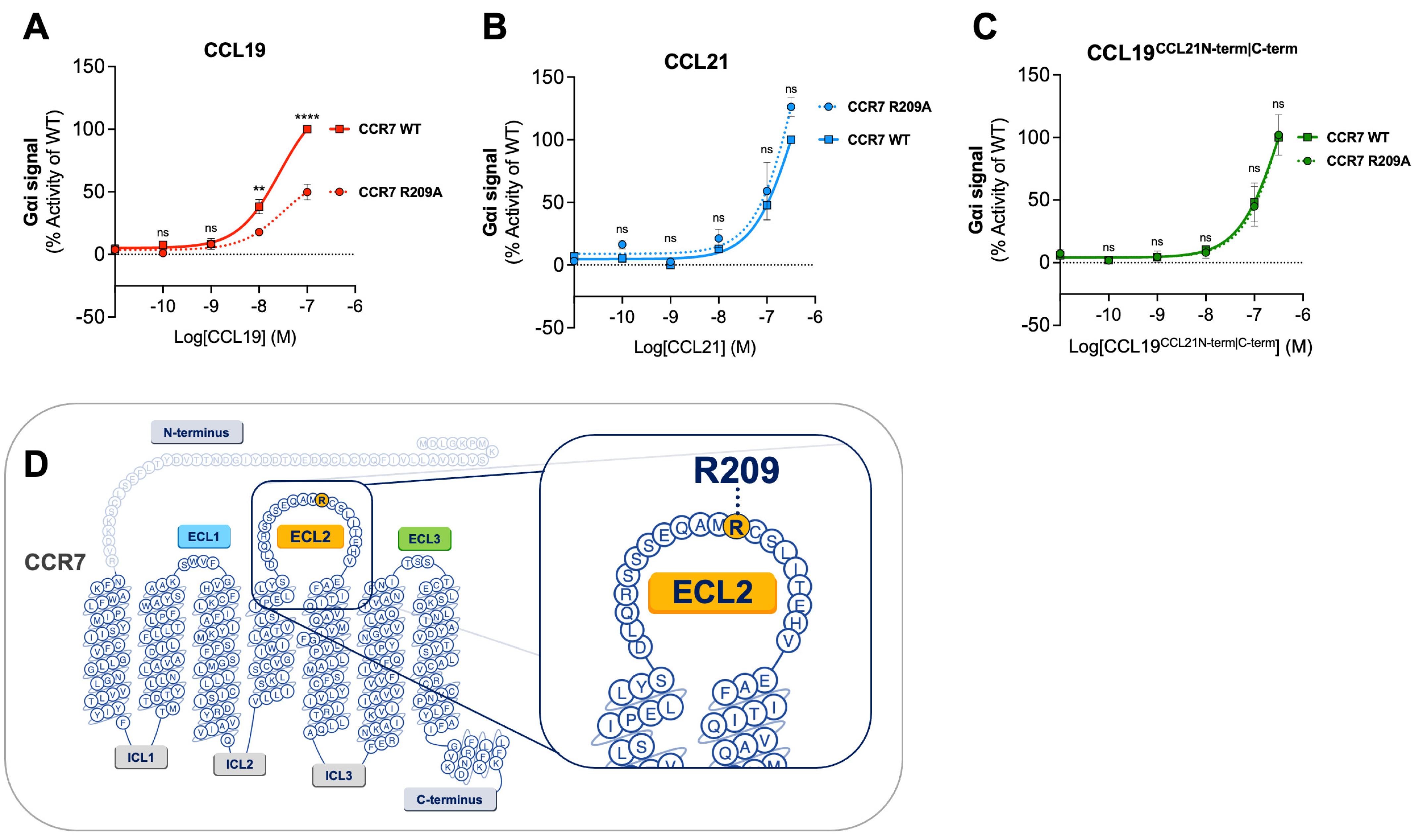
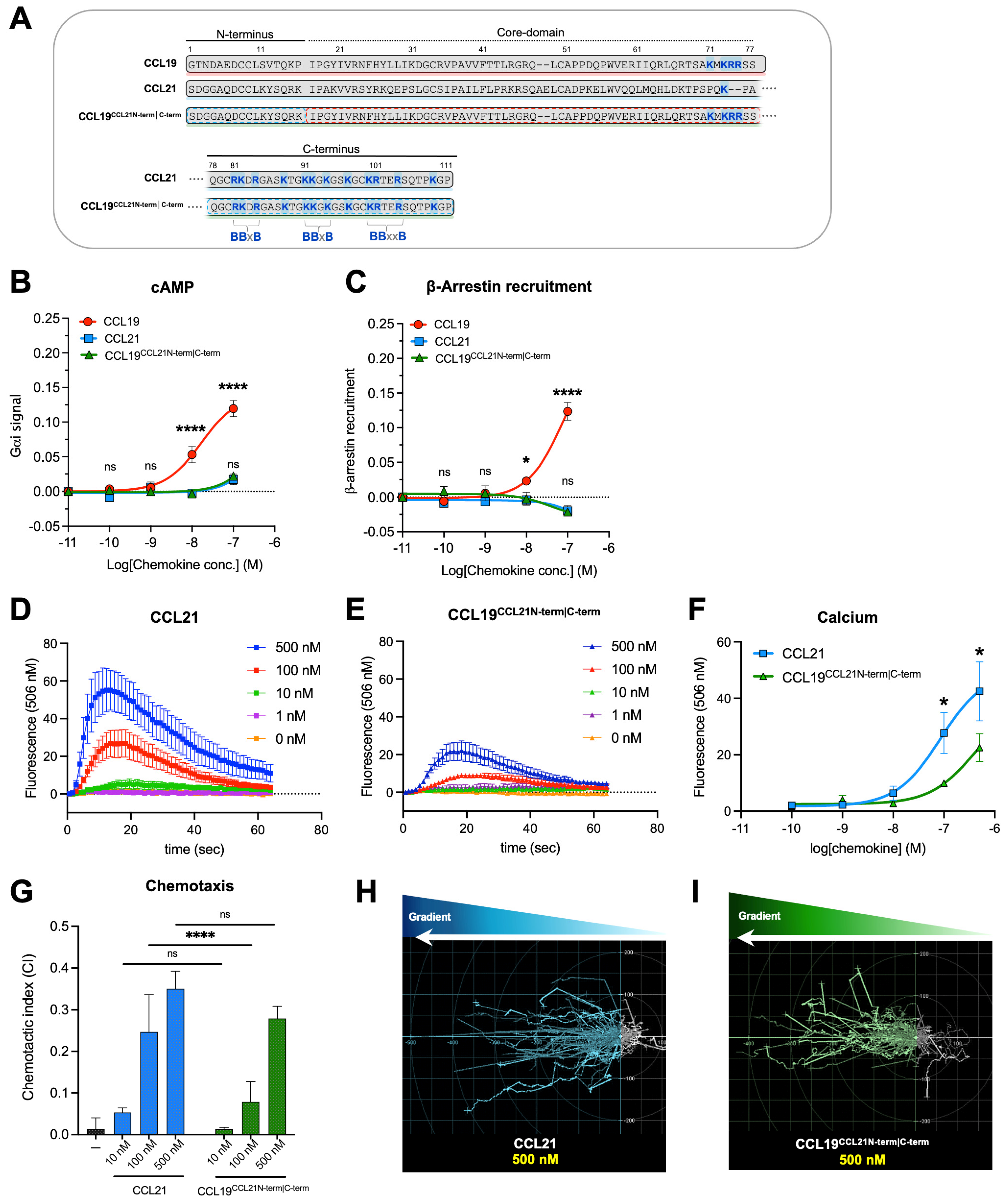
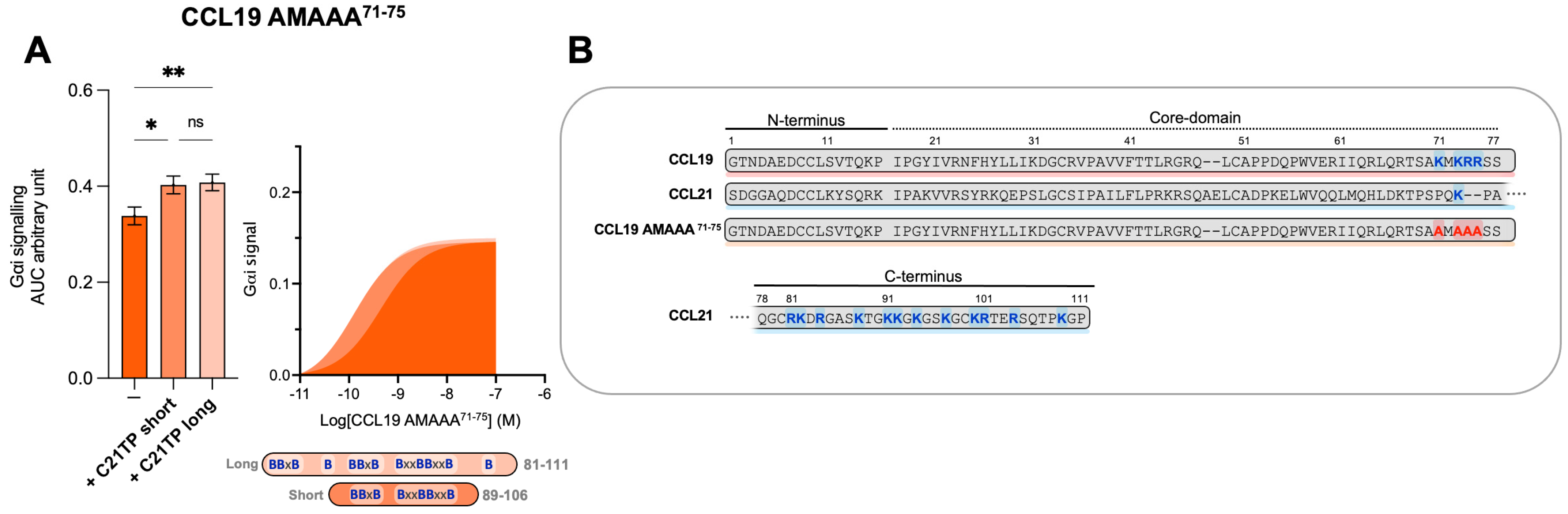
2.2. CCL19 Is Dependent on R209 in CCR7 ECL2 for Proper Receptor Activation—CCL21 and CCL19CCL21N-term|C-term Are Not
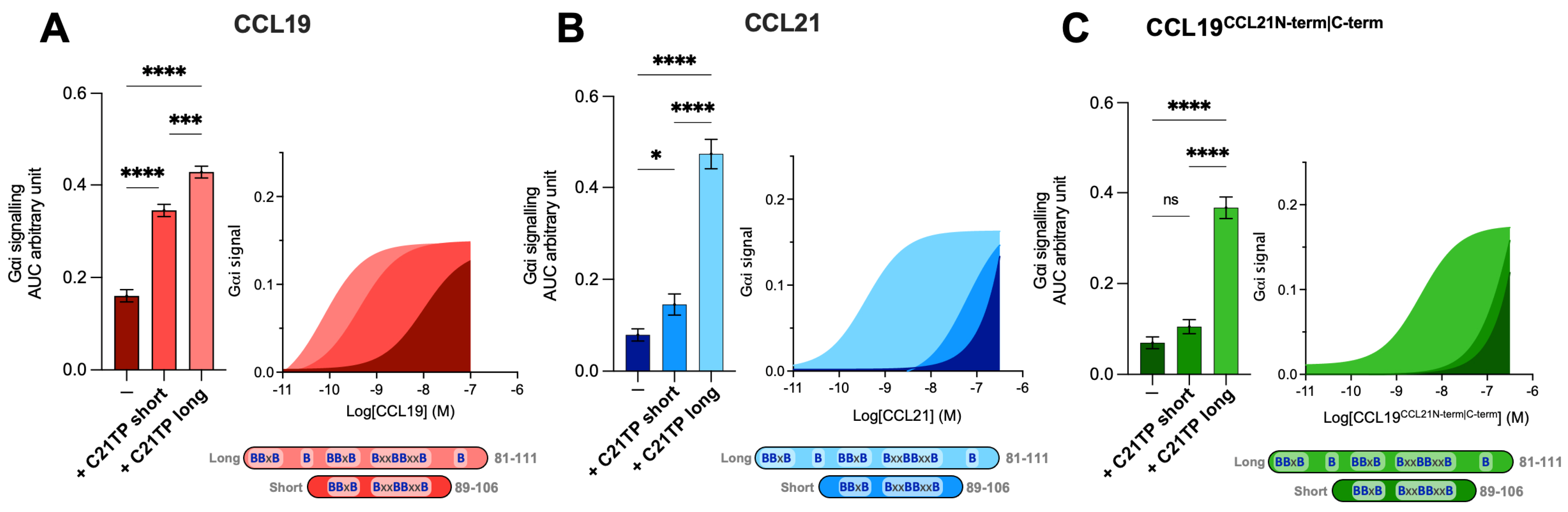
2.3. The Chimeric Chemokine CCL19CCL21N-term|C-term Resembles CCL21 with Regards to C21TP Boosting
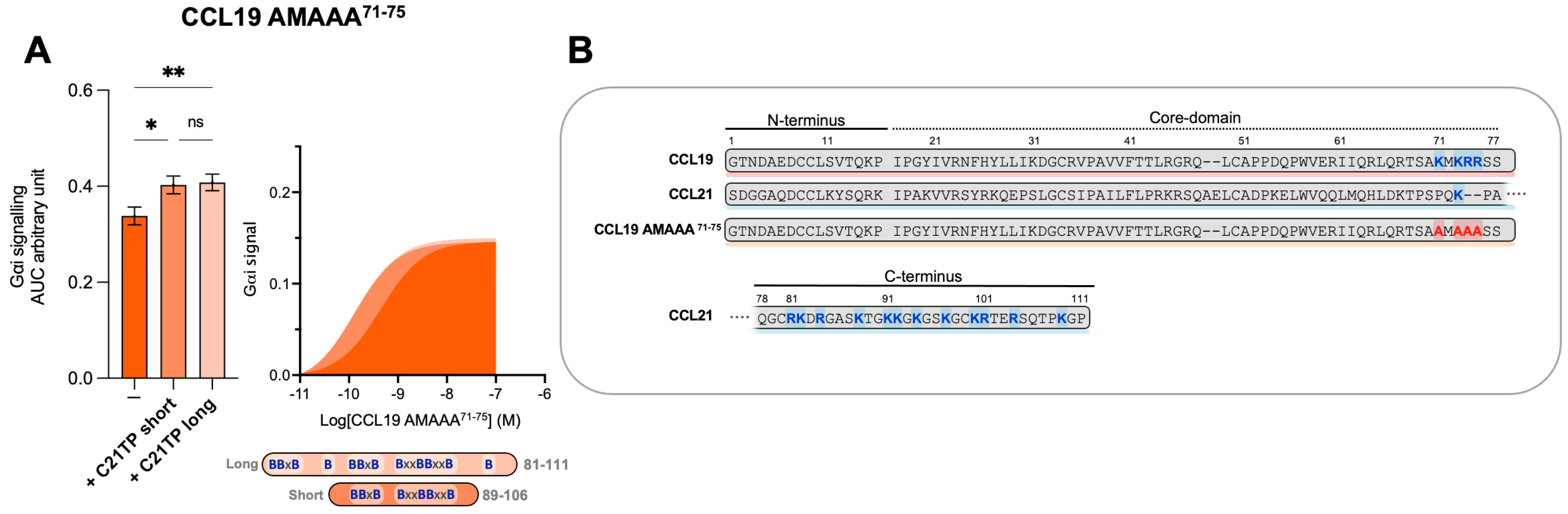
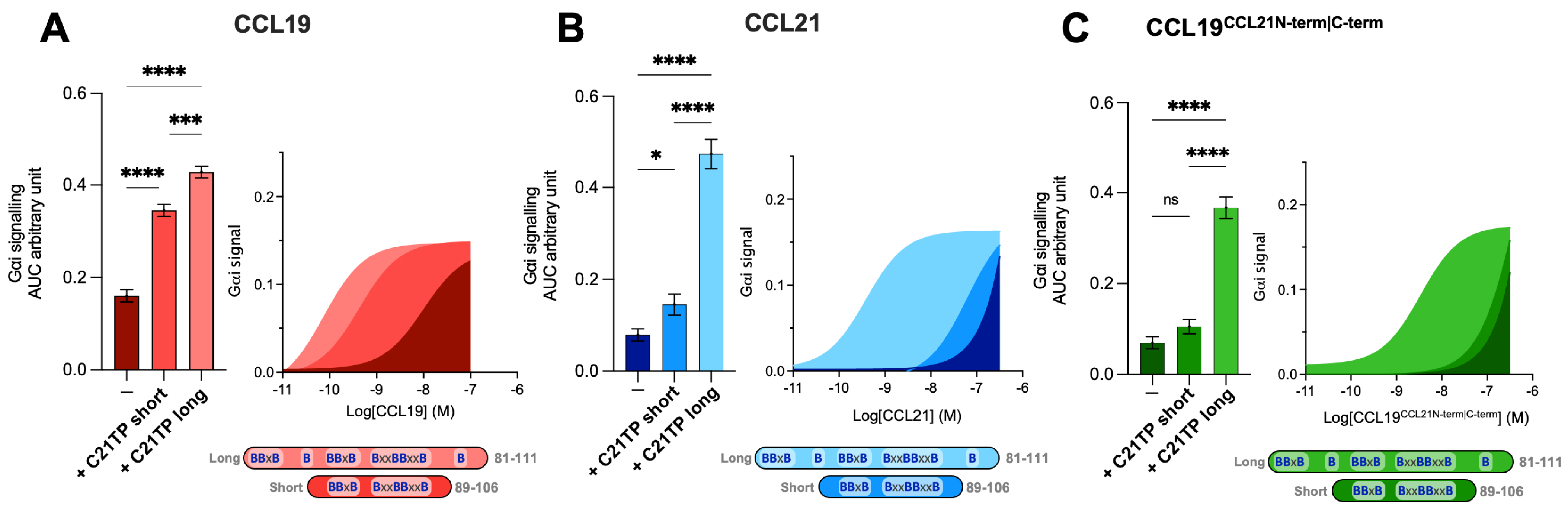
2.4. The Boosting Capacity of C21TPs Is Inversely Correlated with the Length of the Chemokine’s C-Terminus
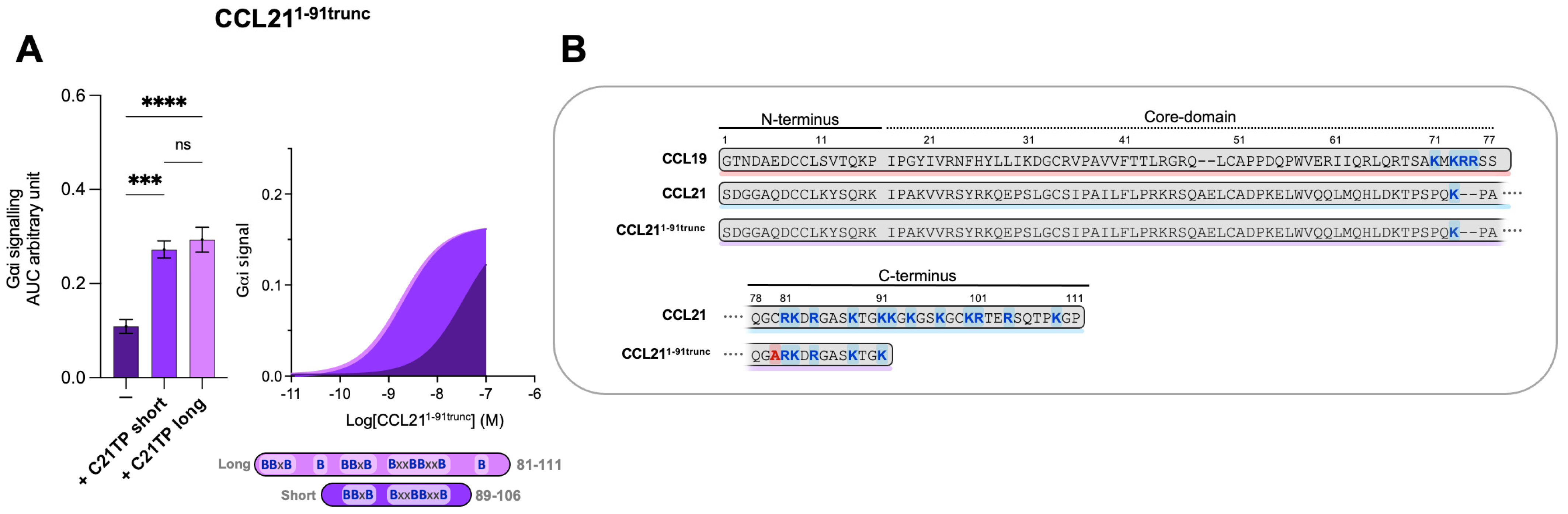
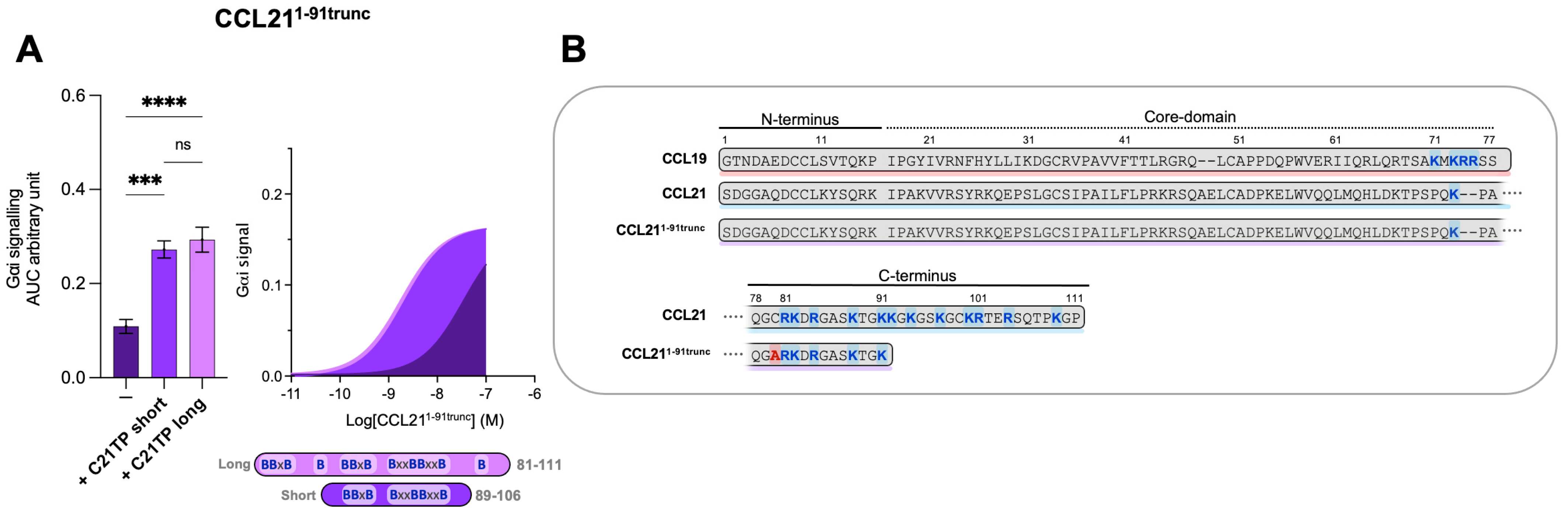
2.5. C-Terminal Basic Residues in Chemokines Highly Influences Basal Potency and Determines the Boosting Ability of C21TPs
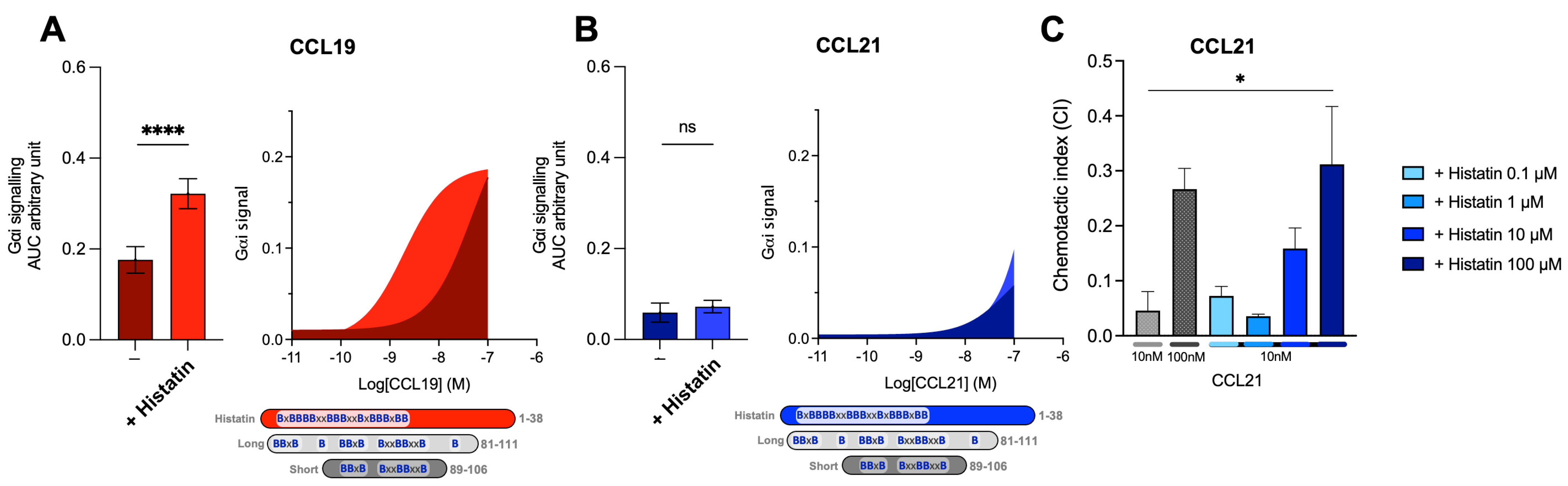
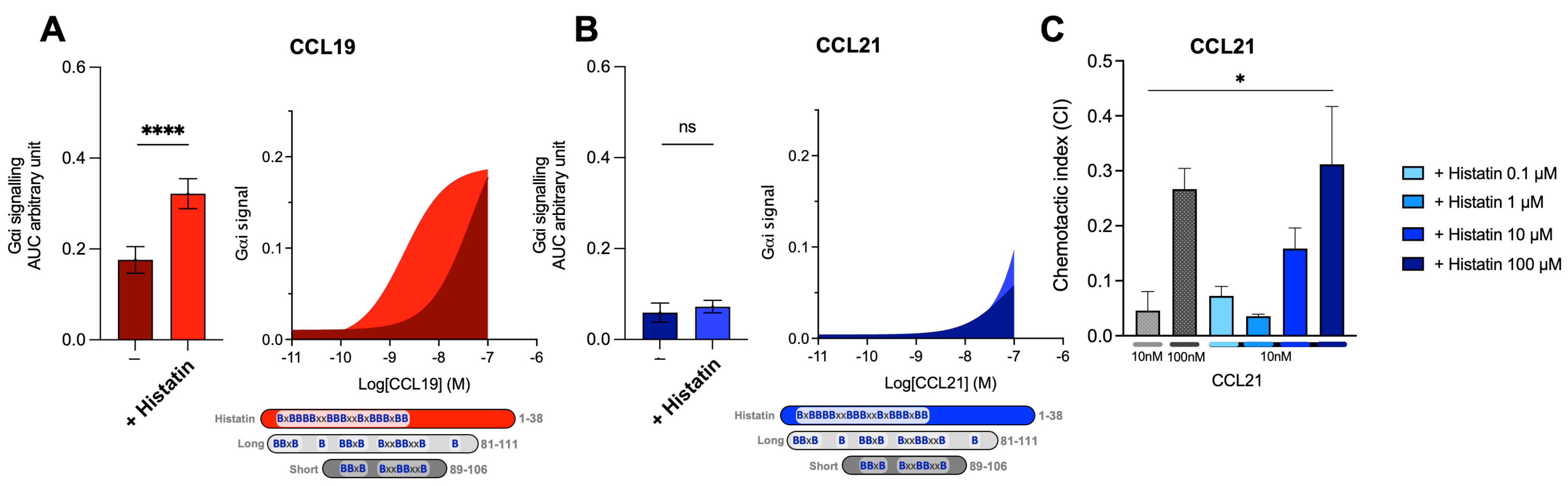
2.6. Host Defense Peptides Similar to Short C21TP Selectively Boosts CCL19, Indicating That Chemokine Boosting Is a Highly Physiological Relevant Mechanism
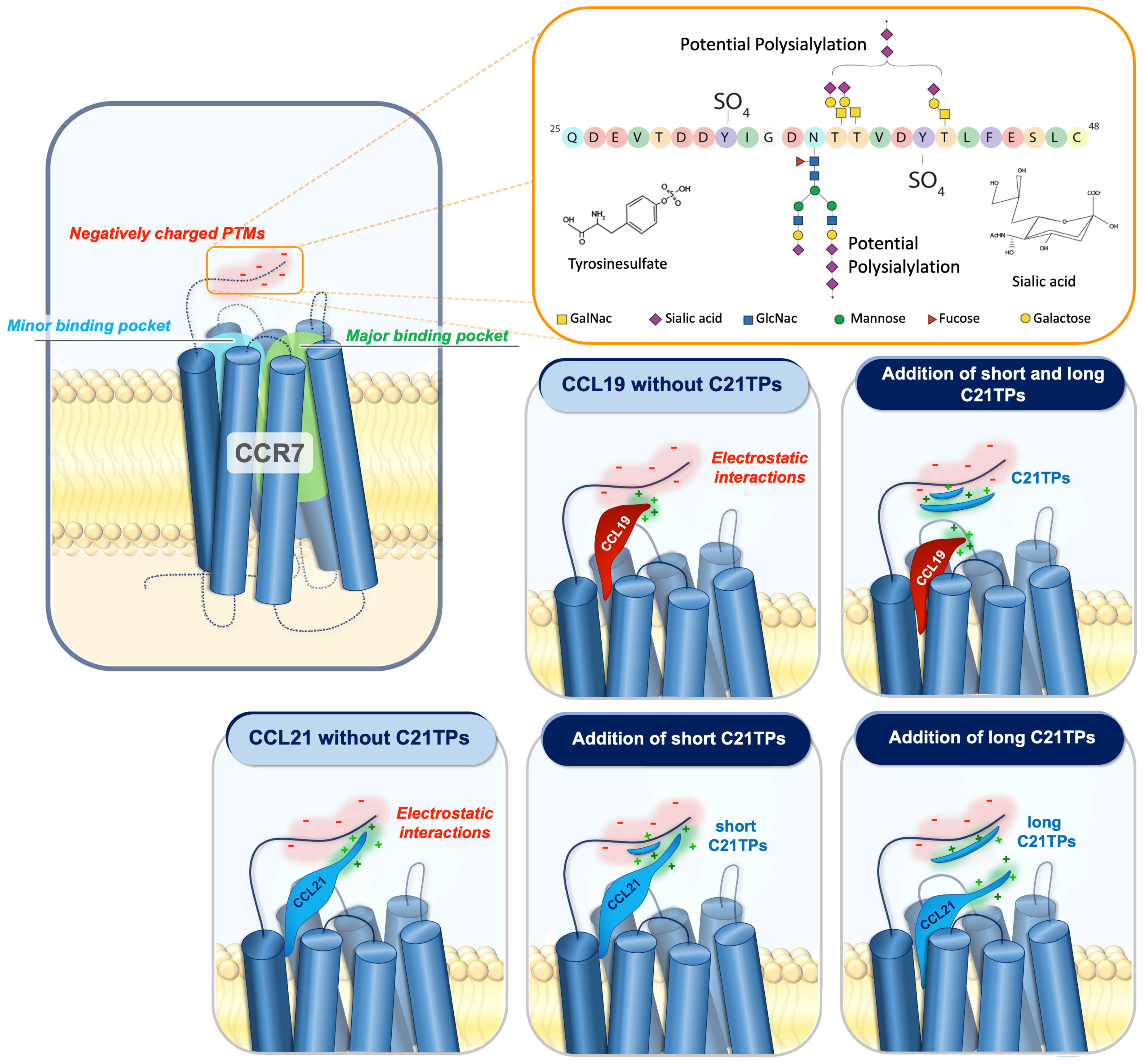
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Monocyte-Derived Dendritic Cell (moDC) Preparation
4.2.2. Cell Culturing
4.2.3. BRET Measurements of Gαi Signaling and β-Arrestin Recruitment
4.2.4. Calcium Signaling
4.2.5. Three-Dimensional (3D) Chemotaxis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteen, A.; Elarsen, O.; Ethiele, S.; Rosenkilde, M.M. Biased and G Protein-Independent Signaling of Chemokine Receptors. Front. Immunol. 2014, 5, 277. [Google Scholar] [CrossRef] [Green Version]
- Schall, T.J.; Proudfoot, A.E.I. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 2011, 11, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Laufer, J.M.; Kindinger, I.; Artinger, M.; Pauli, A.; Legler, D.F. CCR7 Is Recruited to the Immunological Synapse, Acts as Costimulatory Molecule and Drives LFA-1 Clustering for Efficient T Cell Adhesion Through ZAP70. Front. Immunol. 2019, 9, 3115. [Google Scholar] [CrossRef] [Green Version]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [Green Version]
- Hauser, M.A.; Legler, D.F. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J. Leukoc. Biol. 2016, 99, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Kohout, T.A.; Nicholas, S.L.; Perry, S.J.; Reinhart, G.; Junger, S.; Struthers, R.S. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor. J. Biol. Chem. 2004, 279, 23214–23222. [Google Scholar] [CrossRef] [Green Version]
- Hjortø, G.M.; Larsen, O.; Steen, A.; Daugvilaite, V.; Berg, C.; Fares, S.; Hansen, M.; Ali, S.; Rosenkilde, M.M. Differential CCR7 Targeting in Dendritic Cells by Three Naturally Occurring CC-Chemokines. Front. Immunol. 2016, 7, 568. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, R.; Nagira, M.; Kitaura, M.; Imagawa, N.; Imai, T.; Yoshie, O. Secondary Lymphoid-tissue Chemokine Is a Functional Ligand for the CC Chemokine Receptor CCR7. J. Biol. Chem. 1998, 273, 7118–7122. [Google Scholar] [CrossRef] [Green Version]
- Ricart, B.G.; John, B.; Lee, D.; Hunter, C.A.; Hammer, D.A. Dendritic Cells Distinguish Individual Chemokine Signals through CCR7 and CXCR4. J. Immunol. 2010, 186, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, A.S.; Adogamhe, P.E.; Laufer, J.M.; Legler, D.F.; Veldkamp, C.T.; Rosenkilde, M.M.; Hjortø, G.M. CCL19 with CCL21-tail displays enhanced glycosaminoglycan binding with retained chemotactic potency in dendritic cells. J. Leukoc. Biol. 2018, 104, 401–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussouras, N.A.; Hjortø, G.M.; Peterson, F.C.; Szpakowska, M.; Chevigné, A.; Rosenkilde, M.M.; Volkman, B.F.; Dwinell, M.B. Structural Features of an Extended C-Terminal Tail Modulate the Function of the Chemokine CCL21. Biochemistry 2020, 59, 1338–1350. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Sandberg, J.L.; Ziarek, J.J.; Gerarden, K.P.; Rode, R.R.; Jensen, D.R.; McCaslin, D.R.; Peterson, F.C.; Veldkamp, C.T. Solution Structure of CCL21 and Identification of a Putative CCR7 Binding Site. Biochemistry 2012, 51, 733–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiermaier, E.; Moussion, C.; Veldkamp, C.T.; Gerardy-Schahn, R.; de Vries, I.; Williams, L.G.; Chaffee, G.R.; Phillips, A.J.; Freiberger, F.; Imre, R.; et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science 2016, 351, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, N.; Loef, E.J.; Kelch, I.D.; Verdon, D.J.; Black, M.M.; Middleditch, M.J.; Greenwood, D.R.; Graham, E.S.; Brooks, A.E.; Dunbar, P.R.; et al. Plasmin and regulators of plasmin activity control the migratory capacity and adhesion of human T cells and dendritic cells by regulating cleavage of the chemokine CCL21. Immunol. Cell Biol. 2016, 94, 955–963. [Google Scholar] [CrossRef]
- Schumann, K.; Lämmermann, T.; Bruckner, M.; Legler, D.F.; Polleux, J.; Spatz, J.P.; Schuler, G.; Forster, R.; Lutz, M.B.; Sorokin, L.; et al. Immobilized Chemokine Fields and Soluble Chemokine Gradients Cooperatively Shape Migration Patterns of Dendritic Cells. Immunity 2010, 32, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Ott, T.R.; Lio, F.M.; Olshefski, D.; Liu, X.-J.; Ling, N.; Struthers, R.S. The N-terminal domain of CCL21 reconstitutes high affinity binding, G protein activation, and chemotactic activity, to the C-terminal domain of CCL19. Biochem. Biophys. Res. Commun. 2006, 348, 1089–1093. [Google Scholar] [CrossRef]
- Jørgensen, A.S.; Larsen, O.; Allmen, E.U.-V.; Lückmann, M.; Legler, D.F.; Frimurer, T.M.; Veldkamp, C.T.; Hjortø, G.M.; Rosenkilde, M.M. Biased Signaling of CCL21 and CCL19 Does Not Rely on N-Terminal Differences, but Markedly on the Chemokine Core Domains and Extracellular Loop 2 of CCR7. Front. Immunol. 2019, 10, 2156. [Google Scholar] [CrossRef]
- Jørgensen, A.S.; Brandum, E.P.; Mikkelsen, J.M.; Orfin, K.A.; Boilesen, D.R.; Egerod, K.L.; Moussouras, N.A.; Vilhardt, F.; Kalinski, P.; Basse, P.; et al. The C-terminal peptide of CCL21 drastically augments CCL21 activity through the dendritic cell lymph node homing receptor CCR7 by interaction with the receptor N-terminus. Cell. Mol. Life Sci. 2021, 78, 6963–6978. [Google Scholar] [CrossRef]
- Sugiyama, K.; Ogino, T.; Ogata, K. Rapid purification and characterization of histatins (histidine-rich polypeptides) from human whole saliva. Arch. Oral Biol. 1990, 35, 415–419. [Google Scholar] [CrossRef]
- Bax, M.; van Vliet, S.J.; Litjens, M.; García-Vallejo, J.J.; van Kooyk, Y. Interaction of Polysialic Acid with CCL21 Regulates the Migratory Capacity of Human Dendritic Cells. PLoS ONE 2009, 4, e6987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goth, C.K.; Petäjä-Repo, U.E.; Rosenkilde, M.M. G Protein-Coupled Receptors in the Sweet Spot: Glycosylation and other Post-translational Modifications. ACS Pharmacol. Transl. Sci. 2020, 3, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Kleist, A.B.; Getschman, A.E.; Ziarek, J.J.; Nevins, A.M.; Gauthier, P.-A.; Chevigné, A.; Szpakowska, M.; Volkman, B.F. New paradigms in chemokine receptor signal transduction: Moving beyond the two-site model. Biochem. Pharmacol. 2016, 114, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, J.; Huma, Z.E.; Lane, J.R.; Liu, X.; Bridgford, J.L.; Payne, R.J.; Canals, M.; Stone, M.J. Evaluation and extension of the two-site, two-step model for binding and activation of the chemokine receptor. J. Biol. Chem. 2019, 294, 3464–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, S.; Rosenkilde, M.M. Interaction of chemokines with their receptors--from initial chemokine binding to receptor activating steps. Curr. Med. Chem. 2014, 21, 3594–3614. [Google Scholar] [CrossRef] [PubMed]
- Gaieb, Z.; Lo, D.D.; Morikis, D. Molecular Mechanism of Biased Ligand Conformational Changes in CC Chemokine Receptor. J. Chem. Inf. Model. 2016, 56, 1808–1822. [Google Scholar] [CrossRef]
- Varki, A.; Schnaar, R.L.; Schauer, R. Sialic Acids. In Essentials of Glycobiology [Internet], 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 179–195. [Google Scholar]
- Glanz, V.Y.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Trans-sialidase Associated with Atherosclerosis: Defining the Identity of a Key Enzyme Involved in the Pathology. Curr. Drug Targets 2019, 20, 938–941. [Google Scholar] [CrossRef]
- Oppenheim, F.G.; Xu, T.; McMillian, F.M.; Levitz, S.M.; Diamond, R.D.; Offner, G.; Troxler, R.F. Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans. J. Biol. Chem. 1988, 263, 7472–7477. [Google Scholar] [CrossRef]
- Wu, R.-Q.; Zhang, D.; Tu, E.; Chen, Q.-M.; Chen, W. The mucosal immune system in the oral cavity—an orchestra of T cell diversity. Int. J. Oral Sci. 2014, 6, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Hovav, A.-H. Dendritic cells of the oral mucosa. Mucosal Immunol. 2013, 7, 27–37. [Google Scholar] [CrossRef]
- Sharawi, H.; Heyman, O.; Mizraji, G.; Horev, Y.; Laviv, A.; Shapira, L.; Yona, S.; Hovav, A.; Wilensky, A. The Prevalence of Gingival Dendritic Cell Subsets in Periodontal Patients. J. Dent. Res. 2021, 100, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- McGrory, K.; Flaitz, C.M.; Klein, J.R. Chemokine changes during oral wound healing. Biochem. Biophys. Res. Commun. 2004, 324, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.; Tabib, Y.; Capucha, T.; Mizraji, G.; Nir, T.; Saba, F.; Salameh, R.; Eli-Berchoer, L.; Wilensky, A.; Burstyn-Cohen, T.; et al. Multiple Regulatory Levels of Growth Arrest-Specific 6 in Mucosal Immunity Against an Oral Pathogen. Front. Immunol. 2018, 9, 1374. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-H.; Kim, J.-I.; Kwon, H.-J.; Shim, D.-H.; Parajuli, N.; Cuburu, N.; Czerkinsky, C.; Kweon, M.-N. CCR7-CCL19/CCL21-regulated dendritic cells are responsible for effectiveness of sublingual vaccination. J. Immunol. 2009, 182, 6851–6860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boink, M.A.; Roffel, S.; Nazmi, K.; Bolscher, J.G.M.; Veerman, E.C.I.; Gibbs, S. Saliva-Derived Host Defense Peptides Histatin1 and LL-37 Increase Secretion of Antimicrobial Skin and Oral Mucosa Chemokine CCL20 in an IL-1α-Independent Manner. J. Immunol. Res. 2017, 2017, 3078194. [Google Scholar] [CrossRef]
- Dieu, M.-C.; Vanbervliet, B.; Vicari, A.; Bridon, J.-M.; Oldham, E.; Aït-Yahia, S.; Brière, F.; Zlotnik, A.; Lebecque, S.; Caux, C. Selective Recruitment of Immature and Mature Dendritic Cells by Distinct Chemokines Expressed in Different Anatomic Sites. J. Exp. Med. 1998, 188, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Torres, P.; Díaz, J.; Arce, M.; Silva, P.; Mendoza, P.; Lois, P.; Molina-Berríos, A.; Owen, G.I.; Palma, V.; Torres, V.A. The salivary peptide histatin-1 promotes endothelial cell adhesion, migration, and angiogenesis. FASEB J. 2017, 31, 4946–4958. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Ao, M.; Hu, Y.; Li, Q.K.; Zhang, H. Mapping the O-glycoproteome using site-specific extraction of O-linked glycopeptides (EXo, O). Mol. Syst. Biol. 2018, 14, e8486. [Google Scholar] [CrossRef]
- Veldkamp, C.T.; Kiermaier, E.; Gabel-Eissens, S.J.; Gillitzer, M.L.; Lippner, D.R.; Di Silvio, F.A.; Mueller, C.J.; Wantuch, P.L.; Chaffee, G.R.; Famiglietti, M.W.; et al. Solution Structure of CCL19 and Identification of Overlapping CCR7 and PSGL-1 Binding Sites. Biochemistry 2015, 54, 4163–4166. [Google Scholar] [CrossRef] [Green Version]
- Veldkamp, C.T.; Koplinski, C.A.; Jensen, D.R.; Peterson, F.C.; Smits, K.M.; Smith, B.L.; Johnson, S.K.; Lettieri, C.; Buchholz, W.G.; Solheim, J.C.; et al. Production of Recombinant Chemokines and Validation of Refolding. Methods Enzymol. 2016, 570, 539–565. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Met, Ö.; Larsen, N.B.; Rosenkilde, M.M.; Andersen, M.H.; Svane, I.M.; Hjortø, G.M. Autocrine CCL19 blocks dendritic cell migration toward weak gradients of CCL21. Cytotherapy 2016, 18, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, S.; Halim, A.; Schulz, M.A.; Frödin, M.; Rahman, S.H.; Vester-Christensen, M.B.; Behrens, C.; Kristensen, C.; Vakhrushev, S.; et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat. Biotechnol. 2015, 33, 842–844. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandum, E.P.; Jørgensen, A.S.; Calvo, M.B.; Spiess, K.; Peterson, F.C.; Yang, Z.; Volkman, B.F.; Veldkamp, C.T.; Rosenkilde, M.M.; Goth, C.K.; et al. Selective Boosting of CCR7-Acting Chemokines; Short Peptides Boost Chemokines with Short Basic Tails, Longer Peptides Boost Chemokines with Long Basic Tails. Int. J. Mol. Sci. 2022, 23, 1397. https://doi.org/10.3390/ijms23031397
Brandum EP, Jørgensen AS, Calvo MB, Spiess K, Peterson FC, Yang Z, Volkman BF, Veldkamp CT, Rosenkilde MM, Goth CK, et al. Selective Boosting of CCR7-Acting Chemokines; Short Peptides Boost Chemokines with Short Basic Tails, Longer Peptides Boost Chemokines with Long Basic Tails. International Journal of Molecular Sciences. 2022; 23(3):1397. https://doi.org/10.3390/ijms23031397
Chicago/Turabian StyleBrandum, Emma Probst, Astrid Sissel Jørgensen, Marina Barrio Calvo, Katja Spiess, Francis C. Peterson, Zhang Yang, Brian F. Volkman, Christopher T. Veldkamp, Mette Marie Rosenkilde, Christoffer Knak Goth, and et al. 2022. "Selective Boosting of CCR7-Acting Chemokines; Short Peptides Boost Chemokines with Short Basic Tails, Longer Peptides Boost Chemokines with Long Basic Tails" International Journal of Molecular Sciences 23, no. 3: 1397. https://doi.org/10.3390/ijms23031397
APA StyleBrandum, E. P., Jørgensen, A. S., Calvo, M. B., Spiess, K., Peterson, F. C., Yang, Z., Volkman, B. F., Veldkamp, C. T., Rosenkilde, M. M., Goth, C. K., & Hjortø, G. M. (2022). Selective Boosting of CCR7-Acting Chemokines; Short Peptides Boost Chemokines with Short Basic Tails, Longer Peptides Boost Chemokines with Long Basic Tails. International Journal of Molecular Sciences, 23(3), 1397. https://doi.org/10.3390/ijms23031397