
p27, The Cell Cycle and Alzheimer´s Disease



Abstract
1. Cell Cycle
1.1. Cell Cycle and Cell Cycle Regulators
1.2. Cell Cycle and Neurogenesis
1.3. Cell Cycle and AD
2. p27Kip1
2.1. p27Kip1 and Its Role in the Cell Cycle Progression
2.2. Extra Cell Cycle Regulatory Functions of p27
2.3. p27 and AD
3. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement:
Conflicts of Interest
References
- Fisher, R.P. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer 2013, 3, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2021, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Skotheim, J.M.; Di Talia, S.; Siggia, E.D.; Cross, F.R. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 2008, 454, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef]
- Kawauchi, T. Cdk5 regulates multiple cellular events in neural development, function and disease. Dev. Growth Differ. 2014, 56, 335–348. [Google Scholar] [CrossRef]
- Łukasik, P.; Baranowska-bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of cyclin-dependent kinases: Types and their mechanism of action. Int. J. Mol. Sci. 2021, 22, 2806. [Google Scholar] [CrossRef]
- Zou, T.; Lin, Z. The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression. Int. J. Mol. Sci. 2021, 22, 5754. [Google Scholar] [CrossRef]
- Frade, J.M.; Ovejero-Benito, M.C. Neuronal cell cycle: The neuron itself and its circumstances. Cell Cycle 2015, 14, 712–720. [Google Scholar] [CrossRef]
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866. [Google Scholar] [CrossRef]
- Liu, N.; Lucibello, F.C.; Engeland, K.; Müller, R. A new model of cell cycle-regulated transcription: Repression of the cyclin A promoter by CDF-1 and anti-repression by E2F. Oncogene 1998, 16, 2957–2963. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-dependent kinases (Cdk) and their role in diseases development—Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef] [PubMed]
- Denicourt, C.; Dowdy, S.F. Cip/Kip proteins: More than just CDKs inhibitors. Genes Dev. 2004, 18, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Graña, X.; Reddy, E. Cell cycle control in mammalian cells: Role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 1995, 11, 211–219. [Google Scholar] [PubMed]
- Coqueret, O. New roles for p21 and p27 cell-cycle inhibitors: A function for each cell compartment? Trends Cell Biol. 2003, 13, 65–70. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Rovillain, E.; Mansfield, L.; Lord, C.J.; Ashworth, A.; Jat, P.S. An RNA interference screen for identifying downstream effectors of the p53 and pRB tumour suppressor pathways involved in senescence. BMC Genom. 2011, 12, 355. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Besso, A.; Roberts, J.; Guillemot, F. Coupling Cell Cycle Exit, Neuronal Differentiation and Migration in Cortical Neurogenesis. Cell Cycle 2006, 5, 2314–2318. [Google Scholar] [CrossRef]
- Legrier, M.; Ducray, A.; Propper, A.; Kastner, A. Region-specific expression of cell cycle inhibitors in the adult brain. Neuroreport 2001, 12, 3127–3131. [Google Scholar] [CrossRef]
- Watanabe, G.; Pena, P.; Shambaugh, G.E.; Haines, G.K.; Pestell, R.G. Regulation of cyclin dependent kinase inhibitor proteins during neonatal cerebella development. Dev. Brain Res. 1998, 108, 77–87. [Google Scholar] [CrossRef]
- Vernon, A.E.; Philpott, A. A single cdk inhibitor, p27Xic1, functions beyond cell cycle regulation to promote muscle differentiation in Xenopus. Development 2003, 130, 71–83. [Google Scholar] [CrossRef]
- Tury, A.; Mairet-Coello, G.; Dicicco-Bloom, E. The multiple roles of the cyclin-dependent kinase inhibitory protein p57(KIP2) in cerebral cortical neurogenesis. Dev. Neurobiol. 2012, 72, 821–842. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef]
- Kempermann, G.; Jessberger, S.; Steiner, B.; Kronenberg, G. Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 2004, 27, 447–452. [Google Scholar] [CrossRef]
- Lois, C.; Alvarez-Buylla, A. Long-Distance Neuronal Migration in the Adult Mammalian Brain. Science 1994, 264, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Yamada, M.; Imayoshi, I.; Kageyama, R.; Suzuki, Y.; Kuniya, T.; Furutachi, S.; Kawaguchi, D.; Gotoh, Y. Cell cycle arrest determines adult neural stem cell ontogeny by an embryonic Notch-nonoscillatory Hey1 module. Nat. Commun. 2021, 12, 6562. [Google Scholar] [CrossRef] [PubMed]
- Furutachi, S.; Miya, H.; Watanabe, T.; Kawai, H.; Yamasaki, N.; Harada, Y.; Imayoshi, I.; Nelson, M.; Nakayama, K.I.; Hirabayashi, Y.; et al. Slowly dividing neural progenitors are an embryonic origin of adult neural stem cells. Nat. Neurosci. 2015, 18, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Lazarini, F.; Gabellec, M.M.; Moigneu, C.; De Chaumont, F.; Olivo-Marin, J.C.; Lledo, P.M. Adult neurogenesis restores dopaminergic neuronal loss in the olfactory bulb. J. Neurosci. 2014, 34, 14430–14442. [Google Scholar] [CrossRef] [PubMed]
- Lledo, P.M.; Valley, M. Adult Olfactory Bulb Neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Luciano, D.; Jo, R.; Abdi, K.; Paez-Gonzalez, P.; Sheng, H.; Warner, D.; Liu, C.; Eroglu, C.; Kuo, C.T. Post-injury protective astrogenesis from SVZ niche is controlled by Notch modulator Thbs4. Nature 2013, 497, 369. [Google Scholar] [CrossRef]
- Brandt, M.D.; Jessberger, S.; Steiner, B.; Kronenberg, G.; Reuter, K.; Bick-Sander, A.; Von Der Behrens, W.; Kempermann, G. Transient calretinin expression defines early postmitotic step of neuronal differentiation in adult hippocampal neurogenesis of mice. Mol. Cell. Neurosci. 2003, 24, 603–613. [Google Scholar] [CrossRef]
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10409–10414. [Google Scholar] [CrossRef]
- Ambrogini, P.; Lattanzi, D.; Ciuffoli, S.; Agostini, D.; Bertini, L.; Stocchi, V.; Santi, S.; Cuppini, R. Morpho-functional characterization of neuronal cells at different stages of maturation in granule cell layer of adult rat dentate gyrus. Brain Res. 2004, 1017, 21–31. [Google Scholar] [CrossRef]
- Van Praag, H.; Schinder, A.F.; Christie, B.R.; Toni, N.; Palmer, T.D.; Gage, F.H. Functional neurogenesis in the adult hippocampus. Nature 2002, 415, 1030–1034. [Google Scholar] [CrossRef]
- Wang, S.; Scott, B.; Wojtowicz, J. Heterogenous properties of dentate granule neurons in the adult rat. J. Neurobiol. 2000, 42, 248–257. [Google Scholar] [CrossRef]
- Drapeau, E.; Montaron, M.-F.; Aguerre, S.; Abrous, D.N. Learning-Induced Survival of New Neurons Depends on the Cognitive Status of Aged Rats. J. Neurosci. 2007, 27, 6037. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Beylin, A.; Tanapat, P.; Reeves, A.; Shors, T.J. Learning enhances adult neurogenesis in the hippocampal formation. Nat. Neurosci. 1999, 2, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Bizon, J.L.; Gallagher, M. Production of new cells in the rat dentate gyrus over the lifespan: Relation to cognitive decline. Eur. J. Neurosci. 2003, 18, 215–219. [Google Scholar] [CrossRef]
- Bizon, J.L.; Lee, H.J.; Gallagher, M. Neurogenesis in a rat model of age-related cognitive decline. Aging Cell 2004, 3, 227–234. [Google Scholar] [CrossRef]
- Drapeau, E.; Abrous, D.N. Stem Cell Review Series: Role of neurogenesis in age-related memory disorders. Aging Cell 2008, 7, 569–589. [Google Scholar] [CrossRef]
- Hollands, C.; Tobin, M.K.; Hsu, M.; Musaraca, K.; Yu, T.S.; Mishra, R.; Kernie, S.G.; Lazarov, O. Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol. Neurodegener. 2017, 12, 64. [Google Scholar] [CrossRef]
- Ben Abdallah, N.M.B.; Slomianka, L.; Vyssotski, A.L.; Lipp, H.P. Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiol. Aging 2010, 31, 151–161. [Google Scholar] [CrossRef]
- Fabel, K.; Kempermann, G. Physical activity and the regulation of neurogenesis in the adult and aging brain. Neuromol. Med. 2008, 10, 59–66. [Google Scholar] [CrossRef]
- Naylor, A.S.; Bull, C.; Nilsson, M.K.L.; Zhu, C.; Björk-Eriksson, T.; Eriksson, P.S.; Blomgren, K.; Kuhn, H.G. From the Cover: Voluntary running rescues adult hippocampal neurogenesis after irradiation of the young mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 14632. [Google Scholar] [CrossRef]
- Ruan, L.; Lau, B.W.M.; Wang, J.; Huang, L.; ZhuGe, Q.; Wang, B.; Jin, K.; So, K.F. Neurogenesis in neurological and psychiatric diseases and brain injury: From bench to bedside. Prog. Neurobiol. 2014, 115, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Arias-Carrion, O.; Freundlieb, N.; Oertel, W.; Hoglinger, G. Adult Neurogenesis and Parkinsons Disease. CNS Neurol. Disord.—Drug Targets 2008, 6, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Martín, M.; Cifuentes, M.; Grondona, J.M.; López-Ávalos, M.D.; Gómez-Pinedo, U.; García-Verdugo, J.M.; Fernández-Llebrez, P. IGF-I stimulates neurogenesis in the hypothalamus of adult rats. Eur. J. Neurosci. 2010, 31, 1533–1548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Momma, S.; Delfani, K.; Carlén, M.; Cassidy, R.M.; Johansson, C.B.; Brismar, H.; Shupliakov, O.; Frisén, J.; Janson, A.M. Evidence for neurogenesis in the adult mammalian substantia nigra. Proc. Natl. Acad. Sci. USA 2003, 100, 7925–7930. [Google Scholar] [CrossRef]
- Bernier, P.J.; Bédard, A.; Vinet, J.; Lévesque, M.; Parent, A. Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc. Natl. Acad. Sci. USA 2002, 99, 11464–11469. [Google Scholar] [CrossRef]
- Dayer, A.G.; Cleaver, K.M.; Abouantoun, T.; Cameron, H.A. New GABAergic interneurons in the adult neocortex and striatum are generated from different precursors. J. Cell Biol. 2005, 168, 415–427. [Google Scholar] [CrossRef]
- Leal-galicia, P.; Chávez-hernández, M.E.; Mata, F.; Mata-luévanos, J.; Rodríguez-serrano, L.M.; Tapia-de-jesús, A.; Buenrostro-jáuregui, M.H. Adult Neurogenesis: A Story Ranging from Controversial New Neurogenic Areas and Human Adult Neurogenesis to Molecular Regulation. Int. J. Mol. Sci. 2021, 22, 11489. [Google Scholar] [CrossRef]
- 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [CrossRef]
- Duyckaerts, C.; Delatour, B.; Potier, M.-C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef]
- Small, S.A.; Perera, G.M.; DeLaPaz, R.; Mayeux, R.; Stern, Y. Differential Regional Dysfunction of the Hippocampal Formation among Elderly with Memory Decline and Alzheimer’s Disease. Ann. Neurol. 1999, 45, 466–472. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Potter, H. Review and hypothesis: Alzheimer disease and Down syndrome—Chromosome 21 nondisjunction may underlie both disorders. Am. J. Hum. Genet. 1991, 48, 1192. [Google Scholar] [PubMed]
- Yang, S.-S.; Zhang, R.; Wang, G.; Zhang, Y.-F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Hooper, C.; Coley, N.; De Souto Barreto, P.; Payoux, P.; Salabert, A.S.; Andrieu, S.; Weiner, M.; Vellas, B. Cortical β-Amyloid in Older Adults Is Associated with Multidomain Interventions with and without Omega 3 Polyunsaturated Fatty Acid Supplementation. J. Prev. Alzheimer’s Dis. 2020, 7, 128–134. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Gärtner, U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J Neural Transm 1998, 105, 949–960. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Morawski, M.; Jäger, C.; Gertz, H.-J. Early neurone loss in Alzheimer’s disease: Cortical or subcortical? Acta Neuropathol. Commun. 2015, 3, 10. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Yurov, Y.B. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiol. Dis. 2009, 34, 212–220. [Google Scholar] [CrossRef]
- Nagy, Z.; Esiri, M.M.; Cato, A.-M.; Smith, A.D. Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol. 1997, 94, 6–15. [Google Scholar] [CrossRef]
- Nagy, Z.; Esiri, M.M.; Smith, A.D. Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions. Acta Neuropathol. 1997, 93, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, L.A.G.; Hoozemans, J.J.M. Physiological and pathophysiological functions of cell cycle proteins in post-mitotic neurons: Implications for Alzheimer’s disease. Acta Neuropathol. 2015, 129, 511. [Google Scholar] [CrossRef] [PubMed]
- Van den Bos, H.; Spierings, D.C.J.; Taudt, A.S.; Bakker, B.; Porubský, D.; Falconer, E.; Novoa, C.; Halsema, N.; Kazemier, H.G.; Hoekstra-Wakker, K.; et al. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. 2016, 17, 116. [Google Scholar] [CrossRef] [PubMed]
- Westra, J.W.; Barral, S.; Chun, J. A Reevaluation of Tetraploidy in the Alzheimer’s Disease Brain. Neurodegener. Dis. 2010, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Asch, A.S.; Carr, D.J.J.; Yamada, H.Y. “Amyloid-beta accumulation cycle” as a prevention and/or therapy target for Alzheimer’s disease. Aging Cell 2020, 19, e13109. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Kosik, K.S. Accelerated neuronal differentiation induced by p53 suppression. J. Cell Sci. 1996, 109 (Pt 6), 1509–1516. [Google Scholar] [CrossRef]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef]
- Hooper, C.; Meimaridou, E.; Tavassoli, M.; Melino, G.; Lovestone, S.; Killick, R. p53 is upregulated in Alzheimer’s disease and induces tau phosphorylation in HEK293a cells. Neurosci. Lett. 2007, 418, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Ohyagi, Y.; Asahara, H.; Chui, D.-H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Abeta42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005, 19, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Slade, N.; Hof, P.R.; Šimić, G. The interactions of p53 with tau and Aß as potential therapeutic targets for Alzheimer’s disease. Prog. Neurobiol. 2018, 168, 104–127. [Google Scholar] [CrossRef] [PubMed]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef]
- Di Meco, A.; Curtis, M.E.; Lauretti, E.; Praticò, D. Autophagy Dysfunction in Alzheimer’s Disease: Mechanistic Insights and New Therapeutic Opportunities. Biol. Psychiatry 2020, 87, 797–807. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Yang, C.; Cai, C.Z.; Song, J.X.; Tan, J.Q.; Durairajan, S.S.K.; Iyaswamy, A.; Wu, M.Y.; Chen, L.L.; Yue, Z.; Li, M.; et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017, 13, 2028–2040. [Google Scholar] [CrossRef]
- Zhu, X.; Rottkamp, C.A.; Raina, A.K.; Brewer, G.J.; Ghanbari, H.A.; Boux, H.; Smith, M.A. Neuronal CDK7 in hippocampus is related to aging and Alzheimer disease. Neurobiol. Aging 2000, 21, 807–813. [Google Scholar] [CrossRef]
- Allnutt, A.B.; Waters, A.K.; Kesari, S.; Yenugonda, V.M. Physiological and Pathological Roles of Cdk5: Potential Directions for Therapeutic Targeting in Neurodegenerative Disease. ACS Chem. Neurosci. 2020, 11, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Leggio, G.M.; Catania, M.V.; Puzzo, D.; Spatuzza, M.; Pellitteri, R.; Gulisano, W.; Torrisi, S.A.; Giurdanella, G.; Piazza, C.; Impellizzeri, A.R.; et al. The antineoplastic drug flavopiridol reverses memory impairment induced by Amyloid-ß1-42 oligomers in mice. Pharmacol. Res. 2016, 106, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, N.; Gupta, R.; Kumar, P. Pharmacological relevance of CDK inhibitors in Alzheimer’s disease. Neurochem. Int. 2021, 148, 105115. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A. Flavopiridol: The first cyclin-dependent kinase inhibitor in human clinical trials. Investig. New Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Gąssowska-Dobrowolska, M.; Strawski, M.; Adamczyk, A.; Czapski, G.A. Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid beta toxicity. J. Neuroinflamm. 2018, 15, 1. [Google Scholar] [CrossRef]
- Zhang, X.; Hernandez, I.; Rei, D.; Mair, W.; Laha, J.K.; Cornwell, M.E.; Cuny, G.D.; Tsai, L.-H.; Steen, J.A.J.; Kosik, K.S. Diaminothiazoles Modify Tau Phosphorylation and Improve the Tauopathy in Mouse Models. J. Biol. Chem. 2013, 288, 22042. [Google Scholar] [CrossRef]
- Polyak, K. The p27Kip1 tumor suppressor gene: Still a suspect or proven guilty? Cancer Cell 2006, 10, 352–354. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McKay, L.K.; White, J.P. The AMPK/p27Kip1 Pathway as a Novel Target to Promote Autophagy and Resilience in Aged Cells. Cells 2021, 10, 1430. [Google Scholar] [CrossRef]
- Andreu, Z.; Khan, M.A.; González-Gómez, P.; Negueruela, S.; Hortigüela, R.; Emeterio, J.S.; Ferrón, S.R.; Martínez, G.; Vidal, A.; Fariñas, I.; et al. The Cyclin-Dependent Kinase Inhibitor p27kip1 Regulates Radial Stem Cell Quiescence and Neurogenesis in the Adult Hippocampus. Stem Cells 2015, 33, 219–229. [Google Scholar] [CrossRef]
- Hörster, H.; Garthe, A.; Walker, T.L.; Ichwan, M.; Steiner, B.; Khan, M.A.; Lie, D.C.; Nicola, Z.; Ramirez-Rodriguez, G.; Kempermann, G. p27kip1 Is Required for Functionally Relevant Adult Hippocampal Neurogenesis in Mice. Stem Cells 2017, 35, 787–799. [Google Scholar] [CrossRef]
- Doetsch, F.; Verdugo, J.M.-G.; Caille, I.; Alvarez-Buylla, A.; Chao, M.V.; Casaccia-Bonnefil, P. Lack of the Cell-Cycle Inhibitor p27Kip1 Results in Selective Increase of Transit-Amplifying Cells for Adult Neurogenesis. J. Neurosci. 2002, 22, 2255. [Google Scholar] [CrossRef] [PubMed]
- Gil-Perotin, S.; Haines, J.D.; Kaur, J.; Marin-Husstege, M.; Spinetta, M.J.; Kim, K.-H.; Duran-Moreno, M.; Schallert, T.; Zindy, F.; Roussel, M.F.; et al. Roles of p53 and p27Kip1 in the regulation of neurogenesis in the murine adult subventricular zone. Eur. J. Neurosci. 2011, 34, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- James, M.K.; Ray, A.; Leznova, D.; Blain, S.W. Differential Modification of p27Kip1 Controls Its Cyclin D-cdk4 Inhibitory Activity. Mol. Cell. Biol. 2008, 28, 498. [Google Scholar] [CrossRef] [PubMed]
- Bencivenga, D.; Stampone, E.; Roberti, D.; Della Ragione, F.; Borriello, A. p27 Kip1, an Intrinsically Unstructured Protein with Scaffold Properties. Cells 2021, 10, 2254. [Google Scholar] [CrossRef]
- Tsytlonok, M.; Hemmen, K.; Hamilton, G.; Kolimi, N.; Felekyan, S.; Seidel, C.A.M.; Tompa, P.; Sanabria, H. Specific Conformational Dynamics and Expansion Underpin a Multi-Step Mechanism for Specific Binding of p27 with Cdk2/Cyclin A. J. Mol. Biol. 2020, 432, 2998–3017. [Google Scholar] [CrossRef]
- Cheng, M.; Sexl, V.; Sherr, C.J.; Roussel, M.F. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc. Natl. Acad. Sci. USA 1998, 95, 1091. [Google Scholar] [CrossRef]
- Akagawa, R.; Nabeshima, Y.I.; Kawauchi, T. Alternative Functions of Cell Cycle-Related and DNA Repair Proteins in Post-mitotic Neurons. Front. Cell Dev. Biol. 2021, 9, 753175. [Google Scholar] [CrossRef]
- Jaiswal, S.; Sharma, P. Role and regulation of p27 in neuronal apoptosis. J. Neurochem. 2017, 140, 576–588. [Google Scholar] [CrossRef]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair 2018, 69, 63–72. [Google Scholar] [CrossRef]
- Serres, M.P.; Zlotek-Zlotkiewicz, E.; Concha, C.; Gurian-West, M.; Daburon, V.; Roberts, J.M.; Besson, A. Cytoplasmic p27 is oncogenic and cooperates with Ras both in vivo and in vitro. Oncogene 2011, 30, 2846–2858. [Google Scholar] [CrossRef]
- Zheng, Y.-L.; Li, B.-S.; Rudrabhatla, P.; Shukla, V.; Amin, N.D.; Maric, D.; Kesavapany, S.; Kanungo, J.; Pareek, T.K.; Takahashi, S.; et al. Phosphorylation of p27Kip1 at Thr187 by Cyclin-dependent Kinase 5 Modulates Neural Stem Cell Differentiation. Mol. Biol. Cell 2010, 21, 3601. [Google Scholar] [CrossRef] [PubMed]
- Cassimere, E.K.; Mauvais, C.; Denicourt, C. p27Kip1 Is Required to Mediate a G1 Cell Cycle Arrest Downstream of ATM following Genotoxic Stress. PLoS ONE 2016, 11, e0162806. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, M.; Gutierrez-Martinez, P.; Swat, A.; Nebreda, A.R.; Fernandez-Capetillo, O. p27kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res. 2009, 69, 8726. [Google Scholar] [CrossRef] [PubMed]
- Godin, J.D.; Thomas, N.; Laguesse, S.; Malinouskaya, L.; Close, P.; Malaise, O.; Purnelle, A.; Raineteau, O.; Campbell, K.; Fero, M.; et al. p27Kip1 Is a Microtubule-Associated Protein that Promotes Microtubule Polymerization during Neuron Migration. Dev. Cell 2012, 23, 729–744. [Google Scholar] [CrossRef]
- Kawauchi, T.; Chihama, K.; Nabeshima, Y.; Hoshino, M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat. Cell Biol. 2005, 8, 17–26. [Google Scholar] [CrossRef]
- Morelli, G.; Even, A.; Gladwyn-Ng, I.; Le Bail, R.; Shilian, M.; Godin, J.D.; Peyre, E.; Hassan, B.A.; Besson, A.; Rigo, J.M.; et al. p27Kip1 Modulates Axonal Transport by Regulating α-Tubulin Acetyltransferase 1 Stability. Cell Rep. 2018, 23, 2429–2442. [Google Scholar] [CrossRef]
- Nowosad, A.; Jeannot, P.; Callot, C.; Creff, J.; Perchey, R.T.; Joffre, C.; Codogno, P.; Manenti, S.; Besson, A. p27 controls Ragulator and mTOR activity in amino acid-deprived cells to regulate the autophagy–lysosomal pathway and coordinate cell cycle and cell growth. Nat. Cell Biol. 2020, 22, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Larrea, M.D.; Hong, F.; Wander, S.A.; da Silva, T.G.; Helfman, D.; Lannigan, D.; Smith, J.A.; Slingerland, J.M. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc. Natl. Acad. Sci. USA 2009, 106, 9268. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Gurian-West, M.; Schmidt, A.; Hall, A.; Roberts, J.M. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004, 18, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, S.; Stringhi, R.; Marcello, E. Dendritic Spines in Alzheimer’s Disease: How the Actin Cytoskeleton Contributes to Synaptic Failure. Int. J. Mol. Sci. 2020, 21, 908. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X.; Tan, L.; Yu, J.-T. Axonal Transport Defects in Alzheimer’s Disease. Mol. Neurobiol. 2014, 51, 1309–1321. [Google Scholar] [CrossRef]
- Ogawa, O.; Lee, H.; Zhu, X.; Raina, A.; Harris, P.L.R.; Castellani, R.J.; Perry, G.; Smith, M.A. Increased p27, an essential component of cell cycle control, in Alzheimer’s disease. Aging Cell 2003, 2, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, Ú.; Bartolomé, F.; Bermejo, F.; Martín-Requero, Á. Enhanced proteasome-dependent degradation of the CDK inhibitor p27kip1 in immortalized lymphocytes from Alzheimer’s dementia patients. Neurobiol. Aging 2008, 29, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Krimpenfort, P.; Leung, C.; van der Korput, H.A.G.M.; Trapman, J.; Camenisch, I.; Berns, A.; Brandner, S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 2002, 129, 3513–3522. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Y.; Zhu, H.; Ma, C.; Dai, X.; Qin, C. Downregulated microRNA-222 is correlated with increased p27Kip1 expression in a double transgenic mouse model of Alzheimer’s disease. Mol. Med. Rep. 2015, 12, 7687–7692. [Google Scholar] [CrossRef]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- White, J.P.; Billin, A.N.; Campbell, M.E.; Russell, A.J.; Huffman, K.M.; Kraus, W.E. The AMPK/p27Kip1 Axis Regulates Autophagy/Apoptosis Decisions in Aged Skeletal Muscle Stem Cells. Stem Cell Rep. 2018, 11, 425. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals for the lysosome: A control center for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Al Mamun, A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Larrea, M.D.; Doughty, C.; Kwiatkowski, D.J.; Squillace, R.; Slingerland, J.M. mTOR-Raptor Binds and Activates SGK1 to Regulate p27 Phosphorylation. Mol. Cell 2008, 30, 701–711. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
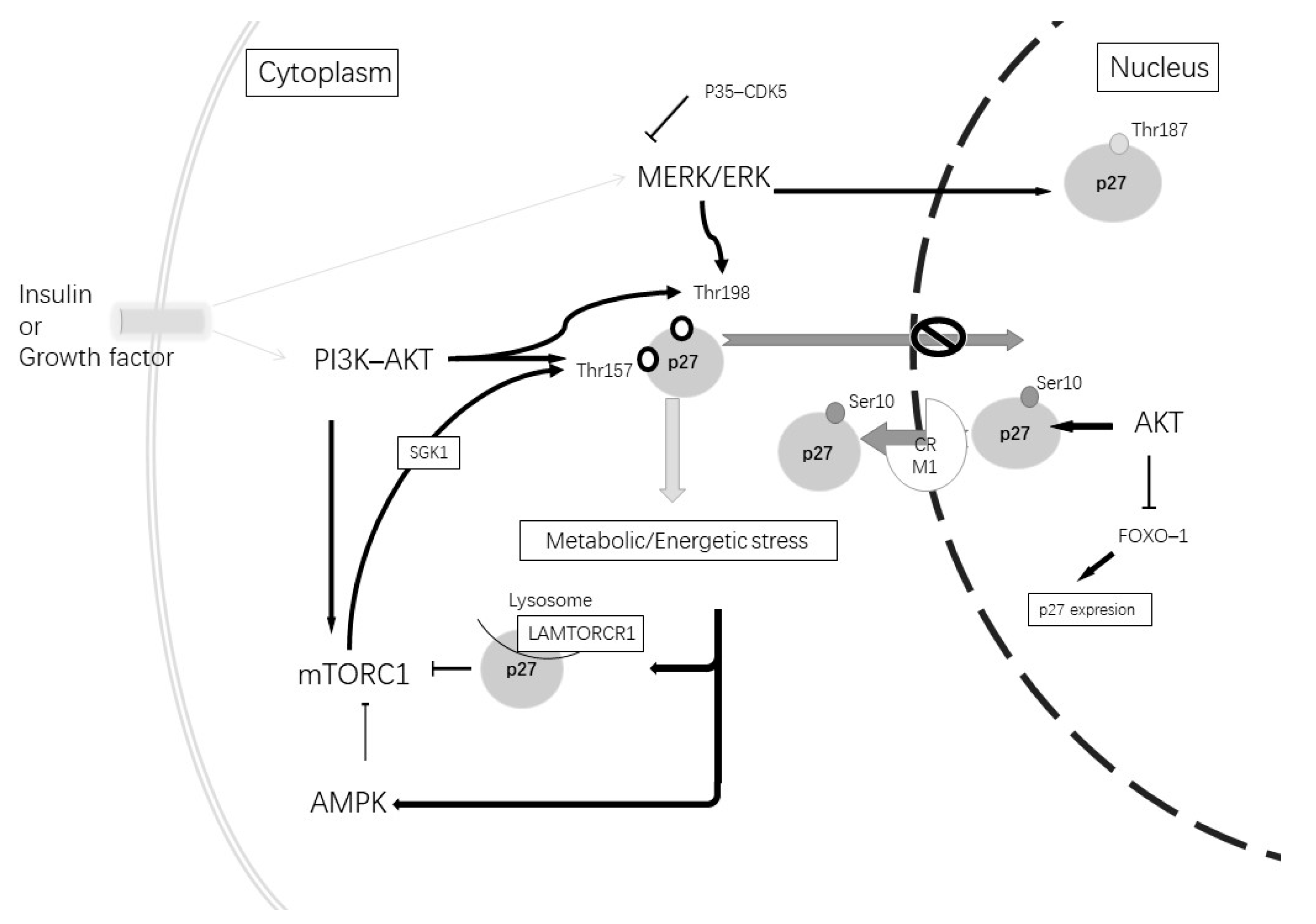{kind=link}
| Subcellular Localization | Phosphorylate Site | Phosphorylated by | Function |
|---|---|---|---|
| Nuclear | Thr187 | CDK2–cyclin E; MERK/EK; CDK5 | Degradation by Skp2 [109] |
| Nuclear, cytoplasmatic | Ser10 | AKT; CDK5 | Nuclear export [110,111] |
| Cytoplasmatic | Thr198 or/and Thr157 | AKT; AMPK; GSK; MERK–ERK | Blocked nuclear import [109] |
| Cytoplasmatic | Thr198 and Ser10 | AKT; AMPK; CDK5; MERK–ERK | Microtubes and actin organization [114,115] |
| Cytoplasmatic | Independent of phosphorylation | – | Axonal transport via stabilization ATA1 [116] |
| Cytoplasmatic | Independent of phosphorylation | – | Increment autophagy via LAMTOR1 [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Osta, A.; Dong, J.; Moreno-Aliaga, M.J.; Ramirez, M.J. p27, The Cell Cycle and Alzheimer´s Disease. Int. J. Mol. Sci. 2022, 23, 1211. https://doi.org/10.3390/ijms23031211
García-Osta A, Dong J, Moreno-Aliaga MJ, Ramirez MJ. p27, The Cell Cycle and Alzheimer´s Disease. International Journal of Molecular Sciences. 2022; 23(3):1211. https://doi.org/10.3390/ijms23031211
Chicago/Turabian StyleGarcía-Osta, Ana, Jinya Dong, María Jesús Moreno-Aliaga, and Maria Javier Ramirez. 2022. "p27, The Cell Cycle and Alzheimer´s Disease" International Journal of Molecular Sciences 23, no. 3: 1211. https://doi.org/10.3390/ijms23031211
APA StyleGarcía-Osta, A., Dong, J., Moreno-Aliaga, M. J., & Ramirez, M. J. (2022). p27, The Cell Cycle and Alzheimer´s Disease. International Journal of Molecular Sciences, 23(3), 1211. https://doi.org/10.3390/ijms23031211