
The Epigenetic Role of Vitamin C in Neurodevelopment

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Regulation of Vitamin C In Vivo
3. Fetal Programming
4. The Fundamentals of Epigenetics
5. Epigenetic Programming in Embryonic Development
6. Nutriepigenomics in Embryonic Development
7. The TET Enzymes
8. Regulation of the TET Enzymes and Vitamin C Dependency
9. TET Expression and Function in Mammalian Development
10. The Histone Lysine Demethylases-Another Vitamin C-Dependent Class of Developmental Regulators
11. Epigenetic Programming and the Role of the TET Enzymes in Neurodevelopment
12. Regulation of Neural Progenitor Cells and Neurogenesis
13. Synaptogenesis, Myelination and TET Function
14. Impact of Tet Mutations in Neurodevelopment
15. Translational Animal Models for Dietary Vitamin C Studies
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Padayatty, S.J.; Levine, M. Vitamin C: The Known and the Unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef]
- Linster, C.L.; van Schaftingen, E. Vitamin C: Biosynthesis, Recycling and Degradation in Mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef]
- Pearson, J.F.; Pullar, J.M.; Wilson, R.; Spittlehouse, J.K.; Vissers, M.C.M.; Skidmore, P.M.L.; Willis, J.; Cameron, V.A.; Carr, A.C. Vitamin C Status Correlates with Markers of Metabolic and Cognitive Health in 50-Year-Olds: Findings of the CHALICE Cohort Study. Nutrients 2017, 9, 831. [Google Scholar] [CrossRef]
- Schleicher, R.L.; Carroll, M.D.; Ford, E.S.; Lacher, D.A. Serum Vitamin C and the Prevalence of Vitamin C Deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am. J. Clin. Nutr. 2009, 90, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.; Carr, A.C. Global Vitamin C Status and Prevalence of Deficiency: A Cause for Concern? Nutrients 2020, 12, 2008. [Google Scholar] [CrossRef]
- De Oliveira, A.M.; de Carvalho Rondó, P.H.; de Moraes Barros, S.B. Concentrations of Ascorbic Acid in the Plasma of Pregnant Smokers and Nonsmokers and Their Newborns. Int. J. Vitam. Nutr. Res. 2004, 74, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Z.; Ren, W.-H.; Liao, W.; Zhang, G.-Y. Concentrations of Antioxidant Vitamins in Maternal and Cord Serum and Their Effect on Birth Outcomes. J. Nutr. Sci. Vitaminol. 2009, 55, 1–8. [Google Scholar] [CrossRef][Green Version]
- Teel, H.M.; Burke, B.S.; Draper, R. Vitamin C in Human Pregnancy and Lactation: I. Studies during Pregnancy. Am. J. Dis. Child. 1938, 56, 1004–1010. [Google Scholar] [CrossRef]
- Schjoldager, J.G.; Tveden-Nyborg, P.; Lykkesfeldt, J. Prolonged Maternal Vitamin C Deficiency Overrides Preferential Fetal Ascorbate Transport but Does Not Influence Perinatal Survival in Guinea Pigs. Br. J. Nutr. 2013, 110, 1573–1579. [Google Scholar] [CrossRef]
- Schjoldager, J.G.; Paidi, M.D.; Lindblad, M.M.; Birck, M.M.; Kjærgaard, A.B.; Dantzer, V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Maternal Vitamin C Deficiency during Pregnancy Results in Transient Fetal and Placental Growth Retardation in Guinea Pigs. Eur. J. Nutr. 2015, 54, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Dobbing, J.; Sands, J. Comparative Aspects of the Brain Growth Spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal Vitamin C Deficiency during Pregnancy Persistently Impairs Hippocampal Neurogenesis in Offspring of Guinea Pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C Deficiency in Early Postnatal Life Impairs Spatial Memory and Reduces the Number of Hippocampal Neurons in Guinea Pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Perez Trueba, G.; Poulsen, H.E.; Christen, S. Vitamin C Deficiency in Weanling Guinea Pigs: Differential Expression of Oxidative Stress and DNA Repair in Liver and Brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Paidi, M.D.; Schjoldager, J.G.; Lykkesfeldt, J.; Tveden-Nyborg, P. Prenatal Vitamin C Deficiency Results in Differential Levels of Oxidative Stress during Late Gestation in Foetal Guinea Pig Brains. Redox Biol. 2014, 2, 361–367. [Google Scholar] [CrossRef]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-Acid Transporter Slc23a1 Is Essential for Vitamin C Transport into the Brain and for Perinatal Survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
- Winkler, B.S.; Orselli, S.M.; Rex, T.S. The Redox Couple between Glutathione and Ascorbic Acid: A Chemical and Physiological Perspective. Free Radic. Biol. Med. 1994, 17, 333–349. [Google Scholar] [CrossRef]
- Washburn, M.P.; Wells, W.W. The Catalytic Mechanism of the Glutathione-Dependent Dehydroascorbate Reductase Activity of Thioltransferase (Glutaredoxin). Biochemistry 1999, 38, 268–274. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.-Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A Family of Mammalian Na+-Dependent L-Ascorbic Acid Transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human Vitamin C (l-Ascorbic Acid) Transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M. Vitamin C Function in the Brain: Vital Role of the Ascorbate Transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal Dehydroascorbic Acid (DHA) Transport Mediated by the Facilitative Sugar Transporters, GLUT2 and GLUT8. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential Compartmentalization of Brain Ascorbate and Glutathione between Neurons and Glia. Neuroscience 1997, 82, 1213–1223. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P. Vitamin C Deficiency in the Young Brain—Findings from Experimental Animal Models. Nutrients 2021, 13, 1685. [Google Scholar] [CrossRef]
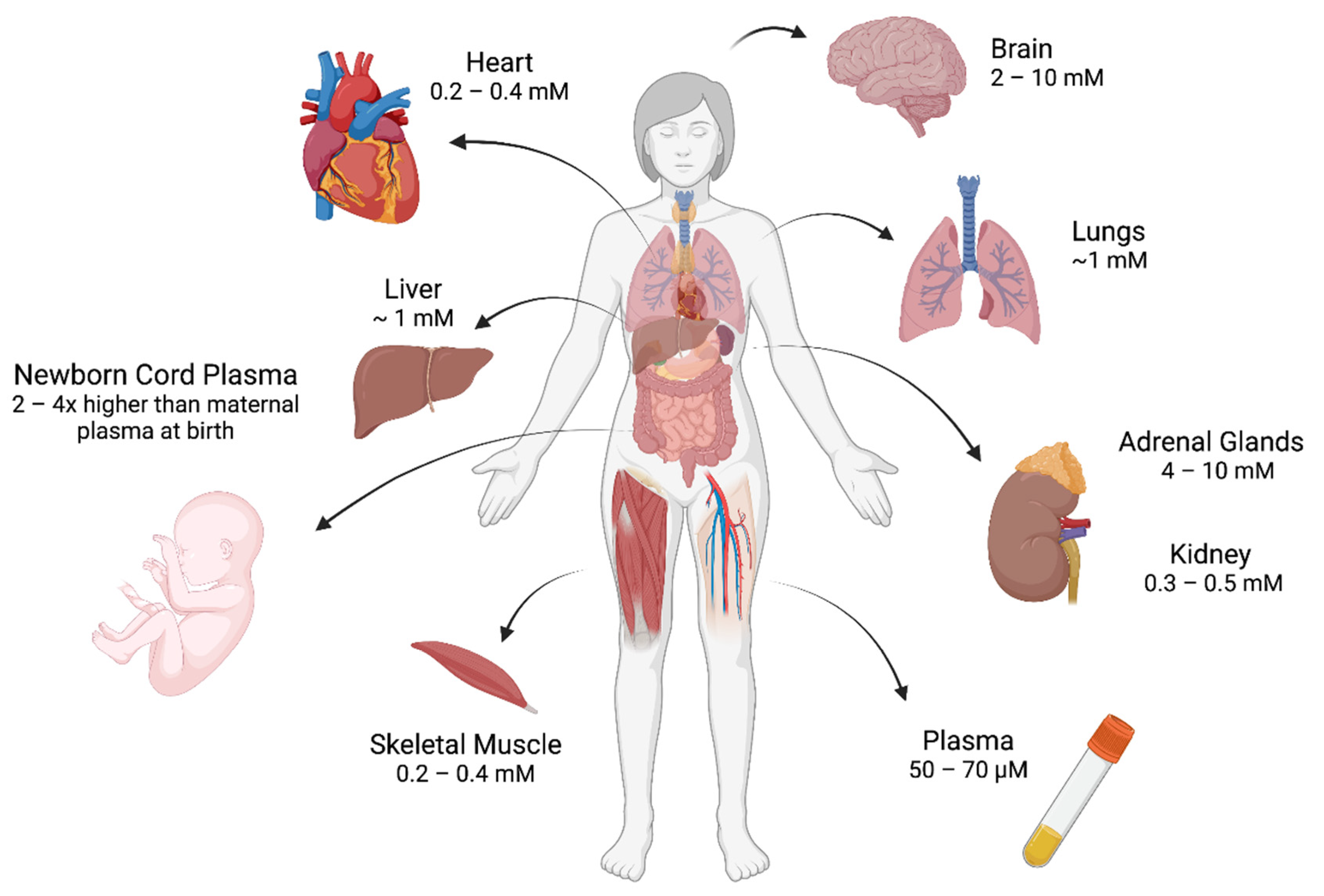
- Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Distribution of Vitamin C Is Tissue Specific with Early Saturation of the Brain and Adrenal Glands Following Differential Oral Dose Regimens in Guinea Pigs. Br. J. Nutr. 2015, 113, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.N.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does Vitamin C Deficiency Affect Cognitive Development and Function? Nutrients 2014, 6, 3818–3846. [Google Scholar] [CrossRef]
- Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of Vitamin C Homeostasis during Deficiency. Nutrients 2013, 5, 2860–2879. [Google Scholar] [CrossRef] [PubMed]
- Spector, R. Micronutrient Homeostasis in Mammalian Brain and Cerebrospinal Fluid. J. Neurochem. 1989, 53, 1667–1674. [Google Scholar] [CrossRef]
- Mun, G.H.; Kim, M.J.; Lee, J.H.; Kim, H.J.; Chung, Y.H.; Chung, Y.B.; Kang, J.S.; Hwang, Y.I.; Oh, S.H.; Kim, J.-G.; et al. Immunohistochemical Study of the Distribution of Sodium-Dependent Vitamin C Transporters in Adult Rat Brain. J. Neurosci. Res. 2006, 83, 919–928. [Google Scholar] [CrossRef]
- Mefford, I.N.; Oke, A.F.; Adams, R.N. Regional Distribution of Ascorbate in Human Brain. Brain Res. 1981, 212, 223–226. [Google Scholar] [CrossRef]
- Rajan, D.P.; Huang, W.; Dutta, B.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V.; Prasad, P.D. Human Placental Sodium-Dependent Vitamin C Transporter (SVCT2): Molecular Cloning and Transport Function. Biochem. Biophys. Res. Commun. 1999, 262, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Scaife, A.R.; McNeill, G.; Campbell, D.M.; Martindale, S.; Devereux, G.; Seaton, A. Maternal Intake of Antioxidant Vitamins in Pregnancy in Relation to Maternal and Fetal Plasma Levels at Delivery. Br. J. Nutr. 2006, 95, 771–778. [Google Scholar] [CrossRef]
- Juhl, B.; Lauszus, F.; Lykkesfeldt, J. Poor Vitamin C Status Late in Pregnancy Is Associated with Increased Risk of Complications in Type 1 Diabetic Women: A Cross-Sectional Study. Nutrients 2017, 9, 186. [Google Scholar] [CrossRef]
- Mikhail, M.S.; Anyaegbunam, A.; Garfinkel, D.; Palan, P.R.; Basu, J.; Romney, S.L. Preeclampsia and Antioxidant Nutrients: Decreased Plasma Levels of Reduced Ascorbic Acid, α-Tocopherol, and Beta-Carotene in Women with Preeclampsia. Am. J. Obstet. Gynecol. 1994, 171, 150–157. [Google Scholar] [CrossRef]
- Siega-Riz, A.M.; Promislow, J.H.; Savitz, D.A.; Thorp, J.M., Jr.; McDonald, T. Vitamin C Intake and the Risk of Preterm Delivery. Am. J. Obstet. Gynecol. 2003, 189, 519–525. [Google Scholar] [CrossRef]
- Jain, S.K.; Wise, R.; Yanamandra, K.; Dhanireddy, R.; Bocchini, J.A. The Effect of Maternal and Cord-Blood Vitamin C, Vitamin E and Lipid Peroxide Levels on Newborn Birth Weight. Mol. Cell. Biochem. 2008, 309, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.E.; Hong, Y.C.; Lee, K.H.; Kim, Y.J.; Kim, W.K.; Chang, N.S.; Park, E.A.; Park, H.S.; Hann, H.J. Influence of Maternal Serum Levels of Vitamins C and E during the Second Trimester on Birth Weight and Length. Eur. J. Clin. Nutr. 2004, 58, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Rumbold, A.R.; Crowther, C.A.; Haslam, R.R.; Dekker, G.A.; Robinson, J.S. Vitamins C and E and the Risks of Preeclampsia and Perinatal Complications. N. Engl. J. Med. 2006, 354, 1796–1806. [Google Scholar] [CrossRef]
- Roberts, J.M.; Myatt, L.; Spong, C.Y.; Thom, E.A.; Hauth, J.C.; Leveno, K.J.; Pearson, G.D.; Wapner, R.J.; Varner, M.W.; Thorp, J.M., Jr.; et al. Vitamins C and E to Prevent Complications of Pregnancy-Associated Hypertension. N. Engl. J. Med. 2010, 362, 1282–1291. [Google Scholar] [CrossRef]
- Chappell, L.C.; Seed, P.T.; CStat; Kelly, F.J.; Briley, A.; Hunt, B.J.; Charnock-Jones, D.S.; Mallet, A.; Poston, L. Vitamin C and E Supplementation in Women at Risk of Preeclampsia Is Associated with Changes in Indices of Oxidative Stress and Placental Function. Am. J. Obstet. Gynecol. 2002, 187, 777–784. [Google Scholar] [CrossRef]
- Chappell, L.C.; Seed, P.T.; Briley, A.L.; Kelly, F.J.; Lee, R.; Hunt, B.J.; Parmar, K.; Bewley, S.J.; Shennan, A.H.; Steer, P.J.; et al. Effect of Antioxidants on the Occurrence of Pre-Eclampsia in Women at Increased Risk: A Randomised Trial. Lancet 1999, 354, 810–816. [Google Scholar] [CrossRef]
- Habibzadeh, N.; Schorah, C.J.; Smithells, R.W. The Effects of Maternal Folic Acid and Vitamin C Nutrition in Early Pregnancy on Reproductive Performance in the Guinea-Pig. Br. J. Nutr. 1986, 55, 23–35. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E.J. Growth in Utero, Blood Pressure in Childhood and Adult Life, and Mortality from Cardiovascular Disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.P.; Osmond, C.; Winter, P.D.; Margetts, B.; Simmonds, S.J. Weight in Infancy and Death from Ischaemic Heart Disease. Lancet 1989, 334, 577–580. [Google Scholar] [CrossRef]
- Barker, D.J.P. Fetal Origins of Coronary Heart Disease. BMJ 1995, 311, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.P. In Utero Programming of Chronic Disease. Clin. Sci. 1998, 95, 115–128. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Low, F.M. The Role of Developmental Plasticity and Epigenetics in Human Health. Birth Defects Res. Part C Embryo Today Rev. 2011, 93, 12–18. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J.P. Type 2 (Non-Insulin-Dependent) Diabetes Mellitus: The Thrifty Phenotype Hypothesis. Diabetologia 1992, 35, 595–601. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic Mechanisms and the Mismatch Concept of the Developmental Origins of Health and Disease. Pediatric. Res. 2007, 61, 5R–10R. [Google Scholar] [CrossRef]
- Hoffman, D.J.; Reynolds, R.M.; Hardy, D.B. Developmental Origins of Health and Disease: Current Knowledge and Potential Mechanisms. Nutr. Rev. 2017, 75, 951–970. [Google Scholar] [CrossRef]
- Kwon, E.J.; Kim, Y.J. What Is Fetal Programming?: A Lifetime Health Is under the Control of in Utero Health. Obstet. Gynecol. Sci. 2017, 60, 506. [Google Scholar] [CrossRef] [PubMed]
- Tammen, S.A.; Friso, S.; Choi, S.-W. Epigenetics: The Link between Nature and Nurture. Mol. Asp. Med. 2013, 34, 753–764. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.; Brenner, C. Epigenetics: Definition, Mechanisms and Clinical Perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Felsenfeld, G. A Brief History of Epigenetics. Cold Spring Harb. Perspect. Biol. 2014, 6, a018200. [Google Scholar] [CrossRef]
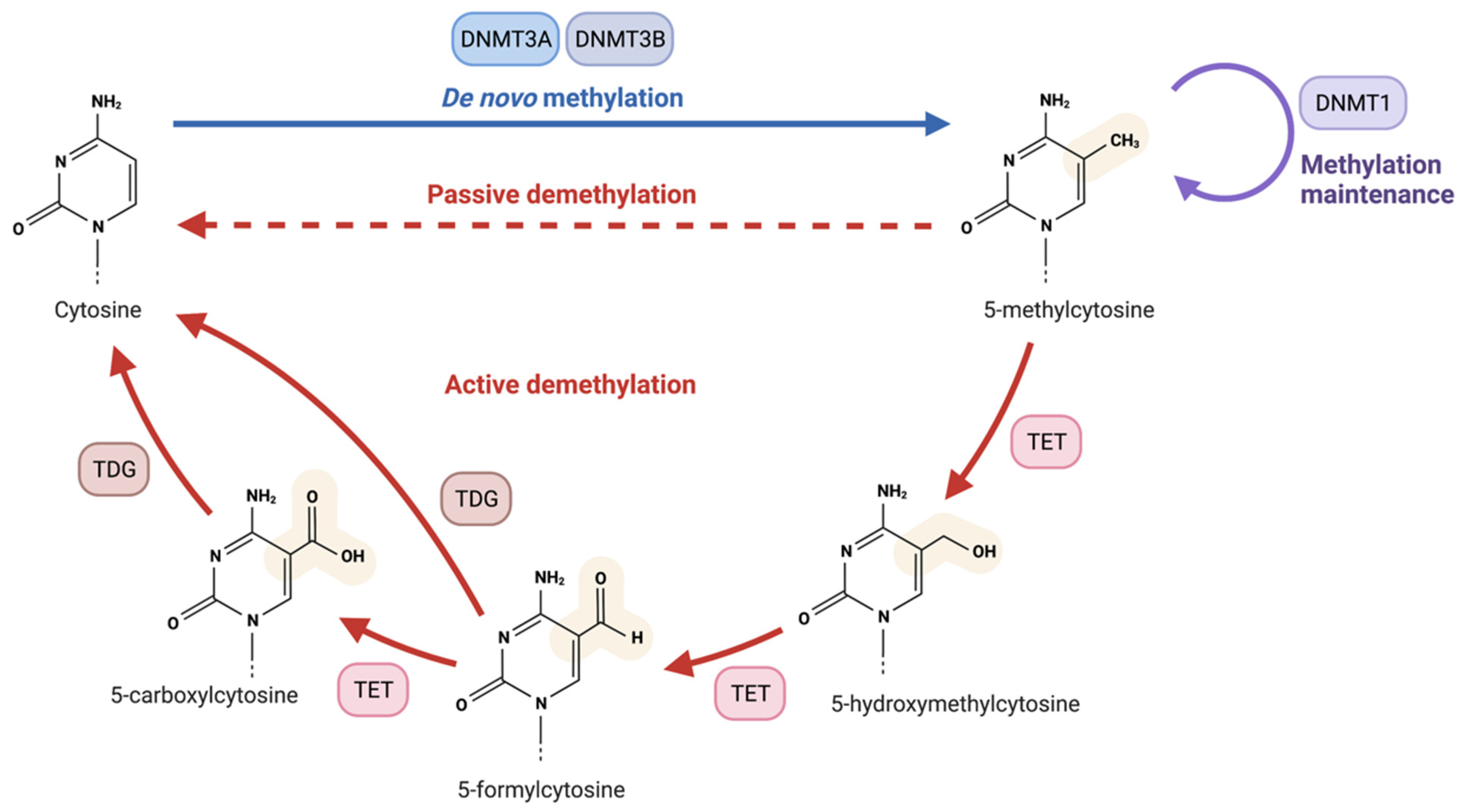
- Rasmussen, K.D.; Helin, K. Role of TET Enzymes in DNA Methylation, Development, and Cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG Islands and the Regulation of Transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Naveh-Many, T.; Cedar, H. Active Gene Sequences Are Undermethylated. Proc. Natl. Acad. Sci. USA 1981, 78, 4246–4250. [Google Scholar] [CrossRef]
- Gillberg, L.; Ørskov, A.D.; Liu, M.; Harsløf, L.B.S.; Jones, P.A.; Grønbæk, K. Vitamin C—A New Player in Regulation of the Cancer Epigenome. Semin. Cancer Biol. 2018, 51, 59–67. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- DesJarlais, R.; Tummino, P.J. Role of Histone-Modifying Enzymes and Their Complexes in Regulation of Chromatin Biology. Biochemistry 2016, 55, 1584–1599. [Google Scholar] [CrossRef]
- Moss, T.J.; Wallrath, L.L. Connections between Epigenetic Gene Silencing and Human Disease. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2007, 618, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M. An Epigenetic Road Map for Histone Lysine Methylation. J. Cell Sci. 2003, 116, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Castelli, G.; Pelosi, E.; Testa, U. Targeting Histone Methyltransferase and Demethylase in Acute Myeloid Leukemia Therapy. OncoTargets Ther. 2017, 11, 131–155. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone Demethylation by a Family of JmjC Domain-Containing Proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-Coding RNAs as Regulators in Epigenetics. Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef]
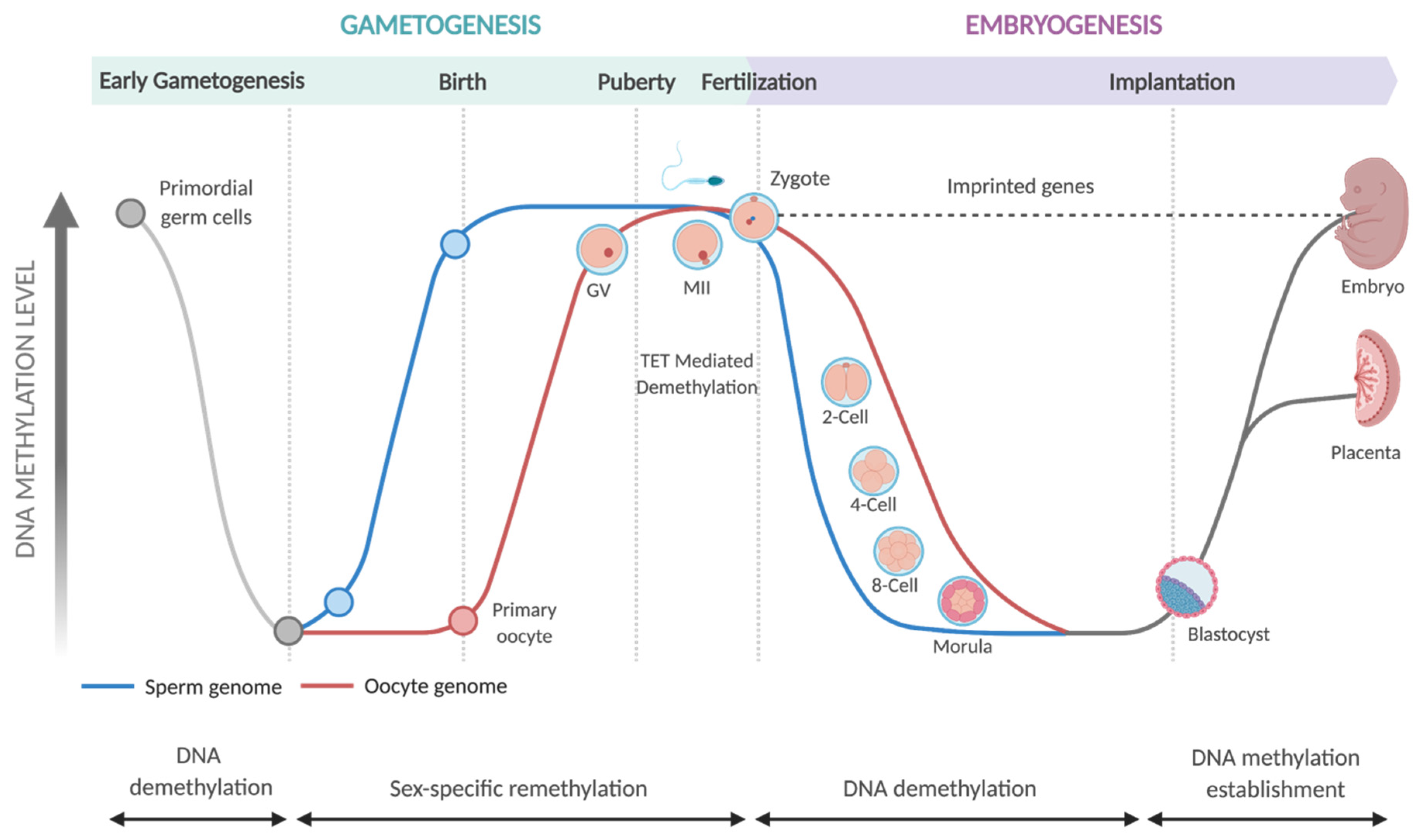
- Smallwood, S.A.; Kelsey, G. De Novo DNA Methylation: A Germ Cell Perspective. Trends Genet. 2012, 28, 33–42. [Google Scholar] [CrossRef]
- Breton-Larrivée, M.; Elder, E.; McGraw, S. DNA Methylation, Environmental Exposures and Early Embryo Development. Anim. Reprod. 2019, 16, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA Methylation Dynamics during Epigenetic Reprogramming in the Germline and Preimplantation Embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef]
- Santos, F.; Hendrich, B.; Reik, W.; Dean, W. Dynamic Reprogramming of DNA Methylation in the Early Mouse Embryo. Dev. Biol. 2002, 241, 172–182. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA Methylation: Mechanisms, Genomics, and Biological Functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef]
- Cambuli, F.; Murray, A.; Dean, W.; Dudzinska, D.; Krueger, F.; Andrews, S.; Senner, C.E.; Cook, S.J.; Hemberger, M. Epigenetic Memory of the First Cell Fate Decision Prevents Complete ES Cell Reprogramming into Trophoblast. Nat. Commun. 2014, 5, 5538. [Google Scholar] [CrossRef] [PubMed]
- Vanhees, K.; Vonhögen, I.G.C.; van Schooten, F.J.; Godschalk, R.W.L. You Are What You Eat, and so Are Your Children: The Impact of Micronutrients on the Epigenetic Programming of Offspring. Cell. Mol. Life Sci. 2014, 71, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Colombo, J.; Gustafson, K.M.; Carlson, S.E. Critical and Sensitive Periods in Development and Nutrition. Ann. Nutr. Metab. 2019, 75, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Banik, A.; Kandilya, D.; Ramya, S.; Stünkel, W.; Chong, Y.; Dheen, S. Maternal Factors That Induce Epigenetic Changes Contribute to Neurological Disorders in Offspring. Genes 2017, 8, 150. [Google Scholar] [CrossRef]
- Laubach, Z.M.; Perng, W.; Dolinoy, D.C.; Faulk, C.D.; Holekamp, K.E.; Getty, T. Epigenetics and the Maintenance of Developmental Plasticity: Extending the Signalling Theory Framework. Biol. Rev. 2018, 93, 1323–1338. [Google Scholar] [CrossRef]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and Epigenetics: An Interplay of Dietary Methyl Donors, One-Carbon Metabolism and DNA Methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef]
- Rochtus, A.; Jansen, K.; Geet, C.; Freson, K. Nutri-Epigenomic Studies Related to Neural Tube Defects: Does Folate Affect Neural Tube Closure Via Changes in DNA Methylation? Mini-Rev. Med. Chem. 2015, 15, 1095–1102. [Google Scholar] [CrossRef]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA Methylation: A Review of Molecular Mechanisms and the Evidence for Folate’s Role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic Gene Promoter Methylation at Birth Is Associated with Child’s Later Adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Craciunescu, C.N.; Zeisel, S.H. Maternal Dietary Choline Deficiency Alters Angiogenesis in Fetal Mouse Hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 12834–12839. [Google Scholar] [CrossRef]
- Konycheva, G.; Dziadek, M.A.; Ferguson, L.R.; Krägeloh, C.U.; Coolen, M.W.; Davison, M.; Breier, B.H. Dietary Methyl Donor Deficiency during Pregnancy in Rats Shapes Learning and Anxiety in Offspring. Nutr. Res. 2011, 31, 790–804. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Zhu, Y.; Yan, J.; Liu, H.; Huang, G.; Li, W. Maternal Folic Acid Impacts DNA Methylation Profile in Male Rat Offspring Implicated in Neurodevelopment and Learning/Memory Abilities. Genes Nutr. 2021, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational Choline Deficiency Causes Global and Igf2 Gene DNA Hypermethylation by Up-Regulation of Dnmt1 Expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.D.; Werler, M.M.; Kelley, K.E.; Vuong, A.M.; Shinde, M.U.; Zheng, Q.; Huber, J.C.; Sharkey, J.R.; Griesenbeck, J.S.; Romitti, P.A.; et al. Nitrosatable Drug Exposure during Early Pregnancy and Neural Tube Defects in Offspring: National Birth Defects Prevention Study. Am. J. Epidemiol. 2011, 174, 1286–1295. [Google Scholar] [CrossRef]
- Schorah, C.J.; Wild, J.; Hartley, R.; Sheppard, S.; Smithells, R.W. The Effect of Periconceptional Supplementation on Blood Vitamin Concentrations in Women at Recurrence Risk for Neural Tube Defect. Br. J. Nutr. 1983, 49, 203–211. [Google Scholar] [CrossRef]
- Lee Chong, T.; Ahearn, E.L.; Cimmino, L. Reprogramming the Epigenome With Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Kanai, T.; Sato, K.; Lee, J.; Jeong, K.S.; Shimokado, K.; Maruyama, N.; Ishigami, A. Insufficient Ascorbic Acid Intake during Gestation Induces Abnormal Cardiac Dilation in Fetal and Neonatal SMP30/GNL Knockout Mice. Pediatric Res. 2013, 73, 578–584. [Google Scholar] [CrossRef]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low Vitamin C and Increased Oxidative Stress and Cell Death in Mice That Lack the Sodium-Dependent Vitamin C Transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef]
- Crawford, D.J.; Liu, M.Y.; Nabel, C.S.; Cao, X.-J.; Garcia, B.A.; Kohli, R.M. Tet2 Catalyzes Stepwise 5-Methylcytosine Oxidation by an Iterative and de Novo Mechanism. J. Am. Chem. Soc. 2016, 138, 730–733. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET Enzymes, TDG and the Dynamics of DNA Demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet Proteins in 5mC to 5hmC Conversion, ES-Cell Self-Renewal and Inner Cell Mass Specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bashkenova, N.; Zang, R.; Huang, X.; Wang, J. The Roles of TET Family Proteins in Development and Stem Cells. Development 2020, 147, dev183129. [Google Scholar] [CrossRef] [PubMed]
- Das, A.B.; Smith-Díaz, C.C.; Vissers, M.C.M.M. Emerging Epigenetic Therapeutics for Myeloid Leukemia: Modulating Demethylase Activity with Ascorbate. Haematologica 2021, 106, 14–25. [Google Scholar] [CrossRef]
- Lu, F.; Liu, Y.; Jiang, L.; Yamaguchi, S.; Zhang, Y. Role of Tet Proteins in Enhancer Activity and Telomere Elongation. Genes Dev. 2014, 28, 2103–2119. [Google Scholar] [CrossRef] [PubMed]
- Brabson, J.P.; Leesang, T.; Mohammad, S.; Cimmino, L. Epigenetic Regulation of Genomic Stability by Vitamin C. Front. Genet. 2021, 12, 675780. [Google Scholar] [CrossRef]
- He, B.; Zhang, C.; Zhang, X.; Fan, Y.; Zeng, H.; Liu, J.; Meng, H.; Bai, D.; Peng, J.; Zhang, Q.; et al. Tissue-Specific 5-Hydroxymethylcytosine Landscape of the Human Genome. Nat. Commun. 2021, 12, 4249. [Google Scholar] [CrossRef]
- Cui, X.L.; Nie, J.; Ku, J.; Dougherty, U.; West-Szymanski, D.C.; Collin, F.; Ellison, C.K.; Sieh, L.; Ning, Y.; Deng, Z.; et al. A Human Tissue Map of 5-Hydroxymethylcytosines Exhibits Tissue Specificity through Gene and Enhancer Modulation. Nat. Commun. 2020, 11, 6161. [Google Scholar] [CrossRef] [PubMed]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) Gene in Hematopoiesis and Hematopoietic Diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic Acid Enhances Tet-Mediated 5-Methylcytosine Oxidation and Promotes DNA Demethylation in Mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C Induces Tet-Dependent DNA Demethylation and a Blastocyst-like State in ES Cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Guo, B.; Wu, H.; Tan, L.; Lu, Q. TET Family of Dioxygenases: Crucial Roles and Underlying Mechanisms. Cytogenet. Genome Res. 2015, 146, 171–180. [Google Scholar] [CrossRef]
- Kuiper, C.; Vissers, M.C.M. Ascorbate as a Co-Factor for Fe- and 2-Oxoglutarate Dependent Dioxygenases: Physiological Activity in Tumor Growth and Progression. Front. Oncol. 2014, 4, 359. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural Studies on Human 2-Oxoglutarate Dependent Oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Mantri, M.; Zhang, Z.; McDonough, M.A.; Schofield, C.J. Autocatalysed Oxidative Modifications to 2-Oxoglutarate Dependent Oxygenases. FEBS J. 2012, 279, 1563–1575. [Google Scholar] [CrossRef]
- Ozer, A.; Bruick, R.K. Non-Heme Dioxygenases: Cellular Sensors and Regulators Jelly Rolled into One? Nat. Chem. Biol. 2007, 3, 144–153. [Google Scholar] [CrossRef]
- Lu, J.; Hu, L.; Cheng, J.; Fang, D.; Wang, C.; Yu, K.; Jiang, H.; Cui, Q.; Xu, Y.; Luo, C. A Computational Investigation on the Substrate Preference of Ten-Eleven-Translocation 2 (TET2). Phys. Chem. Chem. Phys. 2016, 18, 4728–4738. [Google Scholar] [CrossRef]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-Inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef]
- Santiago, M.; Antunes, C.; Guedes, M.; Iacovino, M.; Kyba, M.; Reik, W.; Sousa, N.; Pinto, L.; Branco, M.R.; Marques, C.J. Tet3 Regulates Cellular Identity and DNA Methylation in Neural Progenitor Cells. Cell. Mol. Life Sci. 2020, 77, 2871–2883. [Google Scholar] [CrossRef]
- Hore, T.A.; von Meyenn, F.; Ravichandran, M.; Bachman, M.; Ficz, G.; Oxley, D.; Santos, F.; Balasubramanian, S.; Jurkowski, T.P.; Reik, W. Retinol and Ascorbate Drive Erasure of Epigenetic Memory and Enhance Reprogramming to Naïve Pluripotency by Complementary Mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, 12202–12207. [Google Scholar] [CrossRef]
- Myllylä, R.; Kuutti-Savolainen, E.R.; Kivirikko, K.I. The Role of Ascorbate in the Prolyl Hydroxylase Reaction. Biochem. Biophys. Res. Commun. 1978, 83, 441–448. [Google Scholar] [CrossRef]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate Induces Ten-Eleven Translocation (Tet) Methylcytosine Dioxygenase-Mediated Generation of 5-Hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef]
- Chen, J.; Guo, L.; Zhang, L.; Wu, H.; Yang, J.; Liu, H.; Wang, X.; Hu, X.; Gu, T.; Zhou, Z.; et al. Vitamin C Modulates TET1 Function during Somatic Cell Reprogramming. Nat. Genet. 2013, 45, 1504–1509. [Google Scholar] [CrossRef]
- He, X.; Kim, M.; Kim, S.; Yi, S.; Rhee, Y.; Lee, E.; Park, C.; Dixit, S.; Harrison, F.E. Vitamin C Facilitates Dopamine Neuron Jmjd3-Dependent Epigenetic Control Manner. Stem Cells 2015, 33, 1320–1332. [Google Scholar] [CrossRef]
- Liu, M.; Ohtani, H.; Zhou, W.; Ørskov, A.D.; Charlet, J.; Zhang, Y.W.; Shen, H.; Baylin, S.B.; Liang, G.; Grønbæk, K.; et al. Vitamin C Increases Viral Mimicry Induced by 5-Aza-2’-Deoxycytidine. Proc. Natl. Acad. Sci. USA 2016, 113, 10238–10244. [Google Scholar] [CrossRef]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.-C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.-W.; Page, D.C.; et al. Tet1 Is Dispensable for Maintaining Pluripotency and Its Loss Is Compatible with Embryonic and Postnatal Development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar] [CrossRef]
- Khoueiry, R.; Sohni, A.; Thienpont, B.; Luo, X.; Velde, J.V.; Bartoccetti, M.; Boeckx, B.; Zwijsen, A.; Rao, A.; Lambrechts, D.; et al. Lineage-Specific Functions of TET1 in the Postimplantation Mouse Embryo. Nat. Genet. 2017, 49, 1061–1072. [Google Scholar] [CrossRef]
- Mingay, M.; Chaturvedi, A.; Bilenky, M.; Cao, Q.; Jackson, L.; Hui, T.; Moksa, M.; Heravi-Moussavi, A.; Humphries, R.K.; Heuser, M.; et al. Vitamin C-Induced Epigenomic Remodelling in IDH1 Mutant Acute Myeloid Leukaemia. Leukemia 2018, 32, 11–20. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate Regulates Haematopoietic Stem Cell Function and Leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Gillberg, L.; Ørskov, A.D.; Nasif, A.; Ohtani, H.; Madaj, Z.; Hansen, J.W.; Rapin, N.; Mogensen, J.B.; Liu, M.; Dufva, I.H.; et al. Oral Vitamin C Supplementation to Patients with Myeloid Cancer on Azacitidine Treatment: Normalization of Plasma Vitamin C Induces Epigenetic Changes. Clin. Epigenetics 2019, 11, 143. [Google Scholar] [CrossRef]
- Smith-Díaz, C.C.; Magon, N.J.; McKenzie, J.L.; Hampton, M.B.; Vissers, M.C.M.; Das, A.B. Ascorbate Inhibits Proliferation and Promotes Myeloid Differentiation in TP53-Mutant Leukemia. Front. Oncol. 2021, 11, 709543. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Ferrone, C.K.; Blydt-Hansen, M.; Rauh, M.J. Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 626. [Google Scholar] [CrossRef]
- Gu, T.-P.; Guo, F.; Yang, H.; Wu, H.-P.; Xu, G.-F.; Liu, W.; Xie, Z.-G.; Shi, L.; He, X.; Jin, S.; et al. The Role of Tet3 DNA Dioxygenase in Epigenetic Reprogramming by Oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef]
- Li, X.; Yue, X.; Pastor, W.A.; Lin, L.; Georges, R.; Chavez, L.; Evans, S.M.; Rao, A. Tet Proteins Influence the Balance between Neuroectodermal and Mesodermal Fate Choice by Inhibiting Wnt Signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E8267–E8276. [Google Scholar] [CrossRef]
- Wossidlo, M.; Nakamura, T.; Lepikhov, K.; Marques, C.J.; Zakhartchenko, V.; Boiani, M.; Arand, J.; Nakano, T.; Reik, W.; Walter, J. 5-Hydroxymethylcytosine in the Mammalian Zygote Is Linked with Epigenetic Reprogramming. Nat. Commun. 2011, 2, 241. [Google Scholar] [CrossRef]
- Iqbal, K.; Jin, S.-G.; Pfeifer, G.P.; Szabó, P.E. Reprogramming of the Paternal Genome upon Fertilization Involves Genome-Wide Oxidation of 5-Methylcytosine. Proc. Natl. Acad. Sci. USA 2011, 108, 3642–3647. [Google Scholar] [CrossRef]
- Myllyla, R.; Majamaa, K.; Gunzler, V. Ascorbate Is Consumed Stoichiometrically in the Uncoupled Reactions Catalyzed by Prolyl 4-Hydroxylase and Lysyl Hydroxylase. J. Biol. Chem. 1984, 259, 5403–5405. [Google Scholar] [CrossRef]
- Majamaa, K.; Gunzler, V.; Hanauske-Abel, H.M.; Myllylä, R.; Kivirikko, K.I. Partial Identity of the 2-Oxoglutarate and Ascorbate Binding Sites of Prolyl 4-Hydroxylase. J. Biol. Chem. 1986, 261, 7819–7823. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, T. Tet Family of 5-Methylcytosine Dioxygenases in Mammalian Development. J. Hum. Genet. 2013, 58, 421–427. [Google Scholar] [CrossRef]
- Huang, Y.; Chavez, L.; Chang, X.; Wang, X.; Pastor, W.A.; Kang, J.; Zepeda-Martinez, J.A.; Pape, U.J.; Jacobsen, S.E.; Peters, B.; et al. Distinct Roles of the Methylcytosine Oxidases Tet1 and Tet2 in Mouse Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1361–1366. [Google Scholar] [CrossRef]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Raddatz, G.; Barrasa, M.I.; Cheng, A.W.; Gao, Q.; Powell, B.E.; Li, Z.; Xu, M.; et al. Combined Deficiency of Tet1 and Tet2 Causes Epigenetic Abnormalities but Is Compatible with Postnatal Development. Dev. Cell 2013, 24, 310–323. [Google Scholar] [CrossRef]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) Negatively Regulates Homeostasis and Differentiation of Hematopoietic Stem Cells in Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef]
- Ni, K.; Dansranjavin, T.; Rogenhofer, N.; Oeztuerk, N.; Deuker, J.; Bergmann, M.; Schuppe, H.C.; Wagenlehner, F.; Weidner, W.; Steger, K.; et al. TET Enzymes Are Successively Expressed during Human Spermatogenesis and Their Expression Level Is Pivotal for Male Fertility. Hum. Reprod. 2016, 31, 1411–1424. [Google Scholar] [CrossRef]
- DiTroia, S.P.; Percharde, M.; Guerquin, M.-J.; Wall, E.; Collignon, E.; Ebata, K.T.; Mesh, K.; Mahesula, S.; Agathocleous, M.; Laird, D.J.; et al. Maternal Vitamin C Regulates Reprogramming of DNA Methylation and Germline Development. Nature 2019, 573, 271–275. [Google Scholar] [CrossRef]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T.; et al. Ascorbic Acid during the Suckling Period Is Required for Proper DNA Demethylation in the Liver. Sci. Rep. 2020, 10, 21228. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Jambhekar, A.; Anastas, J.N.; Shi, Y. Histone Lysine Demethylase Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026484. [Google Scholar] [CrossRef]
- Das, P.P.; Shao, Z.; Beyaz, S.; Apostolou, E.; Pinello, L.; Angeles, A.D.L.; O’Brien, K.; Atsma, J.M.; Fujiwara, Y.; Nguyen, M.; et al. Distinct and Combinatorial Functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in Mouse Embryonic Stem Cell Identity. Mol. Cell 2014, 53, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Mosammaparast, N.; Shi, Y. Reversal of Histone Methylation: Biochemical and Molecular Mechanisms of Histone Demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef]
- Monfort, A.; Wutz, A. Breathing-in Epigenetic Change with Vitamin C. EMBO Rep. 2013, 14, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.L.; Masson, N.; Dunne, K.; Flashman, E.; Kawamura, A. The Activity of JmjC Histone Lysine Demethylase KDM4A Is Highly Sensitive to Oxygen Concentrations. ACS Chem. Biol. 2017, 12, 1011–1019. [Google Scholar] [CrossRef]
- Kuroki, S.; Matoba, S.; Akiyoshi, M.; Matsumura, Y.; Miyachi, H.; Mise, N.; Abe, K.; Ogura, A.; Wilhelm, D.; Koopman, P.; et al. Epigenetic Regulation of Mouse Sex Determination by the Histone Demethylase Jmjd1a. Science 2013, 341, 1106–1109. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, X.; Zhou, S.; Liao, L.; Jiang, R.; Xu, J. The Histone H3K9 Demethylase Kdm3b Is Required for Somatic Growth and Female Reproductive Function. Int. J. Biol. Sci. 2015, 11, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Oyola, M.G.; Zhou, S.; Chen, X.; Liao, L.; Tien, J.C.-Y.; Mani, S.K.; Xu, J. Knockout of the Histone Demethylase Kdm3b Decreases Spermatogenesis and Impairs Male Sexual Behaviors. Int. J. Biol. Sci. 2015, 11, 1447–1457. [Google Scholar] [CrossRef]
- Schmitz, S.U.; Albert, M.; Malatesta, M.; Morey, L.; Johansen, J.V.; Bak, M.; Tommerup, N.; Abarrategui, I.; Helin, K. Jarid1b Targets Genes Regulating Development and Is Involved in Neural Differentiation. EMBO J. 2011, 30, 4586–4600. [Google Scholar] [CrossRef]
- Albert, M.; Schmitz, S.U.; Kooistra, S.M.; Malatesta, M.; Morales Torres, C.; Rekling, J.C.; Johansen, J.V.; Abarrategui, I.; Helin, K. The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3. PLoS Genet. 2013, 9, e1003461. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.W.; Lee, S.-K. UTX, a Histone H3-Lysine 27 Demethylase, Acts as a Critical Switch to Activate the Cardiac Developmental Program. Dev. Cell 2012, 22, 25–37. [Google Scholar] [CrossRef]
- Klose, R.J.; Yamane, K.; Bae, Y.; Zhang, D.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. The Transcriptional Repressor JHDM3A Demethylates Trimethyl Histone H3 Lysine 9 and Lysine 36. Nature 2006, 442, 312–316. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhu, Z.; Han, G.; Ye, X.; Xu, B.; Peng, Z.; Ma, Y.; Yu, Y.; Lin, H.; Chen, A.P.; et al. JARID1B Is a Histone H3 Lysine 4 Demethylase Up-Regulated in Prostate Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 19226–19231. [Google Scholar] [CrossRef]
- Isles, A.R. Epigenetics, Chromatin and Brain Development and Function. Brain Neurosci. Adv. 2018, 2, 239821281881201. [Google Scholar] [CrossRef]
- Moody, L.; Chen, H.; Pan, Y.-X. Early-Life Nutritional Programming of Cognition—The Fundamental Role of Epigenetic Mechanisms in Mediating the Relation between Early-Life Environment and Learning and Memory Process. Adv. Nutr. Int. Rev. J. 2017, 8, 337–350. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular Mechanisms of Fragile X Syndrome: A Twenty-Year Perspective. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 219–245. [Google Scholar] [CrossRef]
- Potijk, M.R.; de Winter, A.F.; Bos, A.F.; Kerstjens, J.M.; Reijneveld, S.A. Higher Rates of Behavioural and Emotional Problems at Preschool Age in Children Born Moderately Preterm. Arch. Dis. Child. 2012, 97, 112–117. [Google Scholar] [CrossRef]
- Chyi, L.J.; Lee, H.C.; Hintz, S.R.; Gould, J.B.; Sutcliffe, T.L. School Outcomes of Late Preterm Infants: Special Needs and Challenges for Infants Born at 32 to 36 Weeks Gestation. J. Pediatrics 2008, 153, 25–31. [Google Scholar] [CrossRef]
- Leitner, Y.; Heldman, D.; Harel, S.; Pick, C.G. Deficits in Spatial Orientation of Children with Intrauterine Growth Retardation. Brain Res. Bull. 2005, 67, 13–18. [Google Scholar] [CrossRef]
- Matte, T.D.; Bresnahan, M.; Begg, M.D.; Susser, E. Influence of Variation in Birth Weight within Normal Range and within Sibships on IQ at Age 7 Years: Cohort Study. BMJ 2001, 323, 310–314. [Google Scholar] [CrossRef]
- Leitner, Y.; Fattal-Valevski, A.; Geva, R.; Eshel, R.; Toledano-Alhadef, H.; Rotstein, M.; Bassan, H.; Radianu, B.; Bitchonsky, O.; Jaffa, A.J.; et al. Neurodevelopmental Outcome of Children With Intrauterine Growth Retardation: A Longitudinal, 10-Year Prospective Study. J. Child Neurol. 2007, 22, 580–587. [Google Scholar] [CrossRef]
- Ido, P.; Gale, R.; Laor, A.; Danon, Y.; Stevenson, D.; Seidman, D. The Cognitive Outcome of Full-Term Small for Gestational Age Infants at Late Adolescence. Obstet. Gynecol. 1995, 85, 452–456. [Google Scholar] [CrossRef]
- Tau, G.Z.; Peterson, B.S. Normal Development of Brain Circuits. Neuropsychopharmacology 2010, 35, 147–168. [Google Scholar] [CrossRef]
- Stiles, J.; Jernigan, T.L. The Basics of Brain Development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef]
- Konkel, L. The Brain before Birth: Using FMRI to Explore the Secrets of Fetal Neurodevelopment. Environ. Health Perspect. 2018, 126, 112001. [Google Scholar] [CrossRef]
- Jiang, X.; Nardelli, J. Cellular and Molecular Introduction to Brain Development. Neurobiol. Dis. 2016, 92, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Gan, H.; Lam, C.S.; Poonepalli, A.; Ramasamy, S.; Tay, Y.; Tham, M.; Yu, Y.H. Transcription Factors and Neural Stem Cell Self-Renewal, Growth and Differentiation. Cell Adhes. Migr. 2009, 3, 412–424. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Gotoh, Y. Epigenetic Control of Neural Precursor Cell Fate during Development. Nat. Rev. Neurosci. 2010, 11, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, B.; He, Y.; Jin, P. DNA Methylation Dynamics in Neurogenesis. Epigenomics 2016, 8, 401–414. [Google Scholar] [CrossRef]
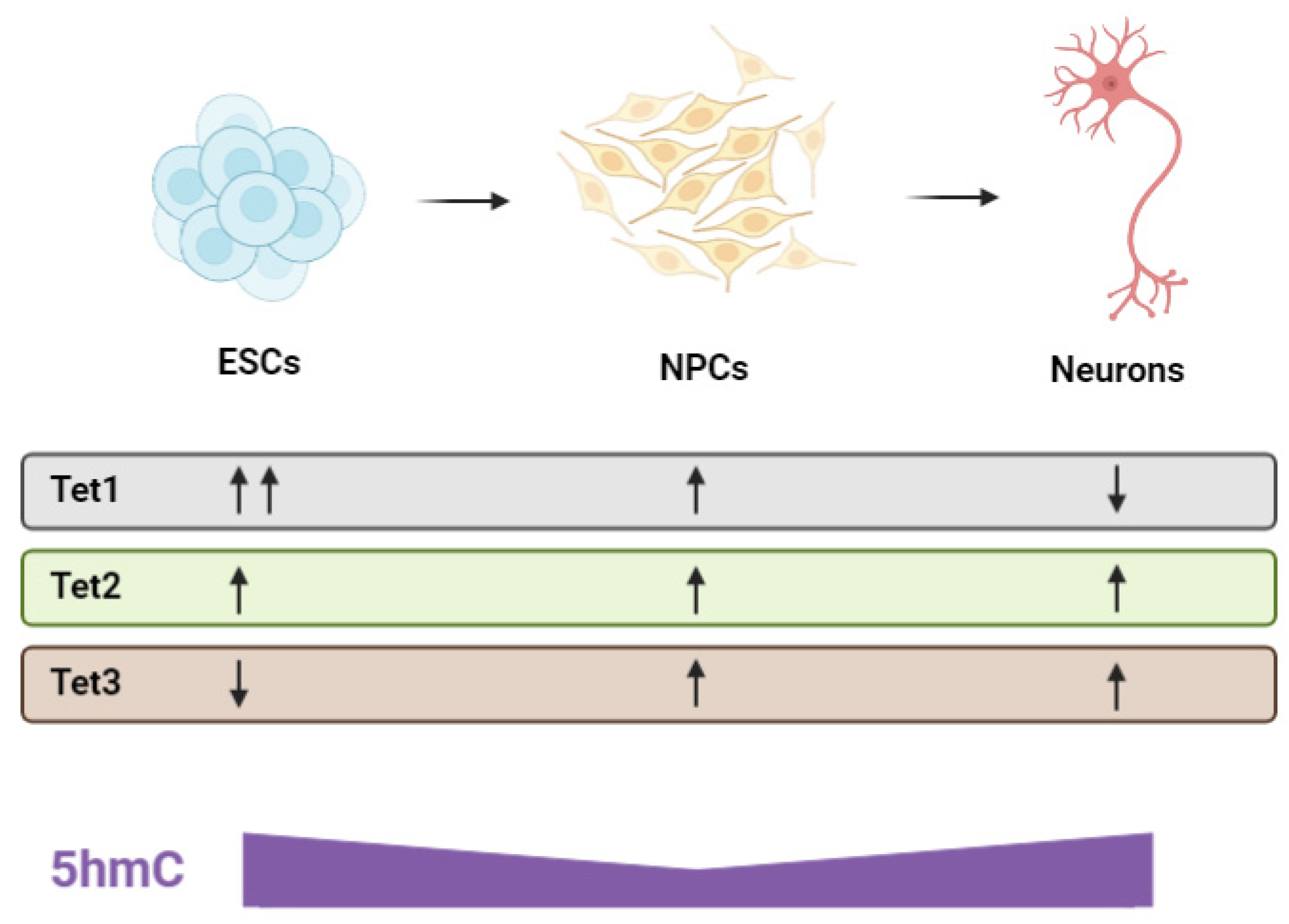
- Tan, L.; Xiong, L.; Xu, W.; Wu, F.; Huang, N.; Xu, Y.; Kong, L.; Zheng, L.; Schwartz, L.; Shi, Y.; et al. Genome-Wide Comparison of DNA Hydroxymethylation in Mouse Embryonic Stem Cells and Neural Progenitor Cells by a New Comparative HMeDIP-Seq Method. Nucleic Acids Res. 2013, 41, e84. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.; Antunes, C.; Guedes, M.; Sousa, N.; Marques, C.J. TET Enzymes and DNA Hydroxymethylation in Neural Development and Function—How Critical Are They? Genomics 2014, 104, 334–340. [Google Scholar] [CrossRef]
- Hon, G.C.; Song, C.X.; Du, T.; Jin, F.; Selvaraj, S.; Lee, A.Y.; Yen, C.A.; Ye, Z.; Mao, S.Q.; Wang, B.A.; et al. 5mC Oxidation by Tet2 Modulates Enhancer Activity and Timing of Transcriptome Reprogramming during Differentiation. Mol. Cell 2014, 56. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration during Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Li, T.; Yang, D.; Li, J.; Tang, Y.; Yang, J.; Le, W. Critical Role of Tet3 in Neural Progenitor Cell Maintenance and Terminal Differentiation. Mol. Neurobiol. 2015, 51, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Chang, M.-Y.; Park, C.-H.; Kim, H.-Y.; Kim, J.-H.; Son, H.; Lee, Y.-S.; Lee, S.-H. Ascorbate-Induced Differentiation of Embryonic Cortical Precursors into Neurons and Astrocytes. J. Neurosci. Res. 2003, 73, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-M.; Ahn, J.-I.; Lee, K.-H.; Lee, Y.-S.; Lee, Y.-S. Ascorbic Acid Responsive Genes during Neuronal Differentiation of Embryonic Stem Cells. NeuroReport 2004, 15, 1959–1963. [Google Scholar] [CrossRef]
- Yao, B.; Christian, K.M.; He, C.; Jin, P.; Ming, G.; Song, H. Epigenetic Mechanisms in Neurogenesis. Nat. Rev. Neurosci. 2016, 17, 537–549. [Google Scholar] [CrossRef]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.-G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-Hydroxymethylcytosine and Chromatin Marks in Mammalian Neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef]
- Li, X.; Yao, B.; Chen, L.; Kang, Y.; Li, Y.; Cheng, Y.; Li, L.; Lin, L.; Wang, Z.; Wang, M.; et al. Ten-Eleven Translocation 2 Interacts with Forkhead Box O3 and Regulates Adult Neurogenesis. Nat. Commun. 2017, 8, 15903. [Google Scholar] [CrossRef] [PubMed]
- Gontier, G.; Iyer, M.; Shea, J.M.; Bieri, G.; Wheatley, E.G.; Ramalho-Santos, M.; Villeda, S.A. Tet2 Rescues Age-Related Regenerative Decline and Enhances Cognitive Function in the Adult Mouse Brain. Cell Rep. 2018, 22, 1974–1981. [Google Scholar] [CrossRef]
- Zhang, R.-R.; Cui, Q.-Y.; Murai, K.; Lim, Y.C.; Smith, Z.D.; Jin, S.; Ye, P.; Rosa, L.; Lee, Y.K.; Wu, H.-P.; et al. Tet1 Regulates Adult Hippocampal Neurogenesis and Cognition. Cell Stem Cell 2013, 13, 237–245. [Google Scholar] [CrossRef]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.H. Tet1 Is Critical for Neuronal Activity-Regulated Gene Expression and Memory Extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef]
- Kang, J.; Lienhard, M.; Pastor, W.A.; Chawla, A.; Novotny, M.; Tsagaratou, A.; Lasken, R.S.; Thompson, E.C.; Azim Surani, M.; Koralov, S.B.; et al. Simultaneous Deletion of the Methylcytosine Oxidases Tet1 and Tet3 Increases Transcriptome Variability in Early Embryogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E4236–E4245. [Google Scholar] [CrossRef]
- Tsukada, Y.; Akiyama, T.; Nakayama, K.I. Maternal TET3 Is Dispensable for Embryonic Development but Is Required for Neonatal Growth. Sci. Rep. 2015, 5, 15876. [Google Scholar] [CrossRef]
- Morrison, J.L.; Botting, K.J.; Darby, J.R.T.; David, A.L.; Dyson, R.M.; Gatford, K.L.; Gray, C.; Herrera, E.A.; Hirst, J.J.; Kim, B.; et al. Guinea Pig Models for Translation of the Developmental Origins of Health and Disease Hypothesis into the Clinic. J. Physiol. 2018, 596, 5535–5569. [Google Scholar] [CrossRef] [PubMed]
- Barra, R.; Morgan, C.; Saez-Briones, P.; Reyes-Parada, M.; Burgos, H.; Morales, B.; Hernandez, A. Facts and Hypotheses about the Programming of Neuroplastic Deficits by Prenatal Malnutrition. Nutr. Rev. 2019, 77, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.P.; Cooke, S.F. Long-Term Potentiation and Long-Term Depression: A Clinical Perspective. Clinics 2011, 66, 3–17. [Google Scholar] [CrossRef]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.-L.; King, J.R.; Song, H.; Sweatt, J.D. TET1 Controls CNS 5-Methylcytosine Hydroxylation, Active DNA Demethylation, Gene Transcription, and Memory Formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Su, Y.; Shin, J.; Zhong, C.; Guo, J.U.; Weng, Y.L.; Gao, F.; Geschwind, D.H.; Coppola, G.; Ming, G.L.; et al. Tet3 Regulates Synaptic Transmission and Homeostatic Plasticity via DNA Oxidation and Repair. Nat. Neurosci. 2015, 18, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-Methylcytosine by TET1 Promotes Active DNA Demethylation in the Adult Brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, W.; Zhao, Q.Y.; Widagdo, J.; Baker-Andresen, D.; Flavell, C.R.; D’Alessio, A.; Zhang, Y.; Bredy, T.W. Neocortical Tet3-Mediated Accumulation of 5-Hydroxymethylcytosine Promotes Rapid Behavioral Adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, 7120–7125. [Google Scholar] [CrossRef]
- MacArthur, I.C.; Dawlaty, M.M. TET Enzymes and 5-Hydroxymethylcytosine in Neural Progenitor Cell Biology and Neurodevelopment. Front. Cell Dev. Biol. 2021, 9, 645335. [Google Scholar] [CrossRef]
- Eldridge, C.F.; Bunge, M.B.; Bunge, R.P.; Wood, P.M. Differentiation of Axon-Related Schwann Cells in Vitro. I. Ascorbic Acid Regulates Basal Lamina Assembly and Myelin Formation. J. Cell Biol. 1987, 105, 1023–1034. [Google Scholar] [CrossRef]
- Guo, Y.E.; Suo, N.; Cui, X.; Yuan, Q.; Xie, X. Vitamin C Promotes Oligodendrocytes Generation and Remyelination. GLIA 2018, 66, 1302–1316. [Google Scholar] [CrossRef]
- Gess, B.; Lohmann, C.; Halfter, H.; Young, P. Sodium-Dependent Vitamin C Transporter 2 (SVCT2) Is Necessary for the Uptake of L-Ascorbic Acid into Schwann Cells. GLIA 2010, 58, 287–299. [Google Scholar] [CrossRef]
- Gess, B.; Röhr, D.; Fledrich, R.; Sereda, M.W.; Kleffner, I.; Humberg, A.; Nowitzki, J.; Strecker, J.K.; Halfter, H.; Young, P. Sodium-Dependent Vitamin C Transporter 2 Deficiency Causes Hypomyelination and Extracellular Matrix Defects in the Peripheral Nervous System. J. Neurosci. 2011, 31, 17180–17192. [Google Scholar] [CrossRef]
- Huff, T.C.; Sant, D.W.; Camarena, V.; Van Booven, D.; Andrade, N.S.; Mustafi, S.; Monje, P.V.; Wang, G. Vitamin C Regulates Schwann Cell Myelination by Promoting DNA Demethylation of Pro-Myelinating Genes. J. Neurochem. 2021, 157, 1759–1773. [Google Scholar] [CrossRef]
- Beck, D.B.; Petracovici, A.; He, C.; Moore, H.W.; Louie, R.J.; Ansar, M.; Douzgou, S.; Sithambaram, S.; Cottrell, T.; Santos-Cortez, R.L.P.; et al. Delineation of a Human Mendelian Disorder of the DNA Demethylation Machinery: TET3 Deficiency. Am. J. Hum. Genet. 2020, 106, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA Methyltransferase Gene DNMT3A Cause an Overgrowth Syndrome with Intellectual Disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef]
- Lumey, L.H.; Khalangot, M.D.; Vaiserman, A.M. Association between Type 2 Diabetes and Prenatal Exposure to the Ukraine Famine of 1932–33: A Retrospective Cohort Study. Lancet Diabetes Endocrinol. 2015, 3, 787–794. [Google Scholar] [CrossRef]
- Huang, C.; Phillips, M.R.; Zhang, Y.; Zhang, J.; Shi, Q.; Song, Z.; Ding, Z.; Pang, S.; Martorell, R. Malnutrition in Early Life and Adult Mental Health: Evidence from a Natural Experiment. Soc. Sci. Med. 2013, 97, 259–266. [Google Scholar] [CrossRef]
- Roseboom, T.J. Coronary Heart Disease after Prenatal Exposure to the Dutch Famine, 1944–1945. Heart 2000, 84, 595–598. [Google Scholar] [CrossRef]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ Perspective: What Is the Optimum Intake of Vitamin C in Humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Poulsen, H.E. Is Vitamin C Supplementation Beneficial? Lessons Learned from Randomised Controlled Trials. Br. J. Nutr. 2010, 103, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Vissers, M.C.M.; Bozonet, S.M.; Pearson, J.F.; Braithwaite, L.J. Dietary Ascorbate Intake Affects Steady State Tissue Concentrations in Vitamin C–Deficient Mice: Tissue Deficiency after Suboptimal Intake and Superior Bioavailability from a Food Source (Kiwifruit). Am. J. Clin. Nutr. 2011, 93, 292–301. [Google Scholar] [CrossRef]
- Burns, J.J.; Evans, C. The synthesis of L-Ascorbic acid in the rat from D-Glucuronolactone and L-Gulonolactone. J. Biol. Chem. 1956, 223, 897–905. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic Vitamin C Deficiency Does Not Accelerate Oxidative Stress in Ageing Brains of Guinea Pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.F.; Taggart, M.J. Are Animal Models Relevant to Key Aspects of Human Parturition? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R525–R545. [Google Scholar] [CrossRef]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef]
- Mess, A. The Guinea Pig Placenta: Model of Placental Growth Dynamics. Placenta 2007, 28, 812–815. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Growth and Development of the Brain and Spinal Cord of the Guinea Pig. Brain Res. 1970, 17, 115–123. [Google Scholar] [CrossRef]
- Mortensen, A.; Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Guinea Pig Ascorbate Status Predicts Tetrahydrobiopterin Plasma Concentration and Oxidation Ratio in Vivo. Nutr. Res. 2013, 33, 859–867. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Effect of Vitamin C | Role in Development |
|---|---|---|
| TET1 | Increased activity in vitro [102,112] and in cell culture [101,102,114,115,116,117] | Highly expressed in mouse embryonic stem cells [93,118]. Expressed in the developing brain [119]. High expression in the early post-implantation stage, regulates differentiation in the extraembryonic ectoderm and epiblast [119]. |
| TET2 | Increased activity in vitro [101,112], in cell culture [101,102,114,117,120], in mice [121,122], and in a clinical setting [123] | Regulation of haematopoiesis, mutations are a driver of leukaemia and other haematopoietic pathologies [95,121,124,125,126]. Expressed in the developing brain [119]. |
| TET3 | Increased activity in vitro [112], in cell culture [114,117], and mice [121] | Indispensable for proper development as knockout results in embryonic lethality [127]. Mediates cell-fate decisions by inhibiting Wnt signaling, regulates neural and mesodermal cell fate [128]. Highly expressed in oocytes and zygotes [127,129,130]. Involved in the demethylation of zygotic paternal DNA following fertilization [127]. Expressed in the developing brain [119]. |
| Enzyme | Roles in Neurodevelopment and Brain Function |
|---|---|
| TET1 | Maintains the pluripotent state of ESCs [93], promotes NPC proliferation and maintenance [185]. Expression becomes downregulated in response to neuronal firing [185,186,192]. Knockout leads to impairments in spatial and short-term memory [185], defective fear memory extinction and enhances LTD [186]. |
| TET2 | Enhances and skews ESC differentiation toward to the neural lineage [176], controls NPC proliferation and promotes terminal neuronal differentiation [183,184]. Combined loss with Tet1 can cause exencephaly [135]. |
| TET3 | Preserves the partially-differentiated identity of NPCs [111] and promotes terminal neuronal differentiation [178]. Expression becomes upregulated in response to neuronal firing [193,195]. Combined loss with Tet1 can cause holoprosencephaly [187]. Deficiency in TET3 function has been linked to a familial intellectual disability disorder in humans [202]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coker, S.J.; Smith-Díaz, C.C.; Dyson, R.M.; Vissers, M.C.M.; Berry, M.J. The Epigenetic Role of Vitamin C in Neurodevelopment. Int. J. Mol. Sci. 2022, 23, 1208. https://doi.org/10.3390/ijms23031208
Coker SJ, Smith-Díaz CC, Dyson RM, Vissers MCM, Berry MJ. The Epigenetic Role of Vitamin C in Neurodevelopment. International Journal of Molecular Sciences. 2022; 23(3):1208. https://doi.org/10.3390/ijms23031208
Chicago/Turabian StyleCoker, Sharna J., Carlos C. Smith-Díaz, Rebecca M. Dyson, Margreet C. M. Vissers, and Mary J. Berry. 2022. "The Epigenetic Role of Vitamin C in Neurodevelopment" International Journal of Molecular Sciences 23, no. 3: 1208. https://doi.org/10.3390/ijms23031208
APA StyleCoker, S. J., Smith-Díaz, C. C., Dyson, R. M., Vissers, M. C. M., & Berry, M. J. (2022). The Epigenetic Role of Vitamin C in Neurodevelopment. International Journal of Molecular Sciences, 23(3), 1208. https://doi.org/10.3390/ijms23031208