
Undifferentiated Sarcomatoid Carcinoma of the Pancreas: From Histology and Molecular Pathology to Precision Oncology

 ,
,  , and
, and 
Abstract
:1. Introduction
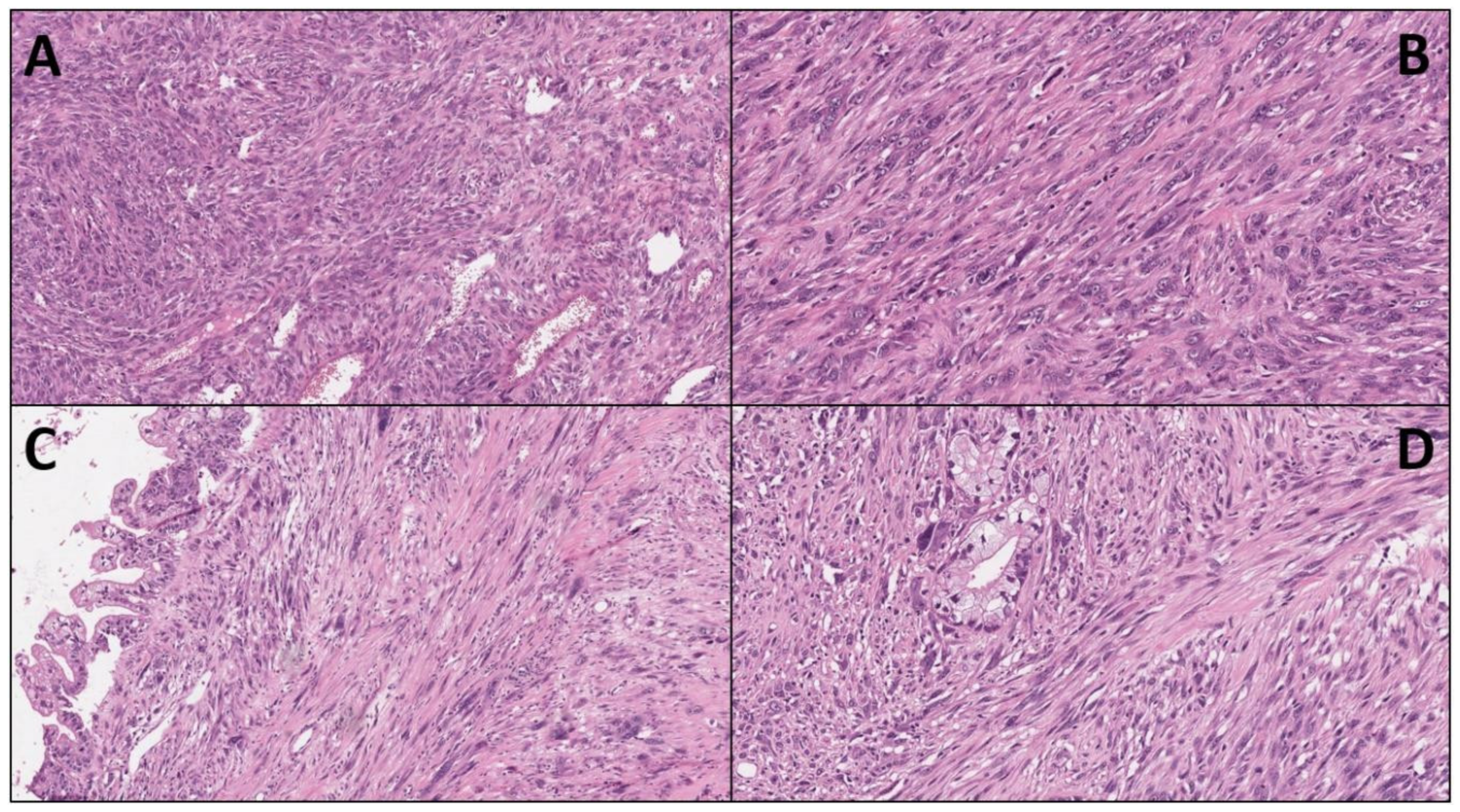
2. Undifferentiated Sarcomatoid Carcinoma of the Pancreas: What Do We Know So Far?
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Pereira, S.P.; Oldfield, L.; Ney, A.; Hart, P.A.; Keane, M.G.; Pandol, S.J.; Li, D.; Greenhalf, W.; Jeon, C.Y.; Koay, E.J.; et al. Early detection of pancreatic cancer. Lancet Gastroenterol. Hepatol. 2020, 5, 698–710. [Google Scholar] [CrossRef]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef] [PubMed]
- Mattiolo, P.; Kryklyva, V.; Brosens, L.A.; Mafficini, A.; Lawlor, R.T.; Milella, M.; Scarpa, A.; Corbo, V.; Luchini, C. Refining targeted therapeutic approaches in pancreatic cancer: From histology and molecular pathology to the clinic. Expert Opin. Ther. Targets 2021. [Google Scholar] [CrossRef]
- Luchini, C.; Paolino, G.; Mattiolo, P.; Piredda, M.L.; Cavaliere, A.; Gaule, M.; Melisi, D.; Salvia, R.; Malleo, G.; Shin, J.I.; et al. KRAS wild-type pancreatic ductal adenocarcinoma: Molecular pathology and therapeutic opportunities. J. Exp. Clin. Cancer Res. 2020, 39, 227. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Luchini, C.; Mafficini, A.; Chatterjee, D.; Piredda, M.L.; Sciammarella, C.; Navale, P.; Malleo, G.; Mattiolo, P.; Marchegiani, G.; Pea, A.; et al. Histo-molecular characterization of pancreatic cancer with microsatellite instability: Intra-tumor heterogeneity, B2M inactivation, and the importance of metastatic sites. Virchows Arch. 2021, 1–8. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchini, C.; Capelli, P.; Scarpa, A. Pancreatic Ductal Adenocarcinoma and Its Variants. Surg. Pathol. Clin. 2016, 9, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Luchini, C.; Conciatori, F.; Vaccaro, V.; Di Cello, I.; Mattiolo, P.; Falcone, I.; Ferretti, G.; Scarpa, A.; Cognetti, F.; et al. Morphologic and Molecular Landscape of Pancreatic Cancer Variants as the Basis of New Therapeutic Strategies for Precision Oncology. Int. J. Mol. Sci. 2020, 21, 8841. [Google Scholar] [CrossRef] [PubMed]
- Quezada-Marin, J.I.; Lam, A.K.; Ochiai, A.; Odze, R.D.; Washington, K.M.; Fukayama, M.; Rugge, M.; Klimstra, D.S.; Nagtegaal, I.D.; Tan, P.H.; et al. Gastrointestinal tissue-based molecular biomarkers: A practical categorisation based on the 2019 World Health Organization classification of epithelial digestive tumours. Histopathology 2020, 77, 340–350. [Google Scholar] [CrossRef]
- Mattiolo, P.; Fiadone, G.; Paolino, G.; Chatterjee, D.; Bernasconi, R.; Piccoli, P.; Parolini, C.; El Aidi, M.; Sperandio, N.; Malleo, G.; et al. Epithelial-mesenchymal transition in undifferentiated carcinoma of the pancreas with and without osteoclast-like giant cells. Virchows Arch. 2021, 478, 319–326. [Google Scholar] [CrossRef]
- Lawlor, R.T.; Veronese, N.; Nottegar, A.; Malleo, G.; Smith, L.; Demurtas, J.; Cheng, L.; Wood, L.D.; Silvestris, N.; Salvia, R.; et al. Prognostic Role of High-Grade Tumor Budding in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis with a Focus on Epithelial to Mesenchymal Transition. Cancers 2019, 11, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjeevi, S.; Ivanics, T.; Dsouza, M.; Ghazi, S.; Del Chiaro, M.; Isaksson, B.; Andrén-Sandberg, Å.; Blomberg, J.; Ansorge, C. An Emergent Case of Pancreatic Cancer; Adenosquamous Carcinoma with Sarcomatoid Change. JOP J. Pancreas 2015, 17, 93–97. [Google Scholar]
- Lu, B.C.; Wang, C.; Yu, J.H.; Shen, Z.H.; Yang, J.H. A huge adenosquamous carcinoma of the pancreas with sarcomatoid change: An unusual case report. World J. Gastroenterol. 2014, 20, 16381–16386. [Google Scholar] [CrossRef]
- Yao, J.; Qian, J.J.; Zhu, C.R.; Bai, D.S.; Miao, Y. Laparoscopic left pancreatectomy for pancreatic sarcomatoid carcinoma: A case report and review of the literature. Oncol. Lett. 2013, 6, 568–570. [Google Scholar] [CrossRef] [Green Version]
- Yepuri, N.; Pruekprasert, N.; Naous, R. High-grade malignant pancreatic neoplasm with sarcomatoid features. AME Case Rep. 2018, 2, 39. [Google Scholar] [CrossRef]
- Xie, Y.; Xiang, Y.; Zhang, D.; Yao, X.; Sheng, J.; Yang, Y.; Zhang, X. Sarcomatoid carcinoma of the pancreas: A case report and review of the literature. Mol. Med. Rep. 2018, 18, 4716–4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhari, N.; Joudeh, A. Early Stage Anaplastic Sarcomatoid Carcinoma of The Pancreas, A Case Report. Am. J. Case Rep. 2019, 20, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Toledo, P.F.; Berger, Z.; Carreno, L.; Cardenas, G.; Castillo, J.; Orellana, O. Sarcomatoid carcinoma of the pancreas-a rare tumor with an uncommon presentation and course: A case report and review of literature. World J. Clin. Cases 2021, 9, 3716–3725. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.B.; Burkhart, R.A.; Griffin, J.F.; Miller, J.A.; Weiss, M.J.; Cameron, J.L.; Wolfgang, C.L.; He, J. Long-term survival after resection of sarcomatoid carcinoma of the pancreas: An updated experience. J. Surg. Res. 2017, 219, 238–243. [Google Scholar] [CrossRef]
- Ren, C.L.; Jin, P.; Han, C.X.; Xiao, Q.; Wang, D.R.; Shi, L.; Wang, D.X.; Chen, H. Unusual early-stage pancreatic sarcomatoid carcinoma. World J. Gastroenterol. 2013, 19, 7820–7824. [Google Scholar] [CrossRef]
- Kimura, T.; Fujimoto, D.; Togawa, T.; Ishida, M.; Iida, A.; Sato, Y.; Goi, T. Sarcomatoid carcinoma of the pancreas with rare long-term survival: A case report. World J. Surg. Oncol. 2020, 18, 105. [Google Scholar] [CrossRef]
- Kimura, T.; Togawa, T.; Iida, A.; Noriki, S.; Sato, Y.; Goi, T. Does cellular senescence play an important role in the prognosis of sarcomatoid carcinoma of the pancreas? World J. Surg. Oncol. 2021, 19, 79. [Google Scholar] [CrossRef]
- Sun, N.; Taguchi, A.; Hanash, S. Switching Roles of TGF-beta in Cancer Development: Implications for Therapeutic Target and Biomarker Studies. J. Clin. Med. 2016, 5, 109. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Kawarada, Y.; Inoue, Y.; Kawasaki, F.; Fukuura, K.; Sato, K.; Tanaka, T.; Itoh, Y.; Hayashi, H. TGF-beta induces p53/Smads complex formation in the PAI-1 promoter to activate transcription. Sci. Rep. 2016, 6, 35483. [Google Scholar] [CrossRef]
- Ding, Y.; Shao, Y.; Na, C.; Yin, J.C.; Hua, H.; Tao, R.; Jiang, Y.; Hu, R.; He, X.; Miao, C.; et al. Genetic characterisation of sarcomatoid carcinomas reveals multiple novel actionable mutations and identifies KRAS mutation as a biomarker of poor prognosis. J. Med. Genet. 2022, 59, 10–17. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Lawlor, R.T.; Mattiolo, P.; Mafficini, A.; Hong, S.M.; Piredda, M.L.; Taormina, S.V.; Malleo, G.; Marchegiani, G.; Pea, A.; Salvia, R.; et al. Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Pancreatic Cancer: Systematic Review and Still-Open Questions. Cancers 2021, 13, 3119. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.C.; Denroche, R.; Jang, G.H.; Nowak, K.M.; Zhang, A.; Borgida, A.; Holter, S.; Topham, J.T.; Wilson, J.; Dodd, A.; et al. Clinical and genomic characterisation of mismatch repair deficient pancreatic adenocarcinoma. Gut 2021, 70, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Grant, R.C.; Scarpa, A.; Gallinger, S. Microsatellite instability/mismatch repair deficiency in pancreatic cancers: The same or different? Gut 2021, 70, 1809–1811. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, K. Pembrolizumab in MSI-high Pancreatic Sarcomatoid Carcinoma. Ann. Hematol Oncol. 2021, 8, 1327. [Google Scholar]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, M.; Tolstykh, T.; Zhang, Y.A.; Bangari, D.S.; Cao, H.; Heyl, K.A.; Lee, J.S.; Malkova, N.V.; Malley, K.; Marquez, E.; et al. An experimental model of anti-PD-1 resistance exhibits activation of TGFss and Notch pathways and is sensitive to local mRNA immunotherapy. Oncoimmunology 2021, 10, 1881268. [Google Scholar] [CrossRef] [PubMed]
- Roper, N.; Velez, M.J.; Chiappori, A.; Kim, Y.S.; Wei, J.S.; Sindiri, S.; Takahashi, N.; Mulford, D.; Kumar, S.; Ylaya, K.; et al. Notch signaling and efficacy of PD-1/PD-L1 blockade in relapsed small cell lung cancer. Nat. Commun. 2021, 12, 3880. [Google Scholar] [CrossRef]
- Silvestris, N.; Argentiero, A.; Brunetti, O.; Sonnessa, M.; Colonna, F.; Delcuratolo, S.; Luchini, C.; Scarpa, A.; Lonardi, S.; Nappo, F.; et al. PD-L1 and Notch as novel biomarkers in pancreatic sarcomatoid carcinoma: A pilot study. Expert Opin. Ther. Targets 2021, 25, 1007–1016. [Google Scholar] [CrossRef]

{kind=link}
| Case/Ref. | Age (Years)/Gender | Pancreas/Tumor Size (cm) | Surgery/Resection Margin Status | (Adjuvant) Therapy | Sarcomatoid Compartment | Follow Up/Survival |
|---|---|---|---|---|---|---|
| 1 [17] | 39/M | Head/7 × 10 | - | - | Vimentin | 1 month/died of disease |
| 2 [18] | 58/F | Tail/16 × 18 | Tumor resection/R0 | Chinese medicine and thymosin | Vimentin, CK7 | 5 months/developed metastases but still alive |
| 3 [19] | 48/M | Tail/10 × 8 × 5 | Left pancreatectomy/R0 | Gemcitabine | Vimentin | 3 months/died of disease |
| 4 [20] | 64/F | Head/3.7 × 3.6 | - | Palliative radiotherapy | Vimentin, CD56 | 3 months/died of disease |
| 5 [21] | 63/M | Head/2.5 × 2 × 1.8 | Pancreatoduodenectomy/R0 | Thymopeptides | Vimentin, CK7, CK19 | 18 months/died of disease |
| 6 [22] | 64/M | Head/2.4 × 2 × 1.9 | Pancreatoduodenectomy with cholecystectomy/R0 | Gemcitabine | Vimentin | 19 months/alive |
| 7 [23] | 61/F | Tail/3.2 × 2.9 | Pancreatectomy with splenectomy/R0 | - | Vimentin, pan-CK | 35 months/alive |
| 8 [24] | 67/F | N/A /4 | Pancreatoduodenectomy/R2 | - | N/A | 2 months/died of disease |
| 9 [24] | 80/F | N/A /5 | Pancreatoduodenectomy/R0 | - | N/A | 1 month/alive |
| 10 [24] | 63/F | N/A /5.7 | Distal pancreatectomy/R1 | - | N/A | 1 month/died of disease |
| 11 [24] | 56/F | N/A /5 | Total pancreatectomy/R0 | Capecitabine | N/A | 3 months/Alive |
| 12 [24] | 79/M | N/A /4 | Pancreatoduodenectomy/R0 | - | N/A | 3 months/died of disease |
| 13 [24] | 54/M | N/A /3 | Distal pancreatectomy/R0 | Gemcitabine, capecitabine+ radiation | N/A | 61 months/alive |
| 14 [24] | 65/M | N/A /15 | Distal pancreatectomy/R0 | - | N/A | 3 months/died of disease |
| 15 [24] | 73/F | N/A /9 | Pancreatoduodenectomy with total gastrectomy/R0 | Radiation | N/A | 188 months/alive |
| 16 [25] | 48/M | Tail/10 × 8 × 3.5 | Tumor resection/R0 | Gemcitabine Oxaliplatin Floxuridine | Vimentin AACT CK18 CK19 pan-CK | >36 months/alive |
| 17 [26,27] | 58/M | Body/ Diameter:5 | Pancreatectomy with splenectomy/ N/A | Gemcitabine | Vimentin CK p-Smad2/3 Snail Fibronectin γ-H2AX p53p21 | 120 months/alive |
| 18 [27] | 68/M | N/A /Diameter:4 | Distal pancreatectomy/ N/A | Chemotherapy | p-Smad2/3 Snail Fibronectin | 18 months/died of disease |
| 19 [27] | 65/F | N/A /Huge tumor | - | Cisplatin | p-Smad2/3 Snail Fibronectin | 2 months/died of disease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkountakos, A.; Simbolo, M.; Bariani, E.; Scarpa, A.; Luchini, C. Undifferentiated Sarcomatoid Carcinoma of the Pancreas: From Histology and Molecular Pathology to Precision Oncology. Int. J. Mol. Sci. 2022, 23, 1283. https://doi.org/10.3390/ijms23031283
Gkountakos A, Simbolo M, Bariani E, Scarpa A, Luchini C. Undifferentiated Sarcomatoid Carcinoma of the Pancreas: From Histology and Molecular Pathology to Precision Oncology. International Journal of Molecular Sciences. 2022; 23(3):1283. https://doi.org/10.3390/ijms23031283
Chicago/Turabian StyleGkountakos, Anastasios, Michele Simbolo, Elena Bariani, Aldo Scarpa, and Claudio Luchini. 2022. "Undifferentiated Sarcomatoid Carcinoma of the Pancreas: From Histology and Molecular Pathology to Precision Oncology" International Journal of Molecular Sciences 23, no. 3: 1283. https://doi.org/10.3390/ijms23031283
APA StyleGkountakos, A., Simbolo, M., Bariani, E., Scarpa, A., & Luchini, C. (2022). Undifferentiated Sarcomatoid Carcinoma of the Pancreas: From Histology and Molecular Pathology to Precision Oncology. International Journal of Molecular Sciences, 23(3), 1283. https://doi.org/10.3390/ijms23031283