
Sesquiterpene Lactones Potentiate Olaparib-Induced DNA Damage in p53 Wildtype Cancer Cells



Abstract
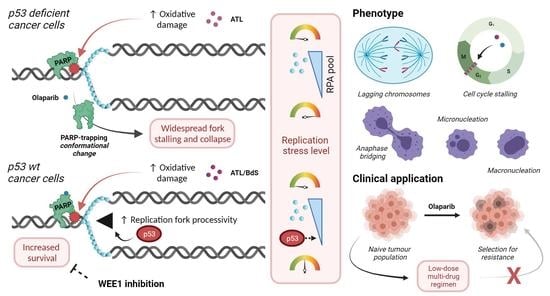
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
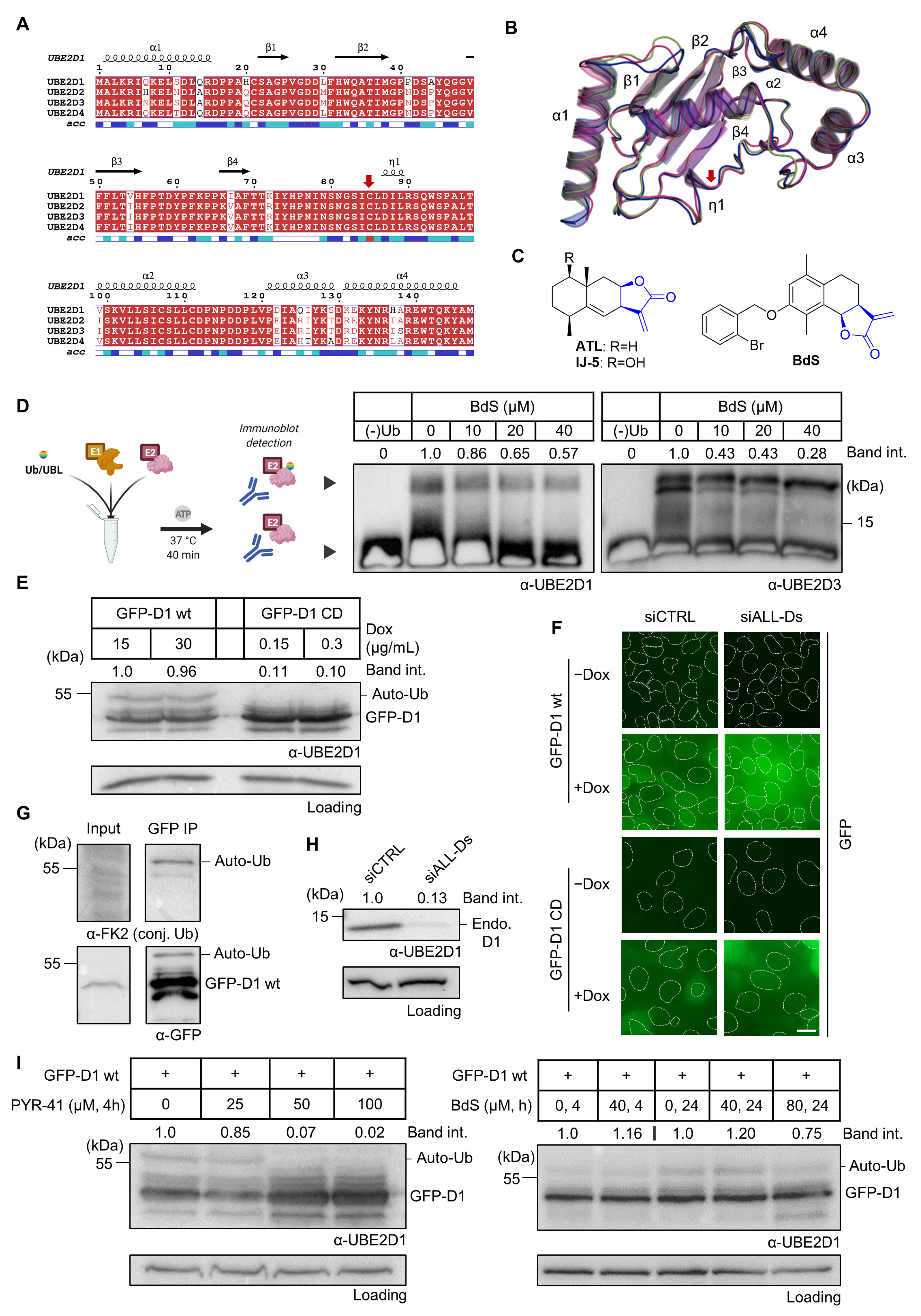
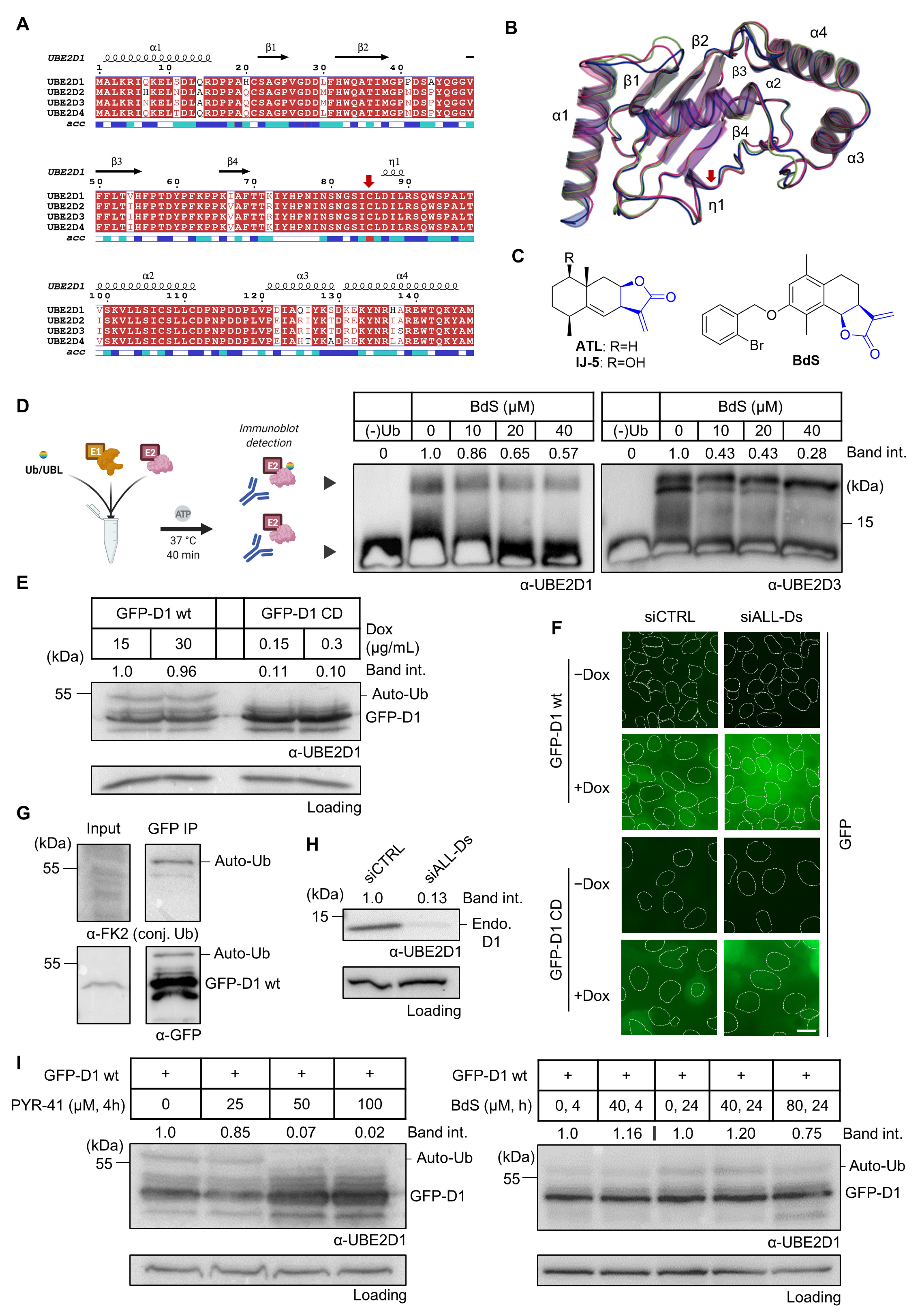
2.1. BdS Activity Is Not Mediated by Cellular Inhibition of UBE2D Enzymes
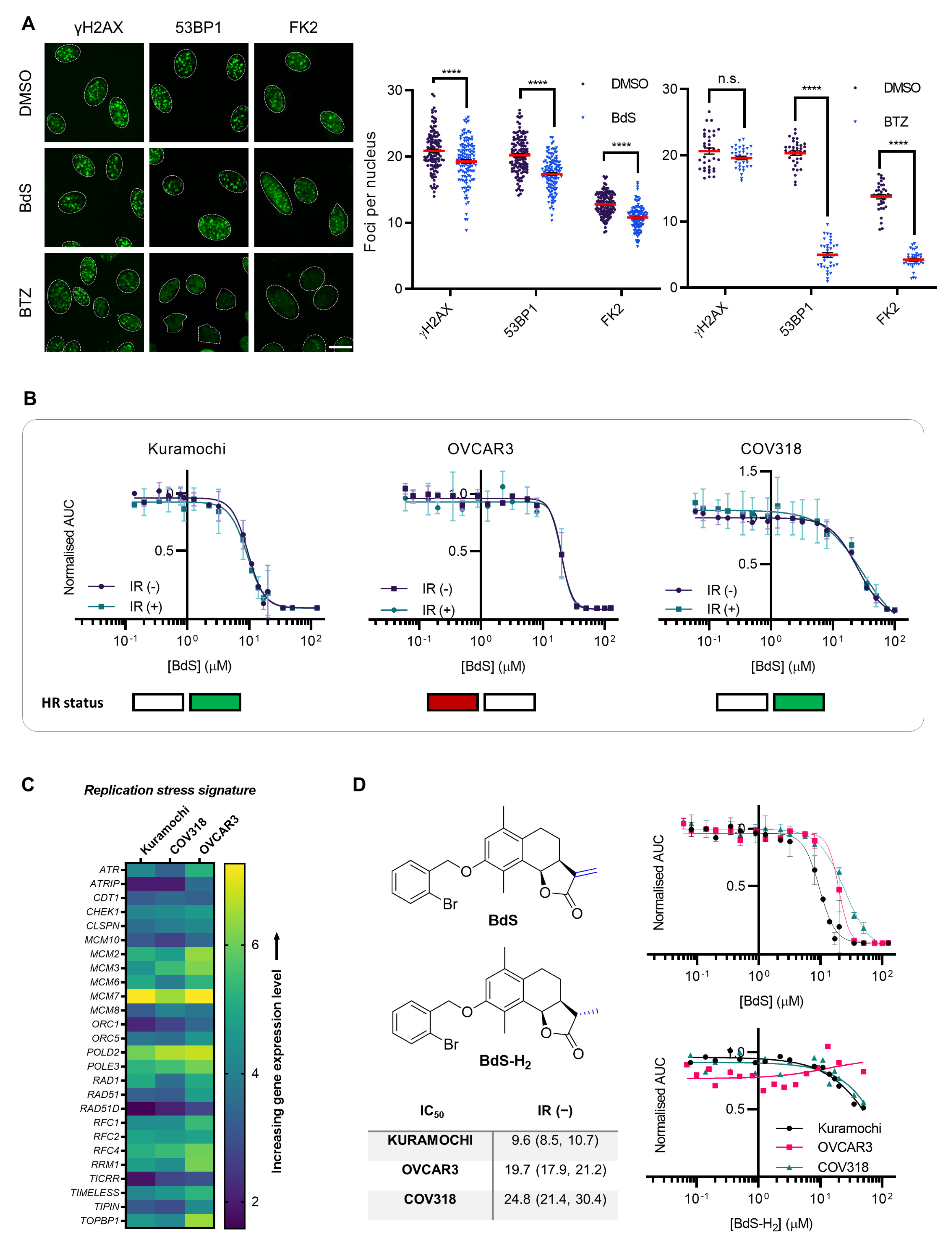
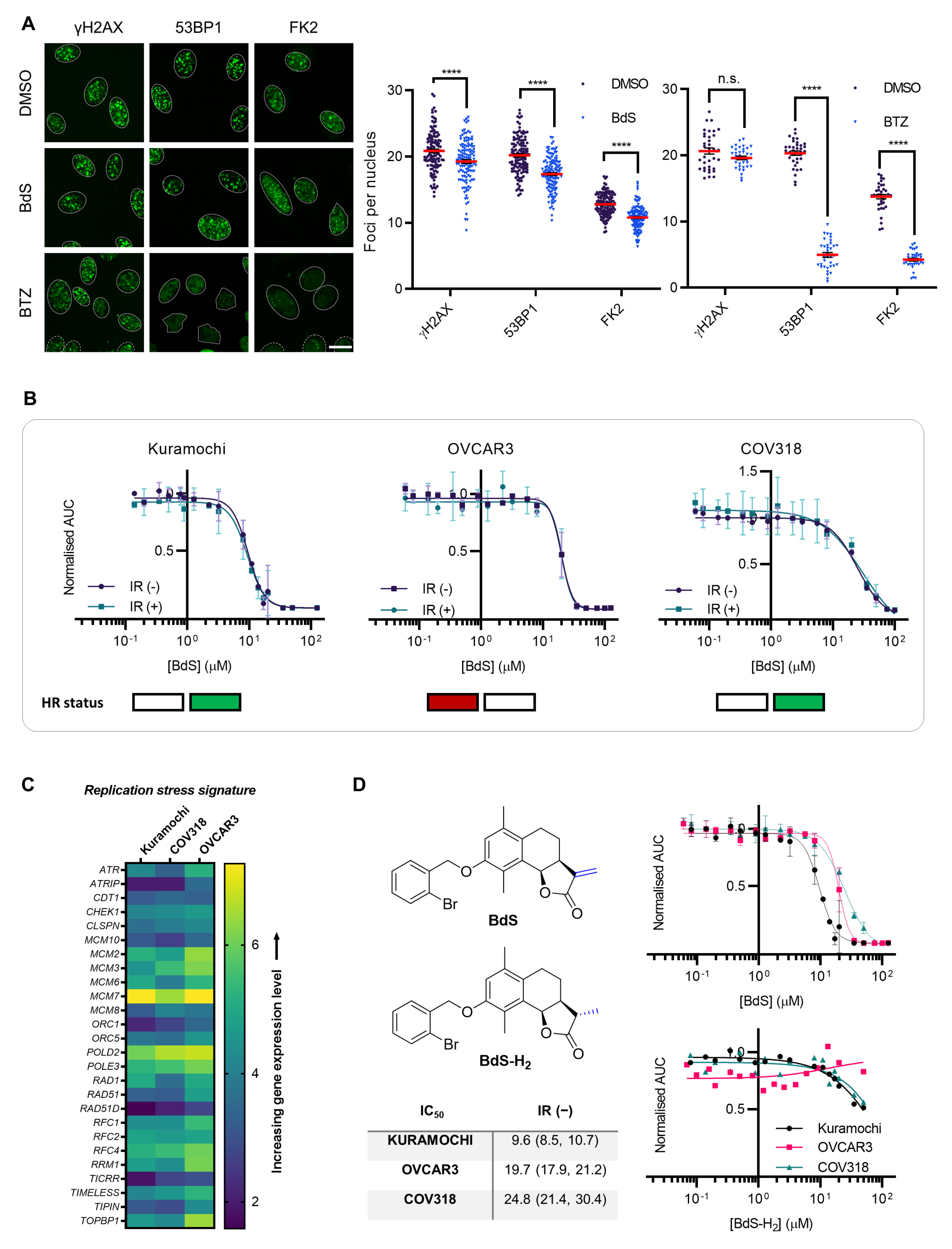
2.2. Homologous Recombination Status Does Not Dictate BdS Growth Inhibition
2.3. Sensitivity of Ovarian Cancer Cell Lines to BdS Correlates with DNA Replication Stress Profiles
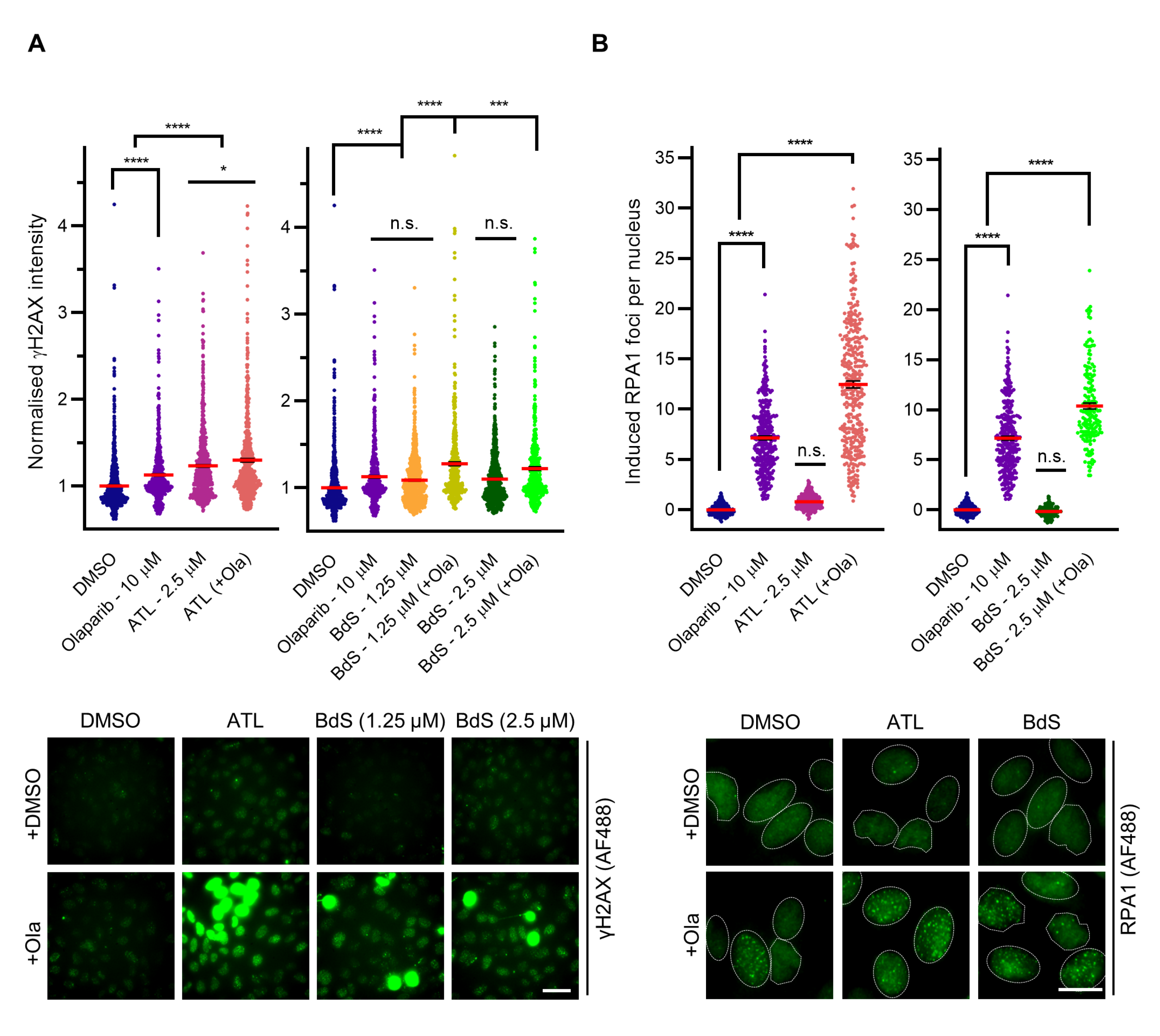
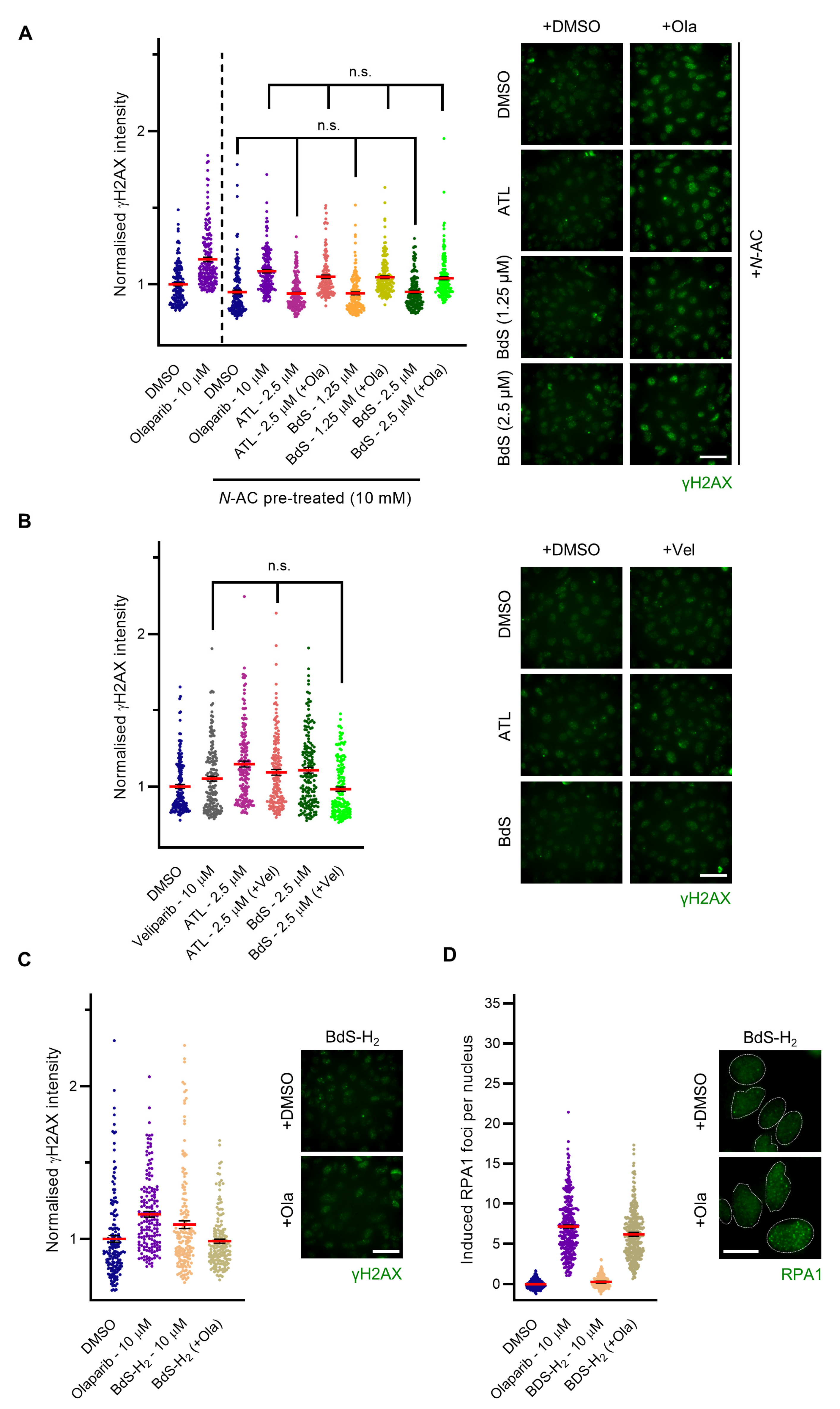
2.4. BdS Synergises with Olaparib to Induce DNA Damage in p53 Wildtype Cancer Cells
2.5. BdS Exacerbates Olaparib-Mediated PARP-Trapping in a Michael Acceptor-Dependent Manner
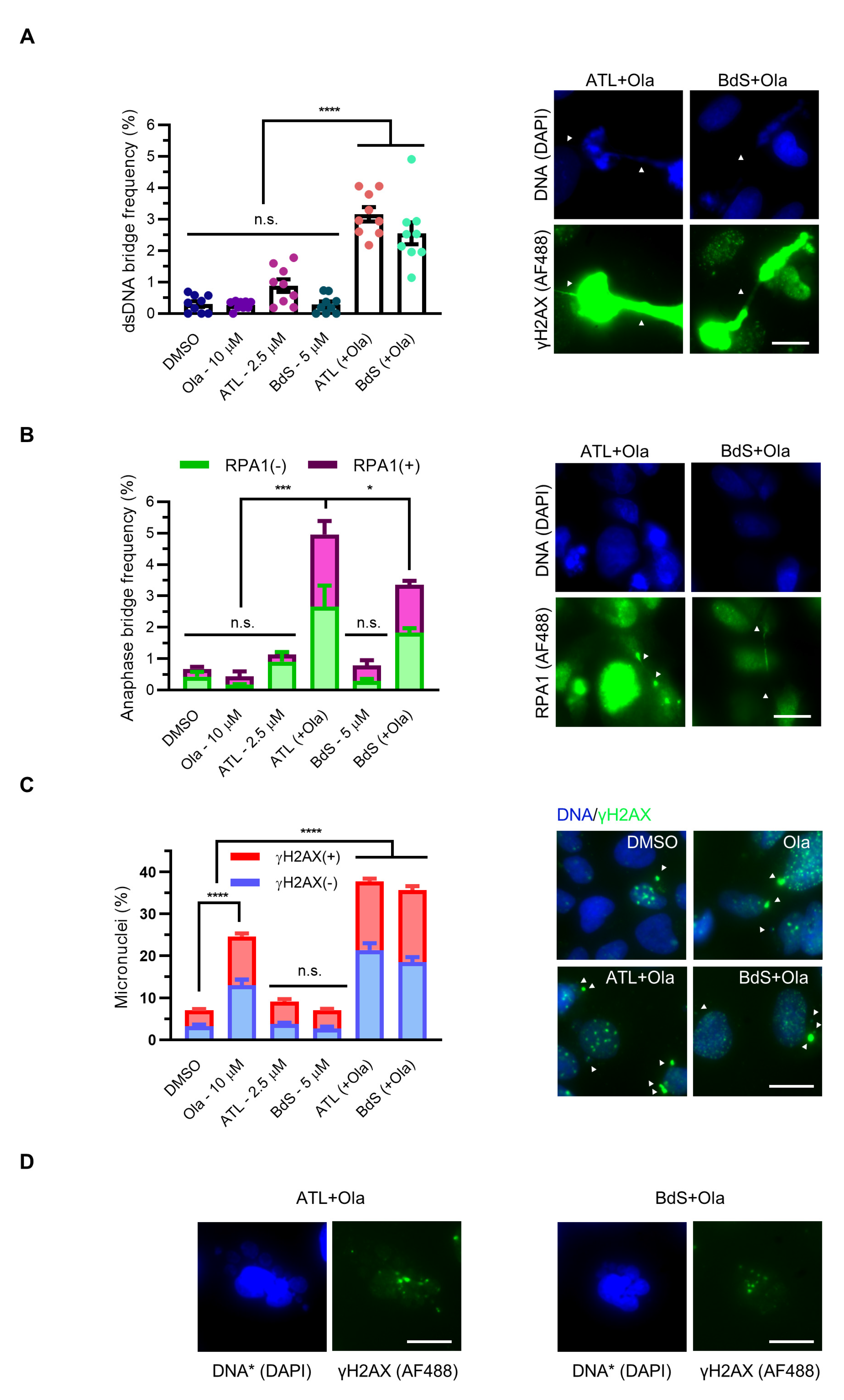
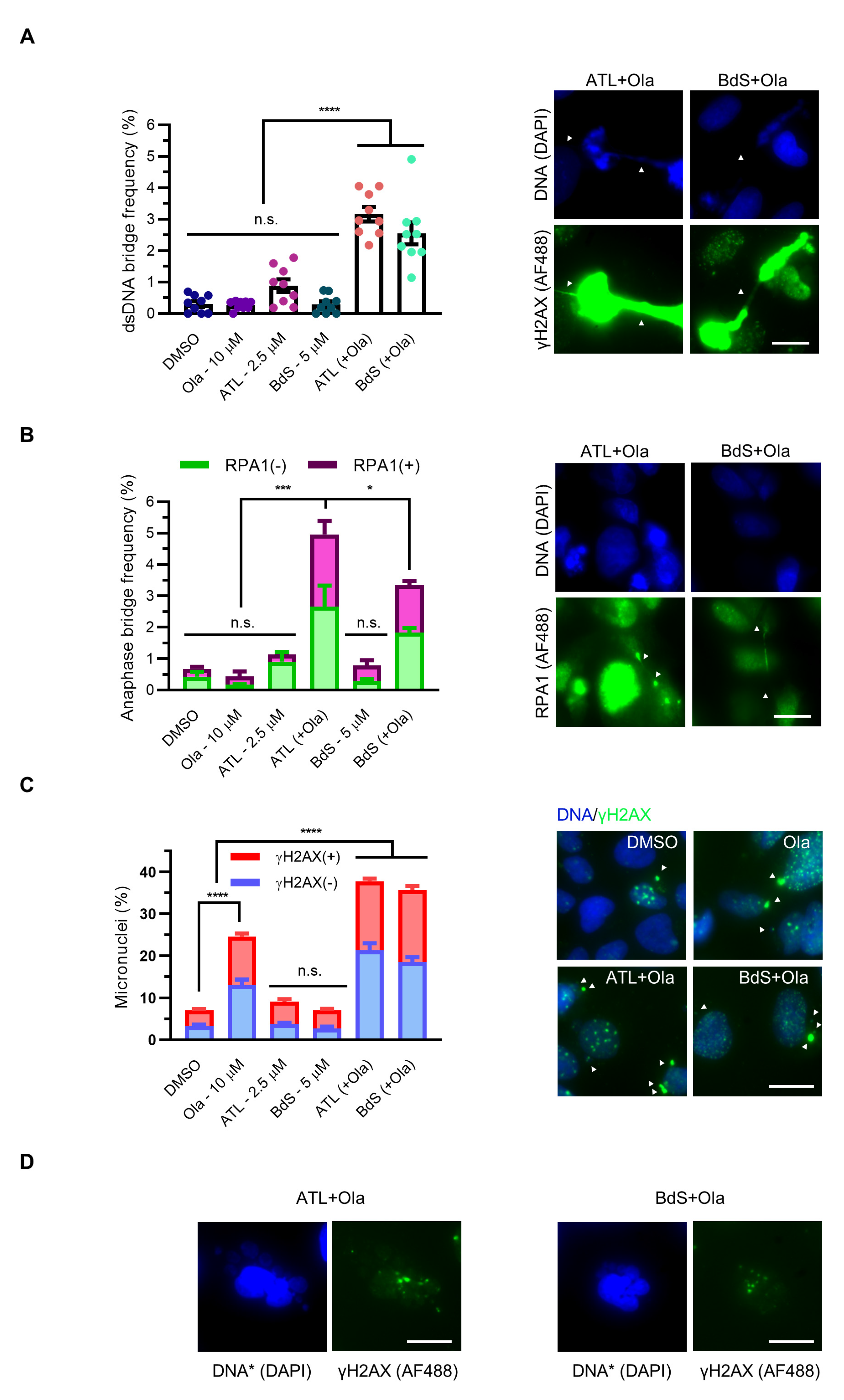
2.6. Combination Treatment of BdS or ATL with Olaparib Induces Pleiotropic Mitotic Defects
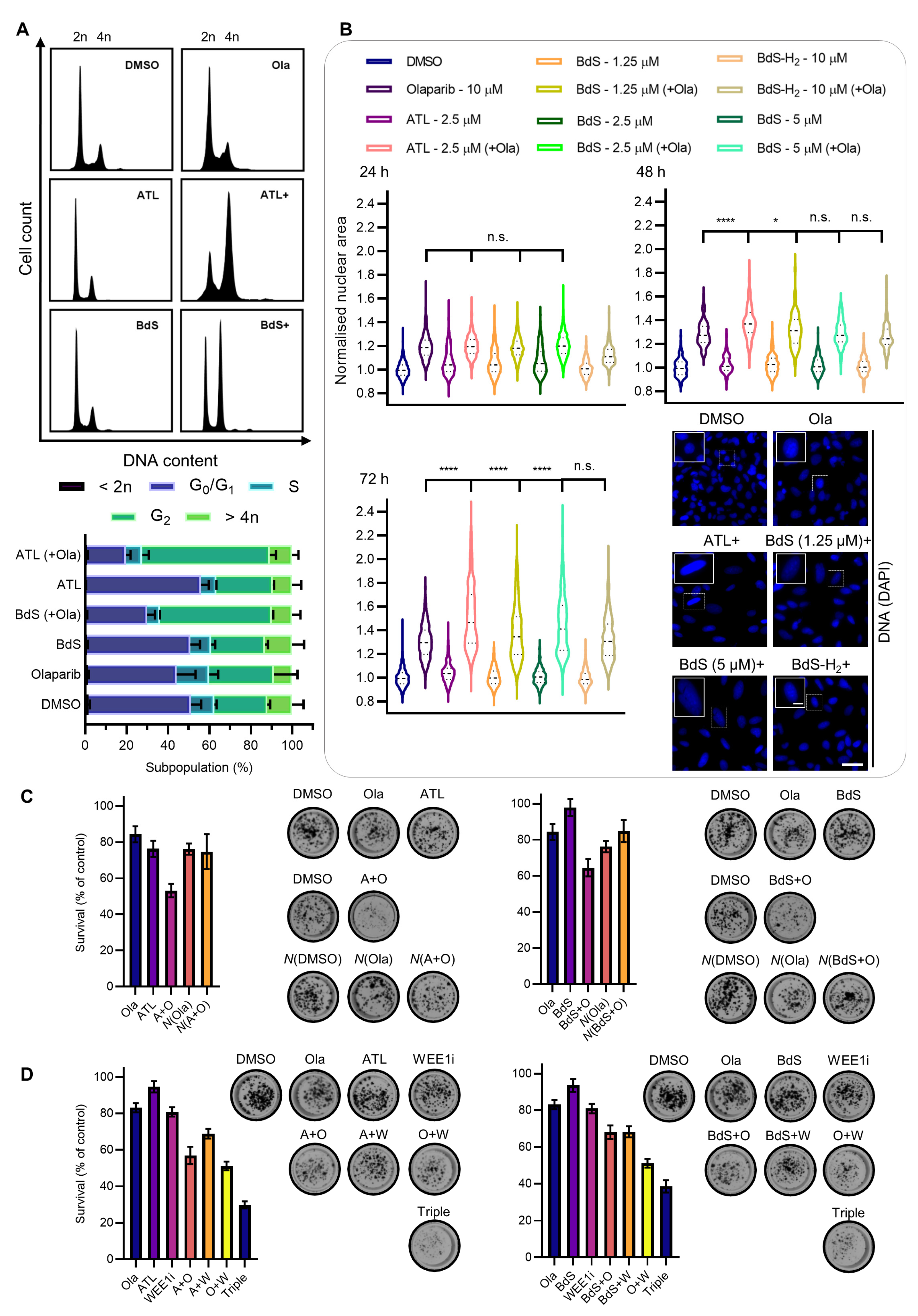
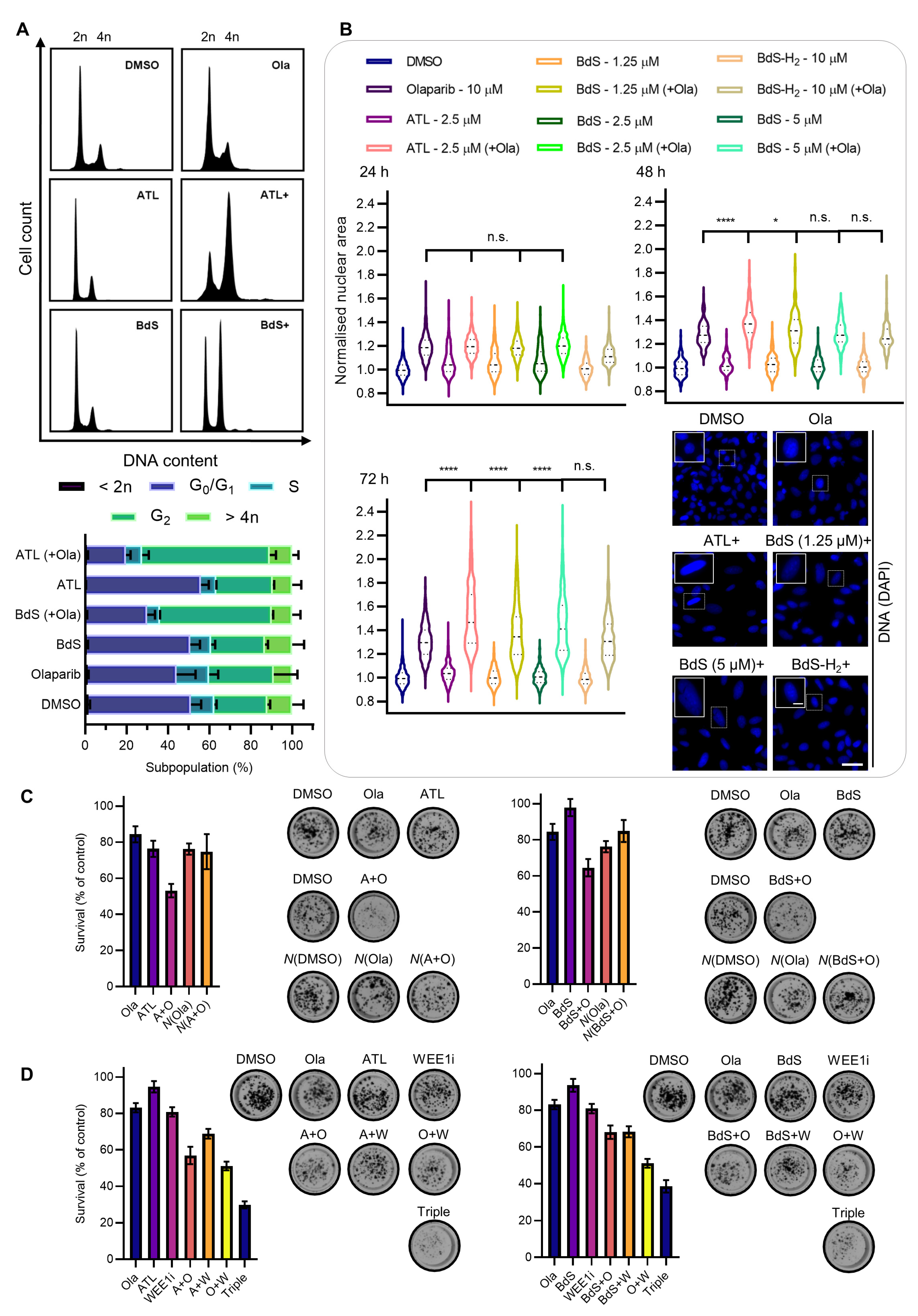
2.7. BdS or ATL Combination Treatment with Olaparib Results in G2 Cell-Cycle Stalling
2.8. BdS/ATL-Enhanced Olaparib-Mediated Cell Death in p53 wt Cancer Cells Is Further Potentiated by WEE1 Inhibition
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
- (3R,3aR,5aS,9bS)-3,5a,9-Trimethyl-3-(phenylselenyl)3a,5,5a,9btetrahydronaphtho[1,2-b]furan-2,8(3H,4H)-dione (2)
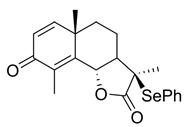
- (3aS,5aS,9bS)-5a,9-Dimethyl-3-methylene-3a,5,5a,9b-tetrahydronaphtho[1,2-b]furan-2,8(3H,4H)-dione (3)
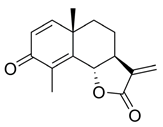
- (3aS,9bR)-6,9-Dimethyl-3-methylene-2-oxo-2,3,3a,4,5,9b-hexahydronaphtho[1,2-b]furan-8-yl acetate (4)

- (3aS,9bR)-8-Hydroxy-6,9-dimethyl-3-methylene-3a,4,5,9btetrahydronaphtho[1,2-b]furan-2(3H)-one (5)

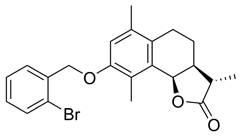
- (3aS,9bR)-8-((2-Bromobenzyl)oxy)-6,9-dimethyl-3-methylene3a,4,5,9b-tetrahydronaphtho[1,2-b] furan-2(3H)-one (BdS, 6)
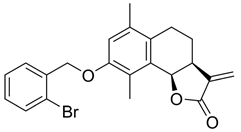
- (3S,3aS,9bR)-3,6,9-Trimethyl-2-oxo-2,3,3a,4,5,9b-hexahydronaphtho[1,2-b]furan-8-yl acetate (7)
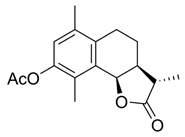
- (3S,3aS,9bR)-8-Hydroxy-3,6,9-trimethyl-3a,4,5,9b-tetrahydronaphtho[1,2-b]furan-2(3H)-one (8)
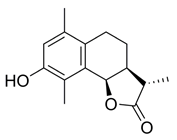
- (3S,3aS,9bR)-8-((2-Bromobenzyl)oxy)-3,6,9-trimethyl-3a,4,5,9b-tetrahydronaphtho[1,2-b]furan-2(3H)-one (BdS-H2, 9)
4.2. Biology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG Inhibitors in Cancer Treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Her, J.; Bunting, S.F. How Cells Ensure Correct Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [Green Version]
- Slade, D. Mitotic Functions of Poly(ADP-Ribose) Polymerases. Biochem. Pharmacol. 2019, 167, 33–43. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Shi, L.Z.; Wong, C.C.L.; Han, X.; Hwang, P.Y.-H.; Truong, L.N.; Zhu, Q.; Shao, Z.; Chen, D.J.; Berns, M.W.; et al. The Interaction of CtIP and Nbs1 Connects CDK and ATM to Regulate HR–Mediated Double-Strand Break Repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ding, Q.; Fujimori, H.; Motegi, A.; Miki, Y.; Masutani, M. Loss of CtIP Disturbs Homologous Recombination Repair and Sensitizes Breast Cancer Cells to PARP Inhibitors. Oncotarget 2016, 7, 7701–7714. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.K.; Galanty, Y.; Sczaniecka-Clift, M.; Coates, J.; Jhujh, S.; Demir, M.; Cornwell, M.; Beli, P.; Jackson, S.P. Systematic E2 Screening Reveals a UBE2D-RNF138-CtIP Axis Promoting DNA Repair. Nat. Cell Biol. 2015, 17, 1458–1470. [Google Scholar] [CrossRef]
- Jonuscheit, S.; Jost, T.; Gajdošová, F.; Wrobel, M.; Hecht, M.; Fietkau, R.; Distel, L. PARP Inhibitors Talazoparib and Niraparib Sensitize Melanoma Cells to Ionizing Radiation. Genes 2021, 12, 849. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, S.; Song, L.; Qu, M.; Zou, Z. Synergistic Lethality between PARP-Trapping and Alantolactone-Induced Oxidative DNA Damage in Homologous Recombination-Proficient Cancer Cells. Oncogene 2020, 39, 2905–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-Based Alignment Tool for Multiple Protein Sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.G.; Wagner, J.M.; Pornillos, O. RCSB PDB—5TUT: UbcH5a-Ub Isopeptide Conjugate. Available online: https://www.rcsb.org/structure/5TUT (accessed on 19 October 2021).
- Ozkan, E.; Yu, H.; Deisenhofer, J. Mechanistic Insight into the Allosteric Activation of a Ubiquitin-Conjugating Enzyme by RING-Type Ubiquitin Ligases. Proc. Natl. Acad. Sci. USA 2005, 102, 18890–18895. [Google Scholar] [CrossRef] [Green Version]
- Benirschke, R.C.; Thompson, J.R.; Nominé, Y.; Wasielewski, E.; Juranić, N.; Macura, S.; Hatakeyama, S.; Nakayama, K.I.; Botuyan, M.V.; Mer, G. Molecular Basis for the Association of Human E4B U Box Ubiquitin Ligase with E2-Conjugating Enzymes UbcH5c and Ubc4. Structure 2010, 18, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Hua, Y.; Wang, D.; Shan, L.; Zhang, Y.; Zhu, J.; Jin, H.; Li, H.; Hu, Z.; Zhang, W. A Sesquiterpene Lactone from a Medicinal Herb Inhibits Proinflammatory Activity of TNF-α by Inhibiting Ubiquitin-Conjugating Enzyme UbcH5. Chem. Biol. 2014, 21, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wu, G.; Gao, S.; Guo, R.; Zhao, Z.; Yuan, H.; Liu, S.; Wu, J.; Lu, X.; Yuan, X.; et al. Discovery of Potent Small-Molecule Inhibitors of Ubiquitin-Conjugating Enzyme UbcH5c from α-Santonin Derivatives. J. Med. Chem. 2017, 60, 6828–6852. [Google Scholar] [CrossRef]
- Gao, F.-H.; Liu, F.; Wei, W.; Liu, L.-B.; Xu, M.-H.; Guo, Z.-Y.; Li, W.; Jiang, B.; Wu, Y.-L. Oridonin Induces Apoptosis and Senescence by Increasing Hydrogen Peroxide and Glutathione Depletion in Colorectal Cancer Cells. Int. J. Mol. Med. 2012, 29, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.V.; Suman, S.; Das, T.P.; Luevano, J.E.; Damodaran, C. Withaferin A, a Steroidal Lactone from Withania Somnifera, Induces Mitotic Catastrophe and Growth Arrest in Prostate Cancer Cells. J. Nat. Prod. 2013, 76, 1909–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fofaria, N.M.; Kim, S.-H.; Srivastava, S.K. Piperine Causes G1 Phase Cell Cycle Arrest and Apoptosis in Melanoma Cells through Checkpoint Kinase-1 Activation. PLoS ONE 2014, 9, e94298. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Levine, D.A. New Insights into PARP Inhibitors’ Effect on Cell Cycle and Homology-Directed DNA Damage Repair. Mol. Cancer Ther. 2014, 13, 1645–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Rape, M. Building Ubiquitin Chains: E2 Enzymes at Work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, H.C.; Irving, E.; Forment, J.V.; Schmidt, C.K. E2 Enzymes in Genome Stability: Pulling the Strings behind the Scenes. Trends Cell Biol. 2021, 31, 628–643. [Google Scholar] [CrossRef]
- Xu, Y.; Meng, X. Molecular Simulation Elaborating the Mechanism of 1β-Hydroxy Alantolactone Inhibiting Ubiquitin-Conjugating Enzyme UbcH5s. Sci. Rep. 2020, 10, 141. [Google Scholar] [CrossRef]
- Moudry, P.; Lukas, C.; Macurek, L.; Hanzlikova, H.; Hodny, Z.; Lukas, J.; Bartek, J. Ubiquitin-Activating Enzyme UBA1 Is Required for Cellular Response to DNA Damage. Cell Cycle 2012, 11, 1573–1582. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Sharma Saha, S.; Gentles, L.; Bradbury, A.; Brecht, D.; Robinson, R.; O’Donnell, R.; Curtin, N.J.; Drew, Y. Genomic, Transcriptomic, and Functional Alterations in DNA Damage Response Pathways as Putative Biomarkers of Chemotherapy Response in Ovarian Cancer. Cancers 2021, 13, 1420. [Google Scholar] [CrossRef]
- Bradbury, A.; O’Donnell, R.; Drew, Y.; Curtin, N.J.; Sharma Saha, S. Characterisation of Ovarian Cancer Cell Line NIH-OVCAR3 and Implications of Genomic, Transcriptomic, Proteomic and Functional DNA Damage Response Biomarkers for Therapeutic Targeting. Cancers 2020, 12, 1939. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Gilmer, C.; Morgan, R.D.; Nelson, L.; Barnes, B.M.; Tighe, A.; Wardenaar, R.; Spierings, D.C.J.; Schlecht, H.; Burghel, G.J.; Foijer, F.; et al. Replication Catastrophe Is Responsible for Intrinsic PAR Glycohydrolase Inhibitor-Sensitivity in Patient-Derived Ovarian Cancer Models. J. Exp. Clin. Cancer Res. 2021, 40, 323. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating Cell Lines as Tumour Models by Comparison of Genomic Profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Pillay, N.; Tighe, A.; Nelson, L.; Littler, S.; Coulson-Gilmer, C.; Bah, N.; Golder, A.; Bakker, B.; Spierings, D.C.J.; James, D.I.; et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.-J.; He, Q.-R.; Dong, S.; Guo, X.; Wang, Y.-G.; Lei, B.-L.; Tian, J.-M.; Gao, J.-M. Diversity Modification and Structure-Activity Relationships of Two Natural Products 1β-Hydroxy Alantolactone and Ivangustin as Potent Cytotoxic Agents. Sci. Rep. 2018, 8, 1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, P.; Xia, Y.; He, W.; Zhang, T.; Hong, L.; Zheng, P.; Shen, X.; Liang, G.; Cui, R.; Zou, P. Enhancement of Oxaliplatin-Induced Colon Cancer Cell Apoptosis by Alantolactone, a Natural Product Inducer of ROS. Int. J. Biol. Sci. 2019, 15, 1676–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Li, P.; Ye, X.; Zhang, F.; Lin, Q.; Wu, K.; Chen, W. Isoalantolactone Increases the Sensitivity of Prostate Cancer Cells to Cisplatin Treatment by Inducing Oxidative Stress. Front. Cell Dev. Biol. 2021, 9, 632779. [Google Scholar] [CrossRef]
- Ji, D.; Zhong, X.; Huang, P.; Kang, P.; Leng, K.; Zheng, W.; Wang, Z.; Xu, Y.; Cui, Y. Deoxyelephantopin Induces Apoptosis via Oxidative Stress and Enhances Gemcitabine Sensitivity in Vitro and in Vivo through Targeting the NF-ΚB Signaling Pathway in Pancreatic Cancer. Aging 2020, 12, 11116–11138. [Google Scholar] [CrossRef]
- Hong, L.; Chen, J.; Wu, F.; Wu, F.; Shen, X.; Zheng, P.; Shao, R.; Lu, K.; Liu, Z.; Chen, D.; et al. Isodeoxyelephantopin Inactivates Thioredoxin Reductase 1 and Activates ROS-Mediated JNK Signaling Pathway to Exacerbate Cisplatin Effectiveness in Human Colon Cancer Cells. Front. Cell Dev. Biol. 2020, 8, 580517. [Google Scholar] [CrossRef]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 Mutation Status in Human Cancer Cell Lines: A Reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, B.; Miao, L.; Mei, Y.; Wu, M. Mutant P53-R273H Gains New Function in Sustained Activation of EGFR Signaling via Suppressing MiR-27a Expression. Cell Death Dis. 2013, 4, e574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, N.; Wang, Y.; Guo, S.; Ou, Y.; Wang, G.; Chen, J.; Li, D.; Zhan, Q. Mutant TP53 G245C and R273H Promote Cellular Malignancy in Esophageal Squamous Cell Carcinoma. BMC Cell Biol. 2018, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Ellison, V.; Annor, G.K.; Freedman, C.; Xiao, G.; Lundine, D.; Freulich, E.; Prives, C.; Bargonetti, J. Frame-Shift Mediated Reduction of Gain-of-Function P53 R273H and Deletion of the R273H C-Terminus in Breast Cancer Cells Result in Replication-Stress Sensitivity. Oncotarget 2021, 12, 1128–1146. [Google Scholar] [CrossRef]
- Klusmann, I.; Rodewald, S.; Müller, L.; Friedrich, M.; Wienken, M.; Li, Y.; Schulz-Heddergott, R.; Dobbelstein, M. P53 Activity Results in DNA Replication Fork Processivity. Cell Rep. 2016, 17, 1845–1857. [Google Scholar] [CrossRef] [Green Version]
- Pappas, K.; Xu, J.; Zairis, S.; Resnick-Silverman, L.; Abate, F.; Steinbach, N.; Ozturk, S.; Saal, L.H.; Su, T.; Cheung, P.; et al. P53 Maintains Baseline Expression of Multiple Tumor Suppressor Genes. Mol. Cancer Res. 2017, 15, 1051–1062. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Feng, H.; Sun, W.; Liu, K.; Lu, J.-J.; Chen, X. Tert-Butyl Hydroperoxide (t-BHP) Induced Apoptosis and Necroptosis in Endothelial Cells: Roles of NOX4 and Mitochondrion. Redox Biol. 2017, 11, 524–534. [Google Scholar] [CrossRef]
- Lukášová, E.; Řezáčová, M.; Bačíková, A.; Šebejová, L.; Vávrová, J.; Kozubek, S. Distinct Cellular Responses to Replication Stress Leading to Apoptosis or Senescence. FEBS Open Bio 2019, 9, 870–890. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved Recombination Intermediates Lead to Ultra-Fine Anaphase Bridges, Chromosome Breaks and Aberrations. Nat. Cell Biol. 2018, 20, 92–103. [Google Scholar] [CrossRef]
- Xu, B.; Sun, Z.; Liu, Z.; Guo, H.; Liu, Q.; Jiang, H.; Zou, Y.; Gong, Y.; Tischfield, J.A.; Shao, C. Replication Stress Induces Micronuclei Comprising of Aggregated DNA Double-Strand Breaks. PLoS ONE 2011, 6, e18618. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Wang, W.; Guo, H.; Sun, Z.; Wei, Z.; Zhang, X.; Liu, Z.; Tischfield, J.A.; Gong, Y.; Shao, C. Oxidative Stress Preferentially Induces a Subtype of Micronuclei and Mediates the Genomic Instability Caused by P53 Dysfunction. Mutat. Res. 2014, 770, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbreit, N.T.; Zhang, C.-Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms Generating Cancer Genome Complexity from a Single Cell Division Error. Science 2020, 368, 6488. [Google Scholar] [CrossRef]
- Wilhelm, T.; Olziersky, A.-M.; Harry, D.; De Sousa, F.; Vassal, H.; Eskat, A.; Meraldi, P. Mild Replication Stress Causes Chromosome Mis-Segregation via Premature Centriole Disengagement. Nat. Commun. 2019, 10, 3585. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.; Adams, S.D.; Draviam, V.M. Multinucleation Associated DNA Damage Blocks Proliferation in P53-Compromised Cells. Commun. Biol. 2021, 4, 451. [Google Scholar] [CrossRef] [PubMed]
- Garcia, T.B.; Snedeker, J.C.; Baturin, D.; Gardner, L.; Fosmire, S.P.; Zhou, C.; Jordan, C.T.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. A Small-Molecule Inhibitor of WEE1, AZD1775, Synergizes with Olaparib by Impairing Homologous Recombination and Enhancing DNA Damage and Apoptosis in Acute Leukemia. Mol. Cancer Ther. 2017, 16, 2058–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxid. Med. Cell. Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef]
- Kuster, A.; Mozaffari, N.L.; Wilkinson, O.J.; Wojtaszek, J.L.; Zurfluh, C.; Przetocka, S.; Zyla, D.; von Aesch, C.; Dillingham, M.S.; Williams, R.S.; et al. A Stapled Peptide Mimetic of the CtIP Tetramerization Motif Interferes with Double-Strand Break Repair and Replication Fork Protection. Sci. Adv. 2021, 7, 6381. [Google Scholar] [CrossRef]
- Mammel, A.E.; Hatch, E.M. Genome Instability from Nuclear Catastrophe and DNA Damage. Semin. Cell Dev. Biol. 2021; in press. [Google Scholar] [CrossRef]
- Schoonen, P.M.; Talens, F.; Stok, C.; Gogola, E.; Heijink, A.M.; Bouwman, P.; Foijer, F.; Tarsounas, M.; Blatter, S.; Jonkers, J.; et al. Progression through Mitosis Promotes PARP Inhibitor-Induced Cytotoxicity in Homologous Recombination-Deficient Cancer Cells. Nat. Commun. 2017, 8, 15981. [Google Scholar] [CrossRef] [PubMed]
- Gach, K.; Długosz, A.; Janecka, A. The Role of Oxidative Stress in Anticancer Activity of Sesquiterpene Lactones. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Coricello, A.; El-Magboub, A.; Luna, M.; Ferrario, A.; Haworth, I.S.; Gomer, C.J.; Aiello, F.; Adams, J.D. Rational Drug Design and Synthesis of New α-Santonin Derivatives as Potential COX-2 Inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 993–996. [Google Scholar] [CrossRef]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ Plugin to Automatically Quantify Colony Formation in Clonogenic Assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osborne, H.C.; Larrosa, I.; Schmidt, C.K. Sesquiterpene Lactones Potentiate Olaparib-Induced DNA Damage in p53 Wildtype Cancer Cells. Int. J. Mol. Sci. 2022, 23, 1116. https://doi.org/10.3390/ijms23031116
Osborne HC, Larrosa I, Schmidt CK. Sesquiterpene Lactones Potentiate Olaparib-Induced DNA Damage in p53 Wildtype Cancer Cells. International Journal of Molecular Sciences. 2022; 23(3):1116. https://doi.org/10.3390/ijms23031116
Chicago/Turabian StyleOsborne, Hugh C., Igor Larrosa, and Christine K. Schmidt. 2022. "Sesquiterpene Lactones Potentiate Olaparib-Induced DNA Damage in p53 Wildtype Cancer Cells" International Journal of Molecular Sciences 23, no. 3: 1116. https://doi.org/10.3390/ijms23031116
APA StyleOsborne, H. C., Larrosa, I., & Schmidt, C. K. (2022). Sesquiterpene Lactones Potentiate Olaparib-Induced DNA Damage in p53 Wildtype Cancer Cells. International Journal of Molecular Sciences, 23(3), 1116. https://doi.org/10.3390/ijms23031116