
The Mitochondrial PHB2/OMA1/DELE1 Pathway Cooperates with Endoplasmic Reticulum Stress to Facilitate the Response to Chemotherapeutics in Ovarian Cancer


Abstract
:1. Introduction
2. Results
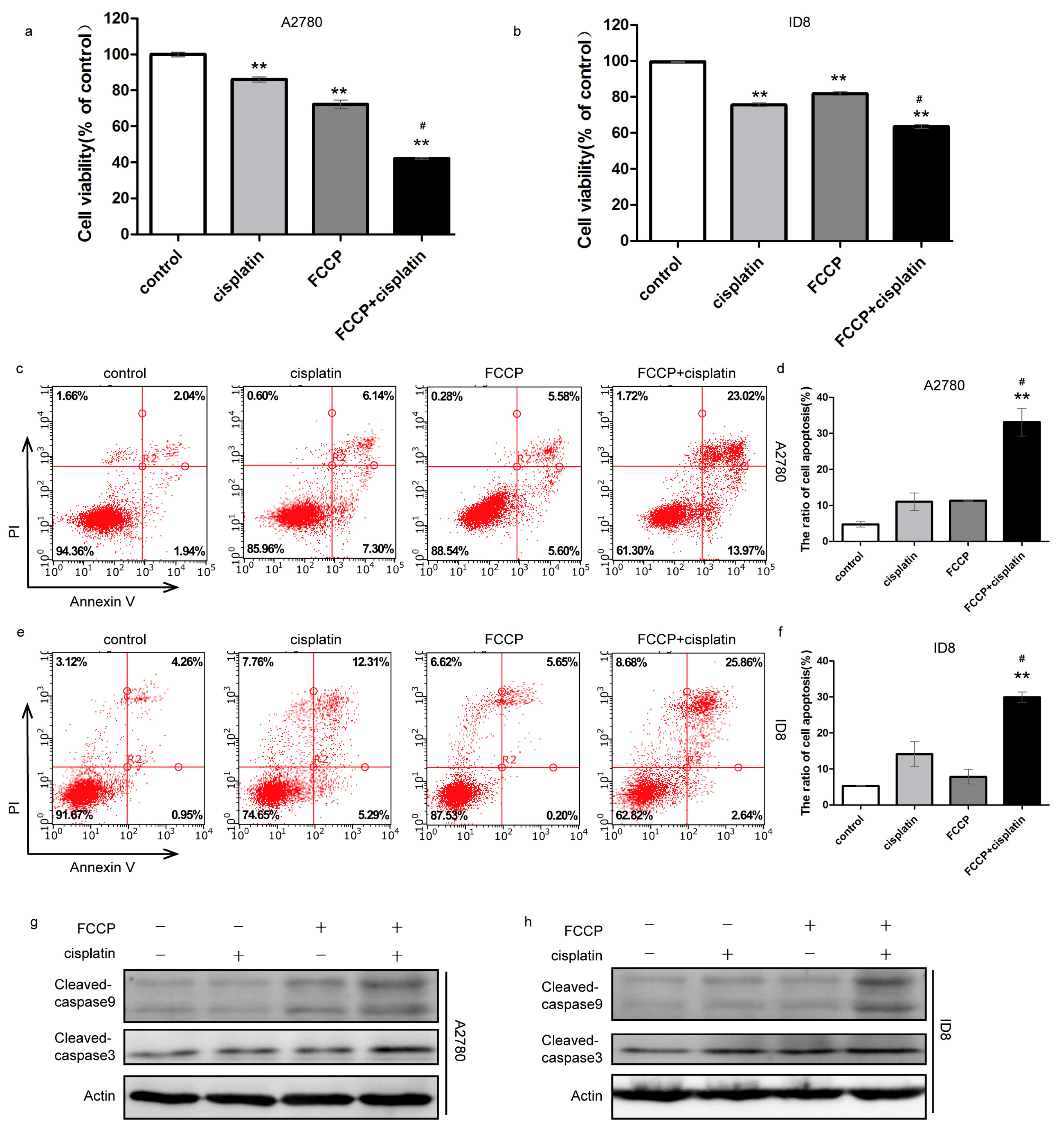
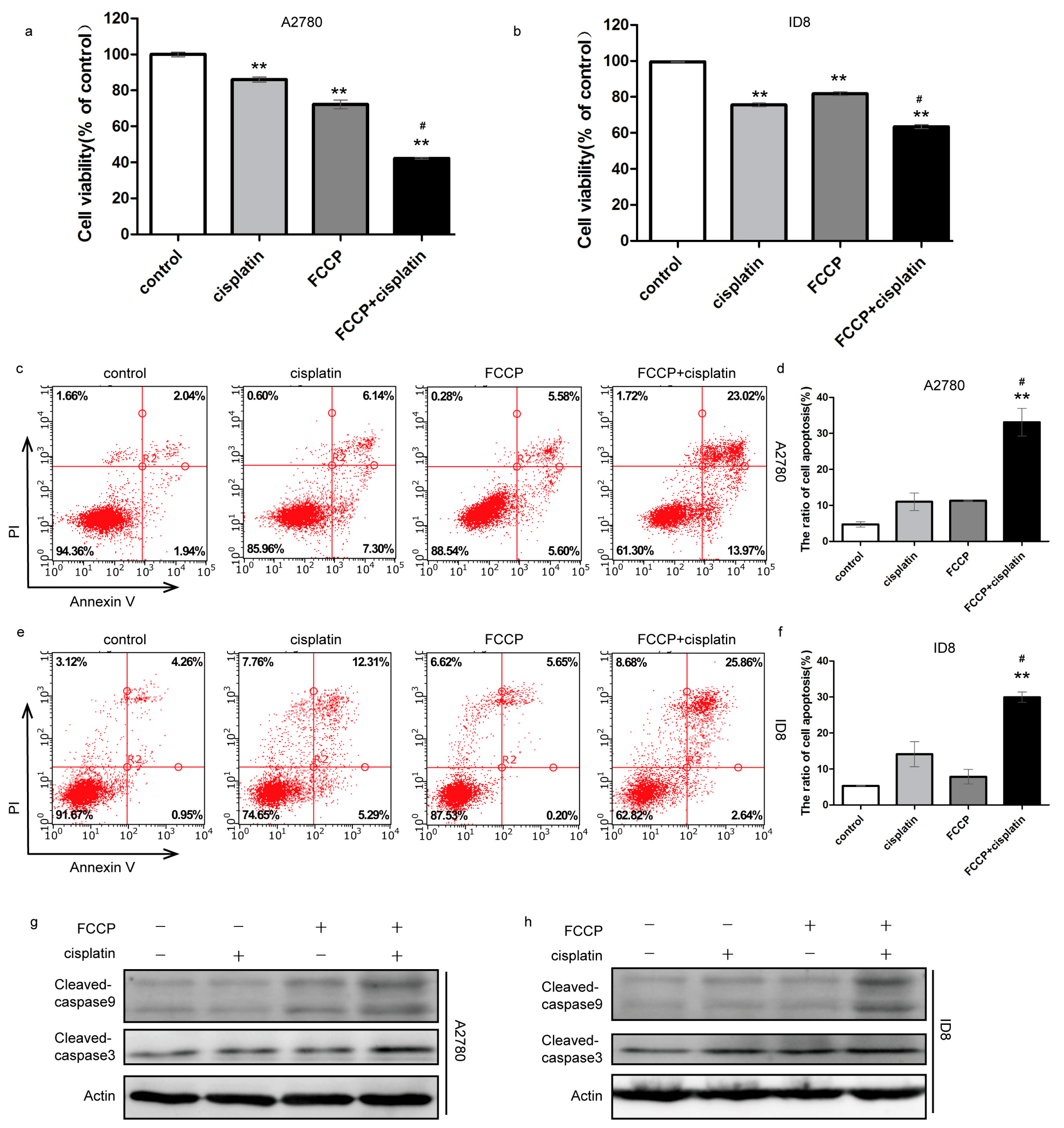
2.1. FCCP Enhanced Cisplatin-Induced Apoptosis in Ovarian Cancer Cells
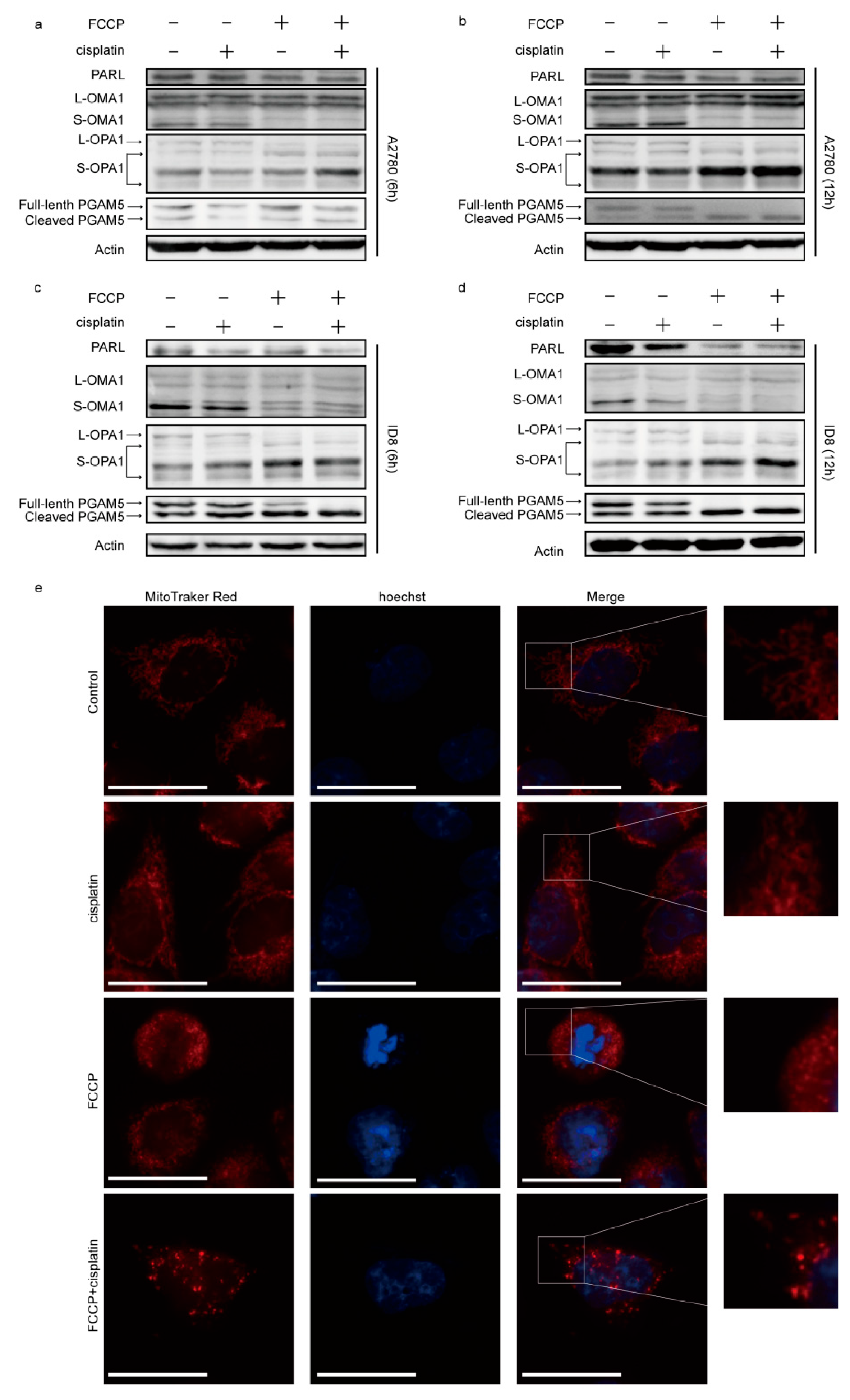
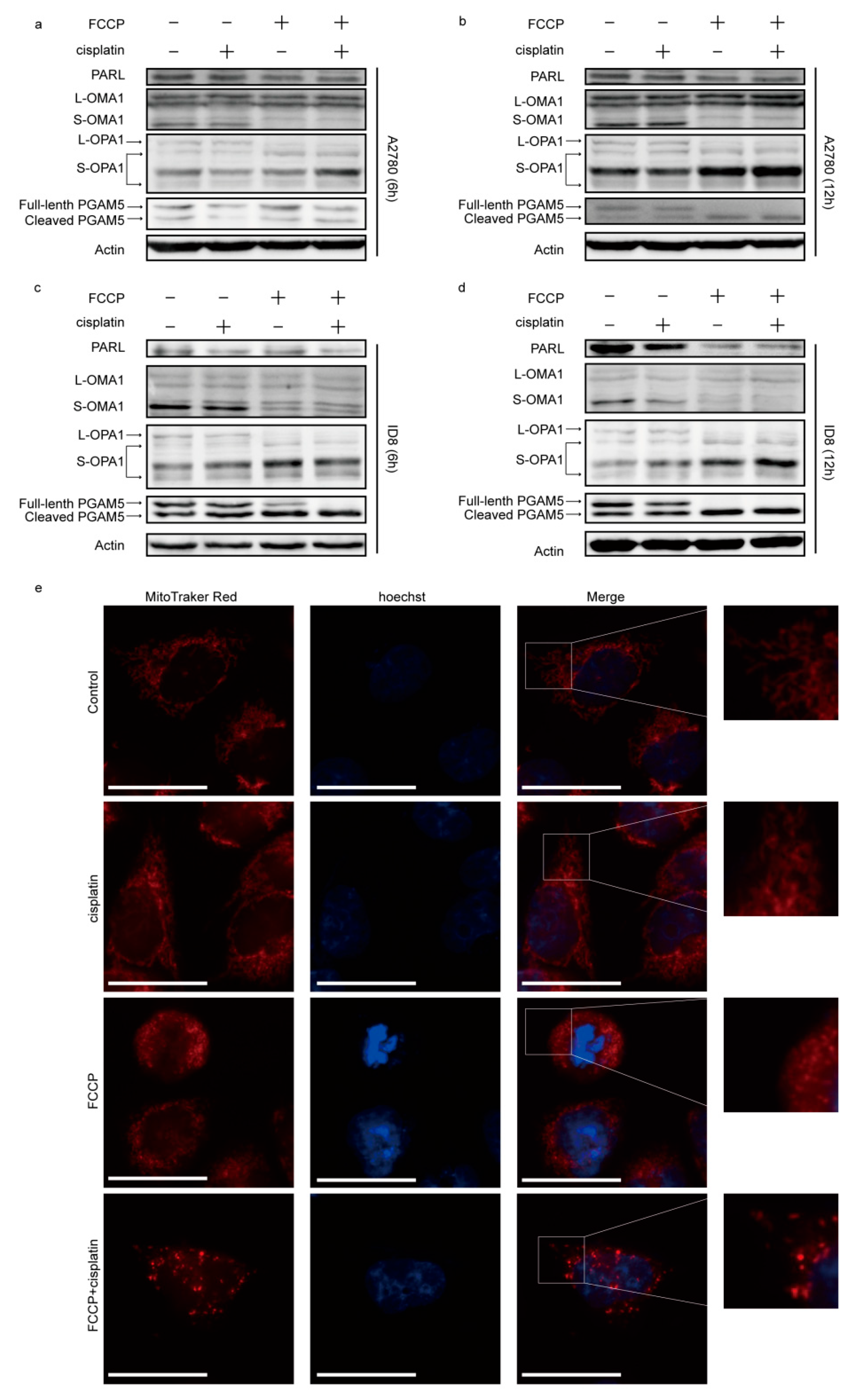
2.2. FCCP Combined with Cisplatin Activated OMA1 and Induced Optic Atrophy 1 (OPA1) Cleavage and Mitochondrial Fragmentation
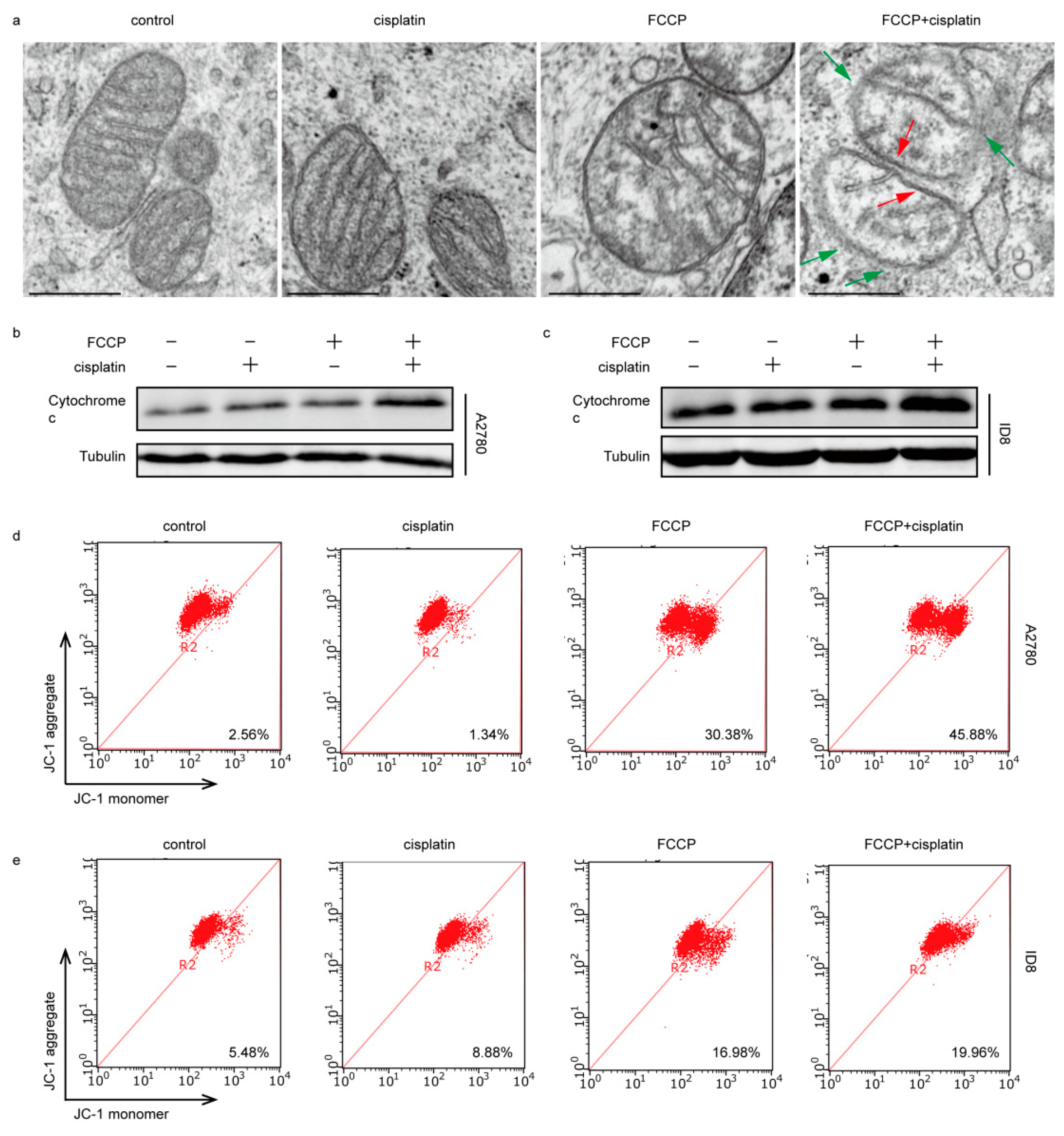
2.3. FCCP Combined with Cisplatin Induced Mitochondrial Inner Membrane (MIM) Cristae Remodeling and Cytochrome c Release
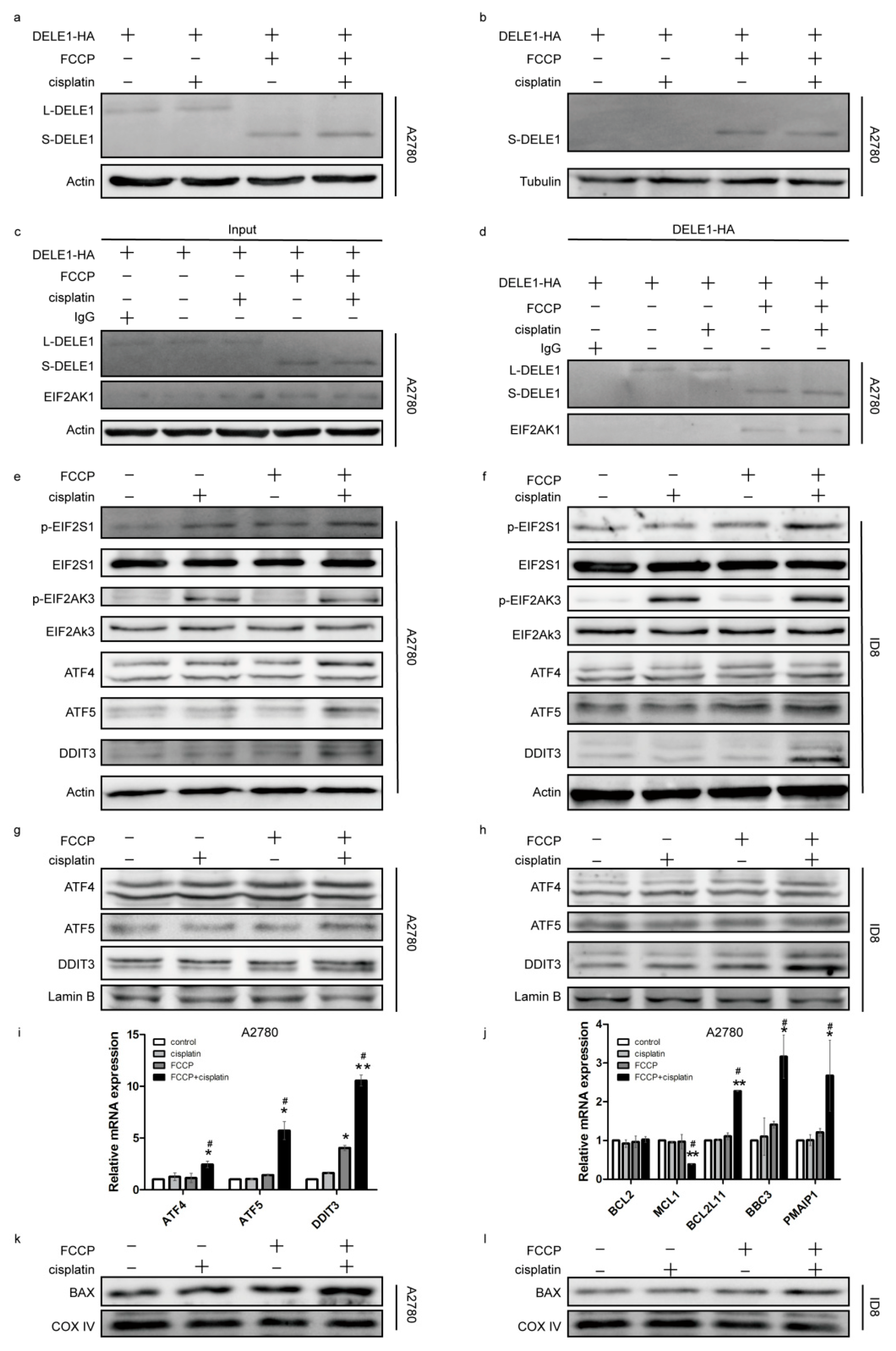
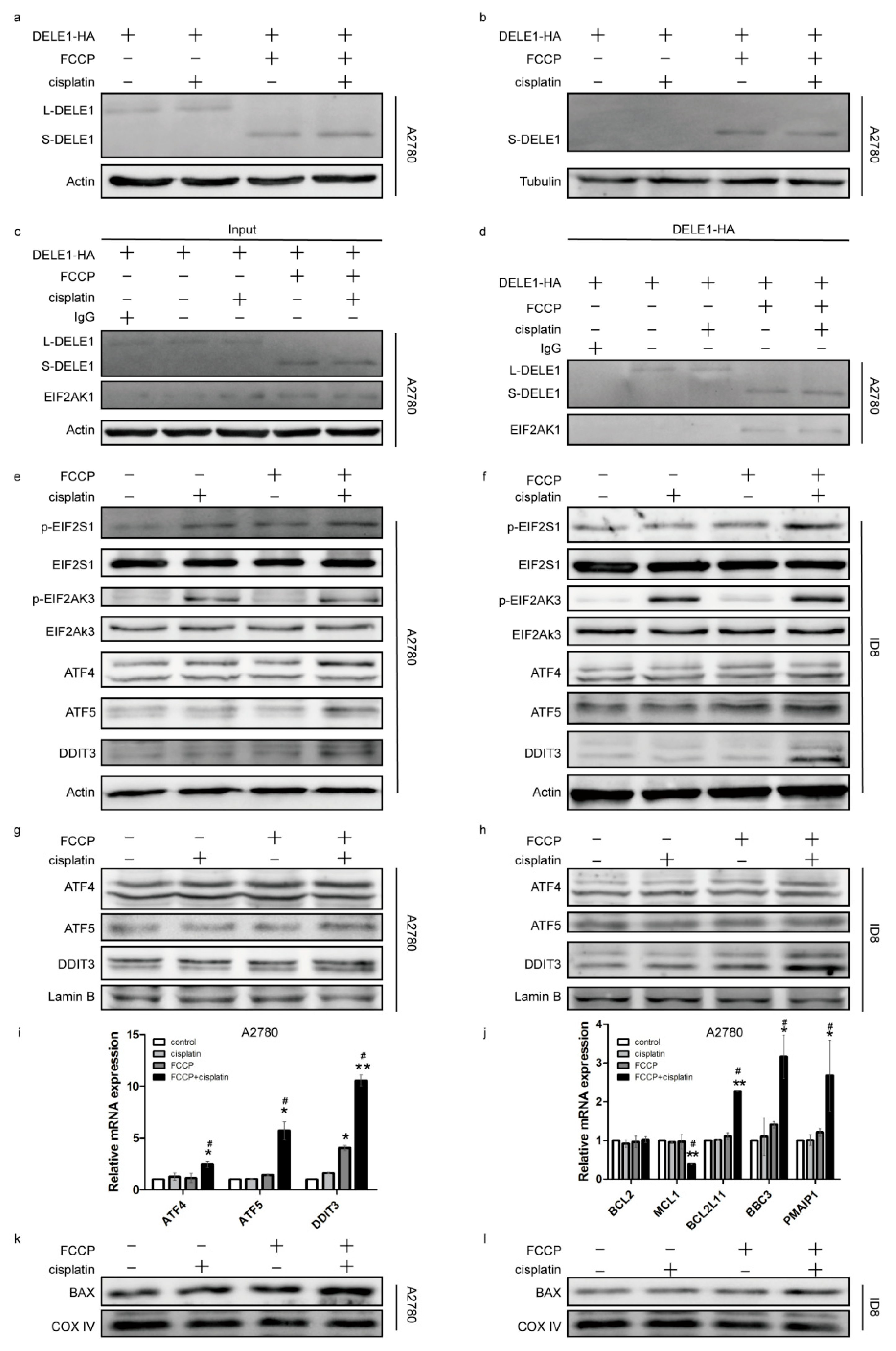
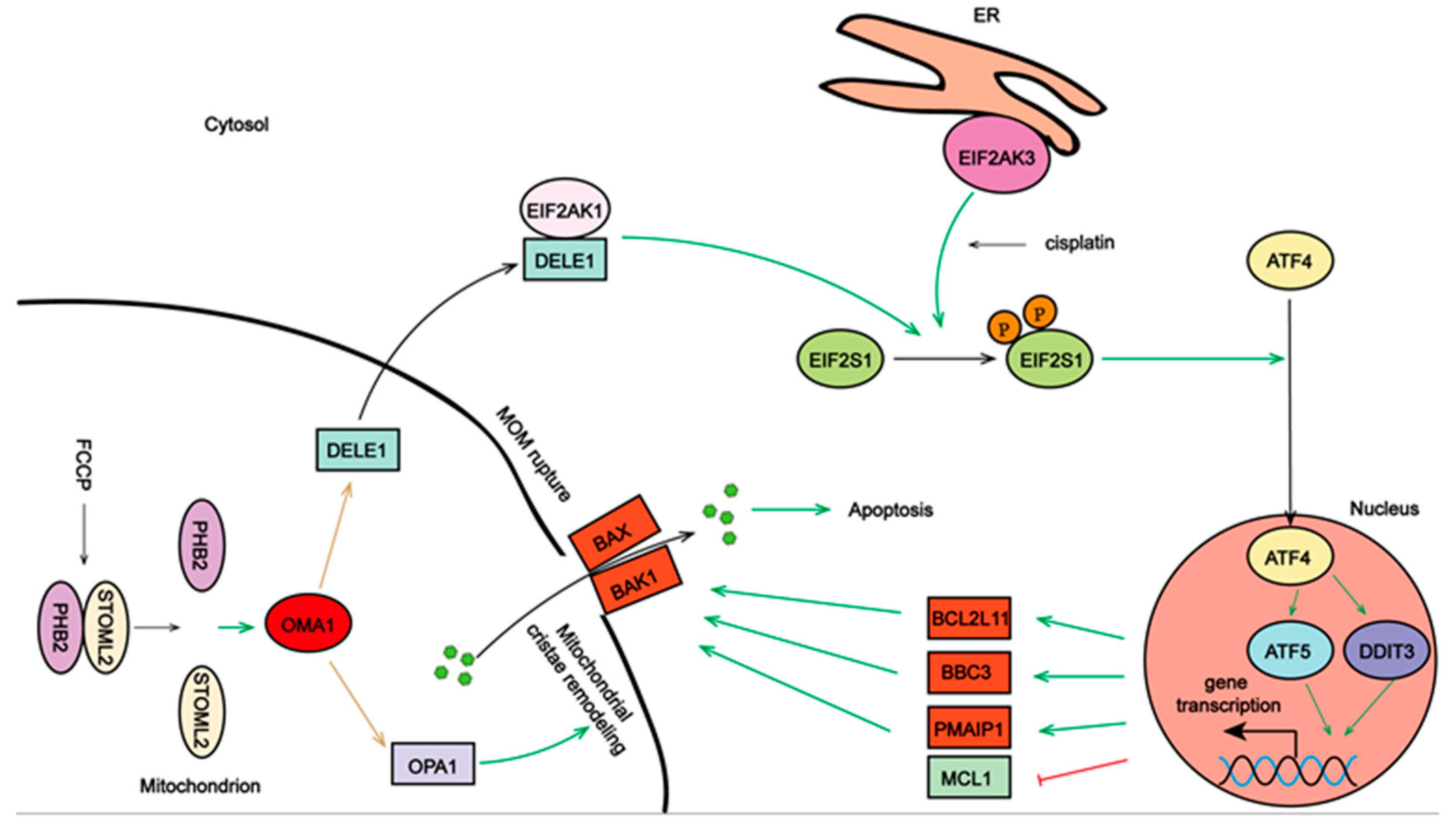
2.4. Activated OMA1 Cooperated with EIF2AK3 to Activate the EIF2S1/ATF4 Pathway by Cleaving DELE1
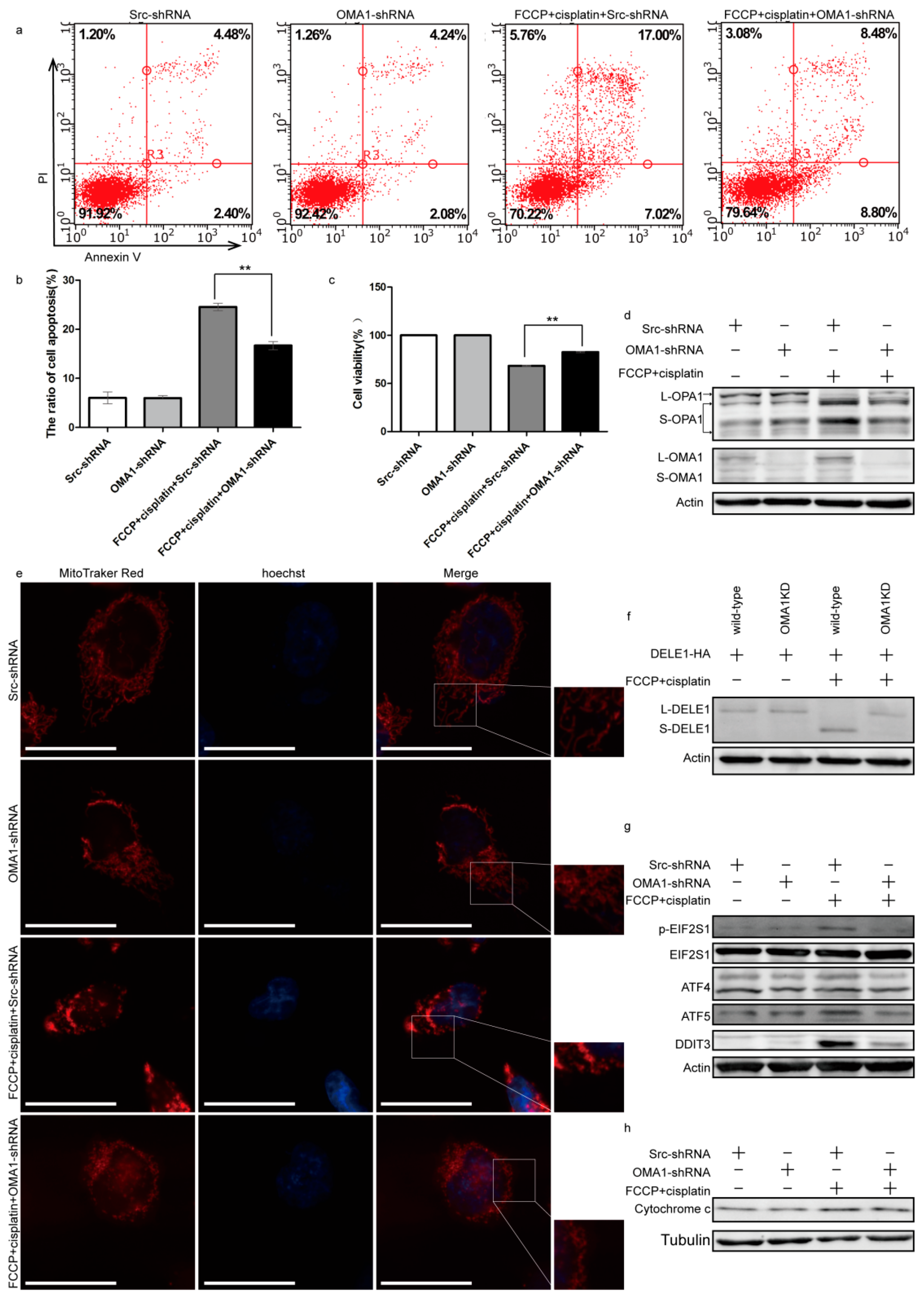
2.5. Knockdown of OMA1 Partially Reversed Apoptosis by Reducing the Mitochondrial Stress and EIF2S1/ATF4 Pathway
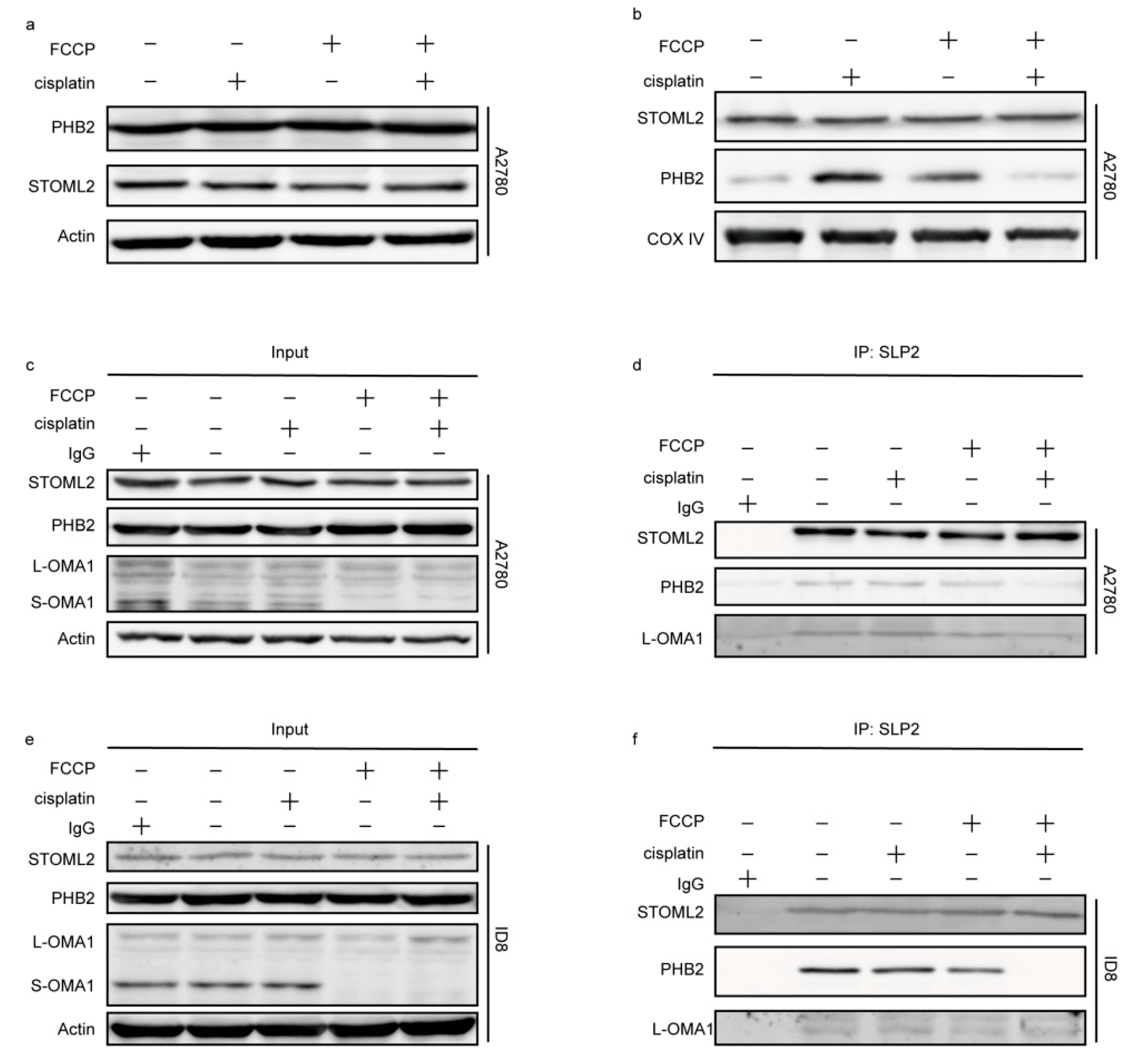
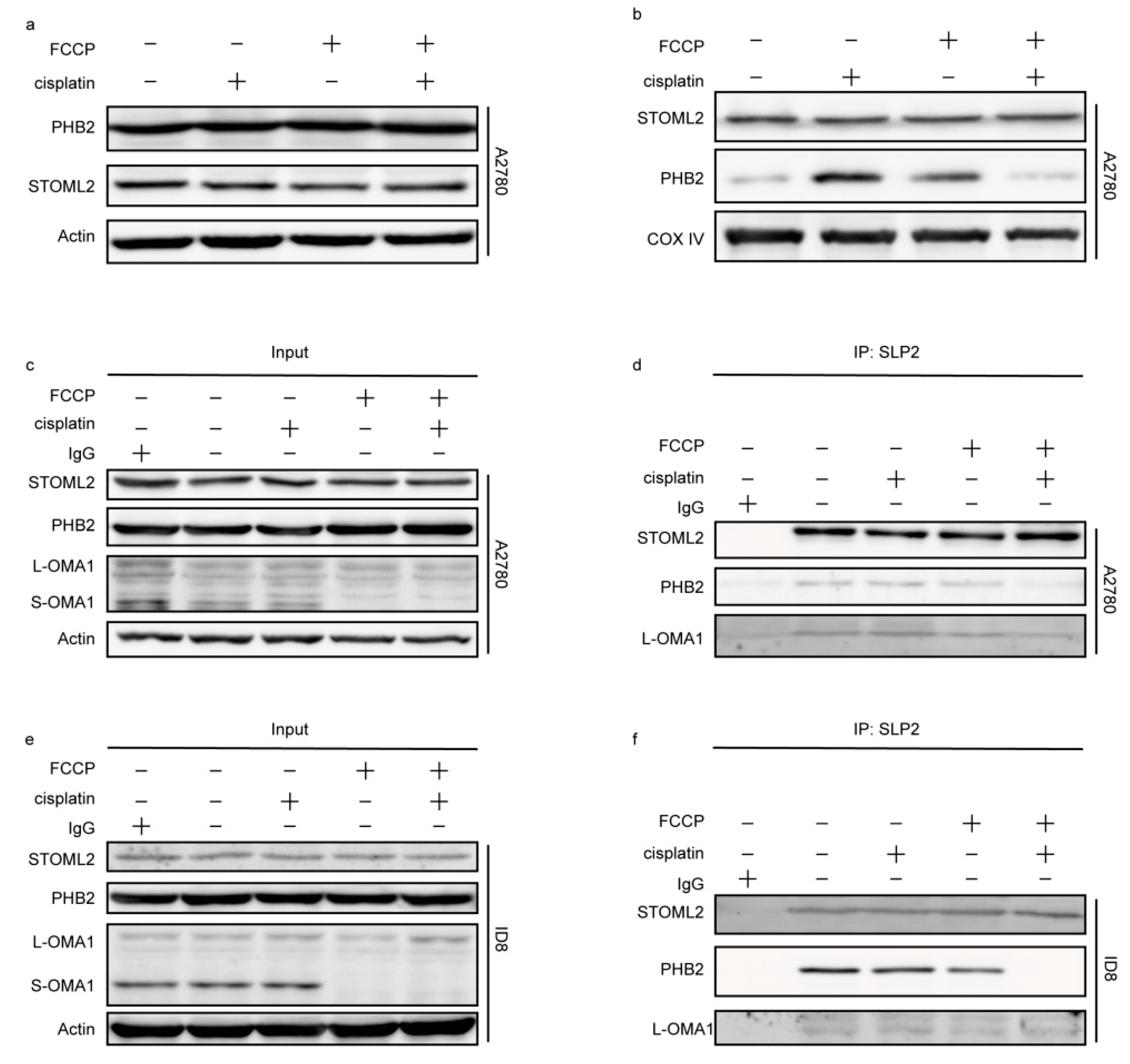
2.6. The OMA1 Protease Activity Was Regulated by the Prohibitin 2 (PHB2)/Stomatin-like Protein 2 (STOML2) Complex
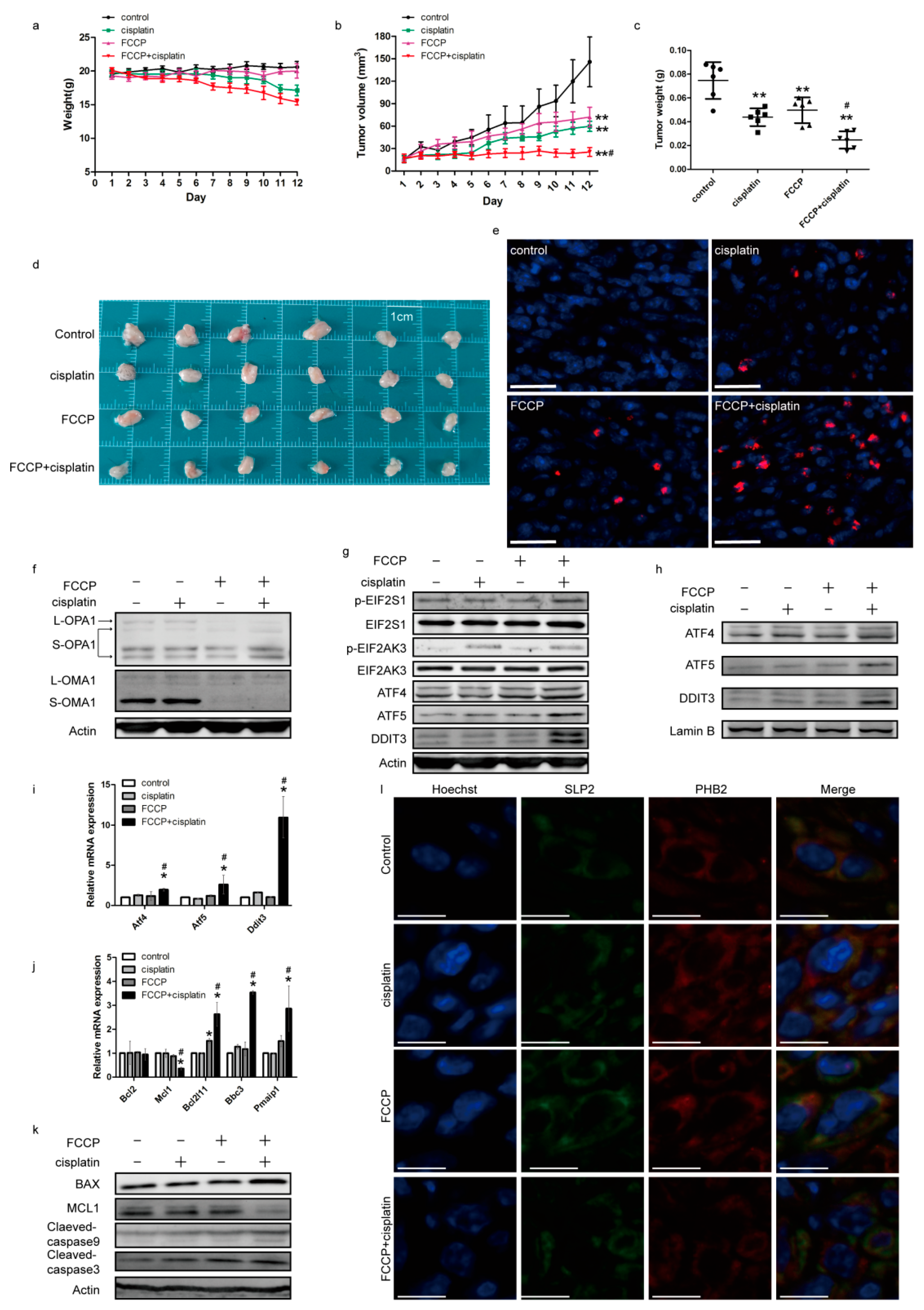
2.7. Effects of FCCP and Cisplatin Treatments in a Mouse Subcutaneous Tumor Model
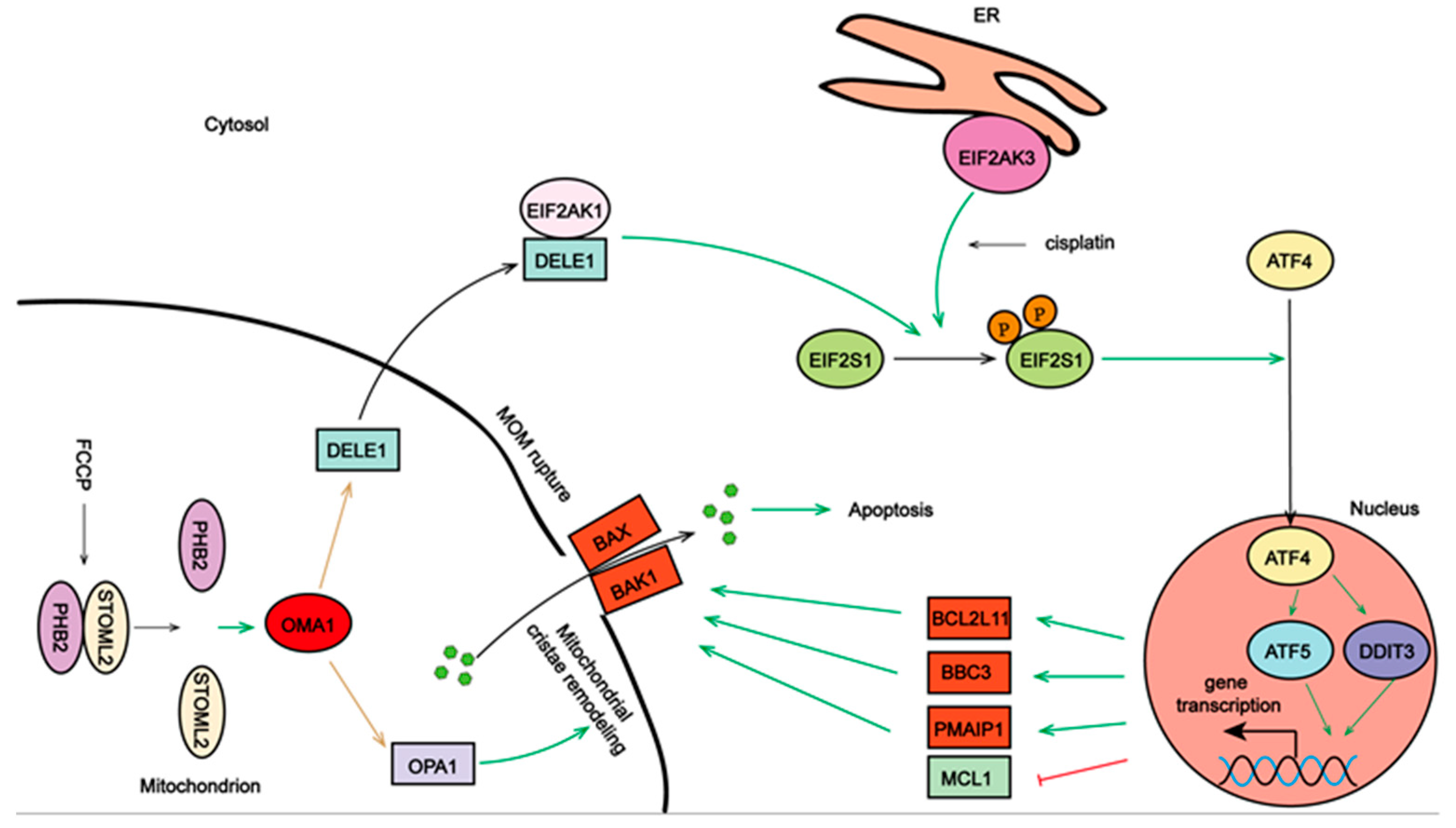
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Reagents and Antibodies
4.3. Plasmids and Transfection
4.4. Cell Viability Assay
4.5. Flow Cytometry
4.6. Western Blotting
4.7. Immunofluorescence and Fluorescence Microscopy
4.8. Transmission Electron Microscopy
4.9. RNA Extraction and qRT-PCR
4.10. Co-Immunoprecipitation
4.11. Mitochondrial Isolation
4.12. Mouse Experiments
4.13. TUNEL Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Bahar, E.; Kim, J.Y.; Kim, Y.S.; Yoon, H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. Int. J. Mol. Sci. 2020, 21, 7613. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, H.; Yin, X.; Liang, H.; Zheng, Z.; Shen, Q.; Hu, W. Exploring cisplatin resistance in ovarian cancer through integrated bioinformatics approach and overcoming chemoresistance with sanguinarine. Am. J. Transl. Res. 2020, 12, 923–939. [Google Scholar] [PubMed]
- Falzone, L.; Scandurra, G.; Lombardo, V.; Gattuso, G.; Lavoro, A.; Distefano, A.B.; Scibilia, G.; Scollo, P. A multidisciplinary approach remains the best strategy to improve and strengthen the management of ovarian cancer (Review). Int. J. Oncol. 2021, 59, 53. [Google Scholar] [CrossRef]
- Saita, S.; Nolte, H.; Fiedler, K.U.; Kashkar, H.; Venne, A.S.; Zahedi, R.P.; Krüger, M.; Langer, T. PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Nat. Cell Biol. 2017, 19, 318–328. [Google Scholar] [CrossRef]
- Zhuang, M.; Guan, S.; Wang, H.; Burlingame, A.L.; Wells, J.A. Substrates of IAP ubiquitin ligases identified with a designed orthogonal E3 ligase, the NEDDylator. Mol. Cell 2013, 49, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavi, M.V. Targeted OMA1 therapies for cancer. Int. J. Cancer 2019, 145, 2330–2341. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Jiang, H.; Shen, Z.; Wang, X. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 14782–14787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavi, M.V. OMA1-An integral membrane protease? Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140558. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zuo, M.; Zeng, L.; Cui, K.; Liu, B.; Yan, C.; Chen, L.; Dong, J.; Shangguan, F.; Hu, W.; et al. OMA1 reprograms metabolism under hypoxia to promote colorectal cancer development. EMBO Rep. 2021, 22, e50827. [Google Scholar] [CrossRef]
- Amini, M.A.; Karimi, J.; Khodadadi, I.; Tavilani, H.; Talebi, S.S.; Afshar, B. Overexpression of ROMO1 and OMA1 are Potentially Biomarkers and Predict Unfavorable Prognosis in Gastric Cancer. J. Gastrointest. Cancer 2020, 51, 939–946. [Google Scholar] [CrossRef]
- Viana, M.P.; Levytskyy, R.M.; Anand, R.; Reichert, A.S.; Khalimonchuk, O. Protease OMA1 modulates mitochondrial bioenergetics and ultrastructure through dynamic association with MICOS complex. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Xie, Q.; Su, J.; Jiao, B.; Shen, L.; Ma, L.; Qu, X.; Yu, C.; Jiang, X.; Xu, Y.; Sun, L. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int. J. Oncol. 2016, 49, 2507–2519. [Google Scholar] [CrossRef] [Green Version]
- Légiot, A.; Céré, C.; Dupoiron, T.; Kaabouni, M.; Camougrand, N.; Manon, S. Mitochondria-Associated Membranes (MAMs) are involved in Bax mitochondrial localization and cytochrome c release. Microb. Cell 2019, 6, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Eckl, E.M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1- DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Hernando-Rodríguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. [Google Scholar] [CrossRef] [Green Version]
- Deshwal, S.; Fiedler, K.U.; Langer, T. Mitochondrial Proteases: Multifaceted Regulators of Mitochondrial Plasticity. Annu. Rev. Biochem. 2020, 89, 501–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorile, A.; Rasmo, D.D.; Cormio, A.; Musicco, C.; Rossi, R.; Fortarezza, F.; Palese, L.L.; Loizzi, V.; Resta, L.; Scillitani, G.; et al. Human Ovarian Cancer Tissue Exhibits Increase of Mitochondrial Biogenesis and Cristae Remodeling. Cancers 2019, 11, 1350. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.Y.; Guo, H.F.; Guo, H.M. Clinical significance of SLP-2 in epithelial ovarian cancer and its regulatory effect on the Notch signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Kahl, A.; Fruitman, H.; Qian, L.; Zhou, P.; Manfredi, G.; Iadecola, C. Prohibitin levels regulate OMA1 activity and turnover in neurons. Cell Death Differ. 2020, 27, 1896–1906. [Google Scholar] [CrossRef]
- Wai, T.; Saita, S.; Nolte, H.; Müller, S.; König, T.; Richter-Dennerlein, R.; Sprenger, H.G.; Madrenas, J.; Mühlmeister, M.; Brandt, U.; et al. The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 2016, 17, 1844–1856. [Google Scholar] [CrossRef] [Green Version]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; Rasmo, D.D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Burwicka, N.; Aktasc, B.H. The eIF2-alpha kinase HRI: A potential target beyond the red blood cell. Expert Opin. Ther. Targets 2017, 21, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease–the long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.; Wang, Q.; Fung, E.; Xue, K.; Tsang, B.K. p53 Is Required for Cisplatin-induced Processing of the Mitochondrial Fusion Protein L-Opa1 That Is Mediated by the Mitochondrial Metallopeptidase OMA1 in Gynecologic Cancers. J. Biol. Chem. 2014, 289, 27134–27145. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Yoon, Y. Mitochondrial Membrane Dynamics—Functional Positioning of OPA1. Antioxidants 2018, 7, 186. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhao, D.; Shah, S.Z.A.; Zhang, X.; Lai, M.; Yang, D.; Wu, X.; Guan, Z.; Li, J.; Zhao, H.; et al. OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases. Cell Death Dis. 2019, 10, 710. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Vázquez, S.; Sánchez-Vera, I.; Saura-Esteller, J.; Cosialls, A.M.; Noisier, A.F.M.; Albericio, F.; Lavilla, R.; Pons, G.; Iglesias-Serret, D.; Gil, J. NOXA upregulation by the prohibitin-binding compound fluorizoline is transcriptionally regulated by integrated stress response-induced ATF3 and ATF4. FEBS J. 2021, 288, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Piazzi, M.; Raffini, M.; Faenza, I.; Blalock, W.L. Prohibitin 2: At a communications crossroads. IUBMB Life 2015, 67, 239–254. [Google Scholar] [CrossRef]
- Yan, X.Y.; Zhang, Y.; Zhang, J.J.; Zhang, L.C.; Liu, Y.N.; Wu, Y.; Xue, Y.N.; Lu, S.Y.; Su, J.; Sun, L.K. p62/SQSTM1 as an oncotarget mediates cisplatin resistance through activating RIP1-NF-κB pathway in human ovarian cancer cells. Cancer Sci. 2017, 108, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name Human | Sequence | Primer Name Mouse | Sequence |
|---|---|---|---|
| ATF4 | 5′-GTTTTGGATTGGTGGGGTGC-3′ 5′-TCTTGGTTCCTGCCACGTTT-3′ | Atf4 | 5′-GCCGGTTTAAGTTGTGTGCT-3′ 5′-CTGGATTCGAGGAATGTGCT-3′ |
| ATF5 | 5′-TCAGGAGGAGGAAACCAGACC-3′ 5′-CCTCGGCTGAAGAAAGAAGGT-3′ | Atf5 | 5′-GGCTGGCTCGTAGACTATGG-3′ 5′-CCAGAGGAAGGAGAGCTGTG-3′ |
| DDIT3 | 5′-GGAAACAGAGTGGTCATTCCC-3′ 5′-CTGCTTGAGCCGTTCATTCTC-3′ | Ddit3 | 5′-CCTAGCTTGGCTGACAGAGG-3′ 5′-CTGCTCCTTCTCCTTCATGC-3′ |
| PMAIP1 | 5′-CACGAGGAACAAGTGCAAGTAG-3′ 5′-TGATGAAACGTGCACCTCCT-3′ | Pmaip1 | 5′-GACAAAGTGAATTTACGGCAGA-3′ 5′-GGTTTCACGTTATCACAGCTCA-3′ |
| BCL2L11 | 5′-TAAGTTCTGAGTGTGACCGAGA-3′ 5′-GCTCTGTCTGTAGGGAGGTAGG-3′ | Bcl2l11 | 5′-ACTTGGATTCACACCACCTCC-3′ 5′-GTCGGGATTACCTTGCGGTT-3′ |
| BBC3 | 5′-GAGCAGGGCAGGAAGTAACAA-3′ 5′-GAGGGCTGAGGACCACAAAT-3′ | Bbc3 | 5′-TGTTTGGCTCCCGAGTTTGT-3′ 5′-CCAGGCAAGCGACAGATACA-3′ |
| MCL1 | 5′-TGCTTCGGAAACTGGACATCA-3′ 5′-TAGCCACAAAGGCACCAAAAG-3′ | Mcl1 | 5′-CAAACTGGGGCAGGATTGTG-3′ 5′-CACATTTCTGATGCCGCCTT-3′ |
| BCL2 | 5′-TTCTTTGAGTTCGGTGGGGT-3′ 5′-GCCCATGCTGAAACTCCCTTA-3′ | Bcl2 | 5′-CGTCGTGACTTCGCAGAGAT-3′ 5′-AGTTCCACAAAGGCATCCCA-3′ |
| ACTB | 5′-TTTCTGAGTTGATTTCCCGGTC-3′ 5′-ACCGAACTTGCATTGATTCCAG-3′ | Actb | 5′-GTATGGAATCCTGTGGCATC-3′ 5′-AAGCACTTGCGGTGCACGAT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.; Yu, H.; Kong, Q.; Wang, B.; Shen, L.; Dong, D.; Sun, L. The Mitochondrial PHB2/OMA1/DELE1 Pathway Cooperates with Endoplasmic Reticulum Stress to Facilitate the Response to Chemotherapeutics in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 1320. https://doi.org/10.3390/ijms23031320
Cheng M, Yu H, Kong Q, Wang B, Shen L, Dong D, Sun L. The Mitochondrial PHB2/OMA1/DELE1 Pathway Cooperates with Endoplasmic Reticulum Stress to Facilitate the Response to Chemotherapeutics in Ovarian Cancer. International Journal of Molecular Sciences. 2022; 23(3):1320. https://doi.org/10.3390/ijms23031320
Chicago/Turabian StyleCheng, Meiyu, Huimei Yu, Qinghuan Kong, Bingrong Wang, Luyan Shen, Delu Dong, and Liankun Sun. 2022. "The Mitochondrial PHB2/OMA1/DELE1 Pathway Cooperates with Endoplasmic Reticulum Stress to Facilitate the Response to Chemotherapeutics in Ovarian Cancer" International Journal of Molecular Sciences 23, no. 3: 1320. https://doi.org/10.3390/ijms23031320
APA StyleCheng, M., Yu, H., Kong, Q., Wang, B., Shen, L., Dong, D., & Sun, L. (2022). The Mitochondrial PHB2/OMA1/DELE1 Pathway Cooperates with Endoplasmic Reticulum Stress to Facilitate the Response to Chemotherapeutics in Ovarian Cancer. International Journal of Molecular Sciences, 23(3), 1320. https://doi.org/10.3390/ijms23031320