
The H. pylori CagA Oncoprotein Induces DNA Double Strand Breaks through Fanconi Anemia Pathway Downregulation and Replication Fork Collapse

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
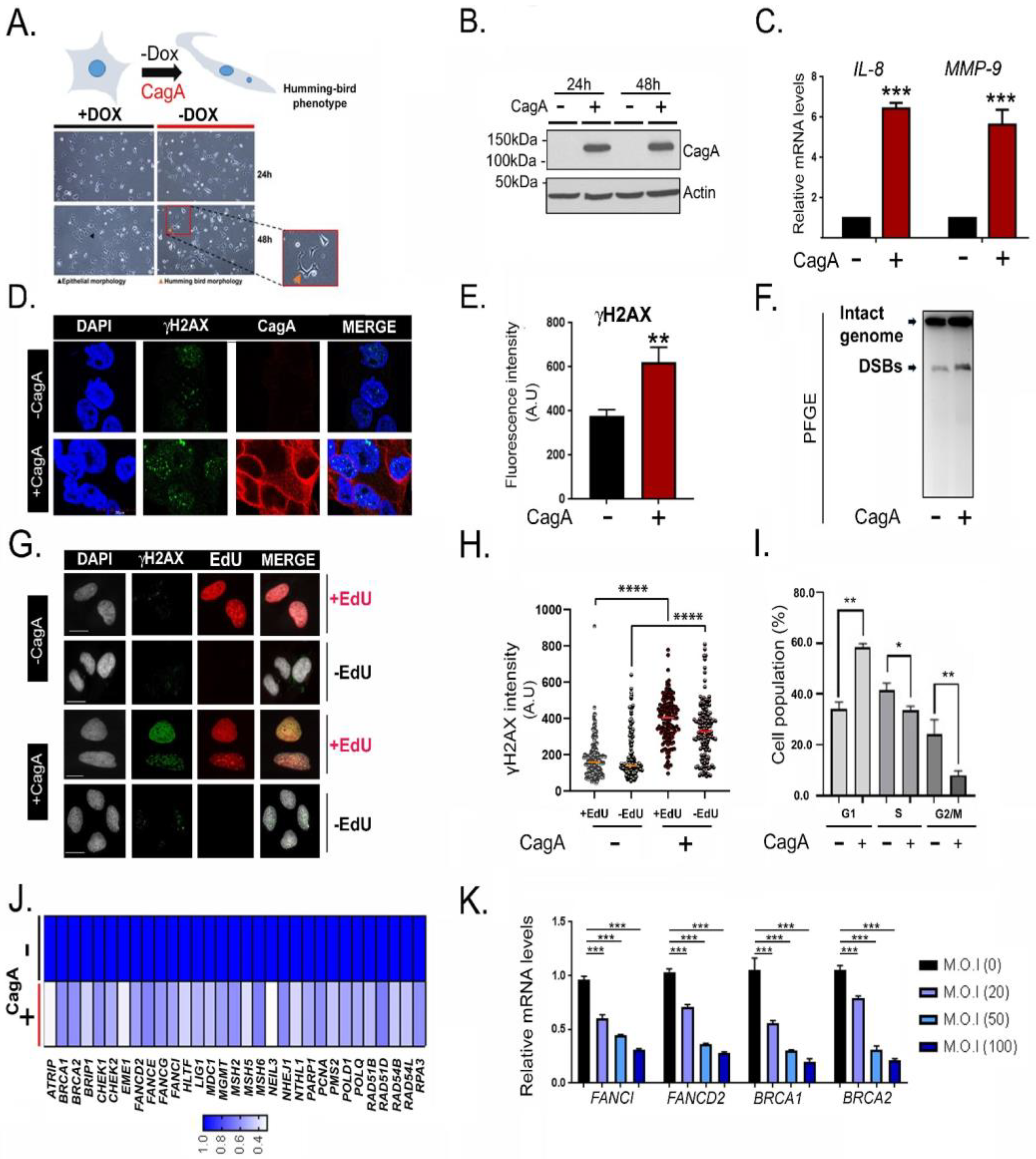
2.1. CagA Induces Host Double Strand Breaks and Downregulates FA Repair Factors
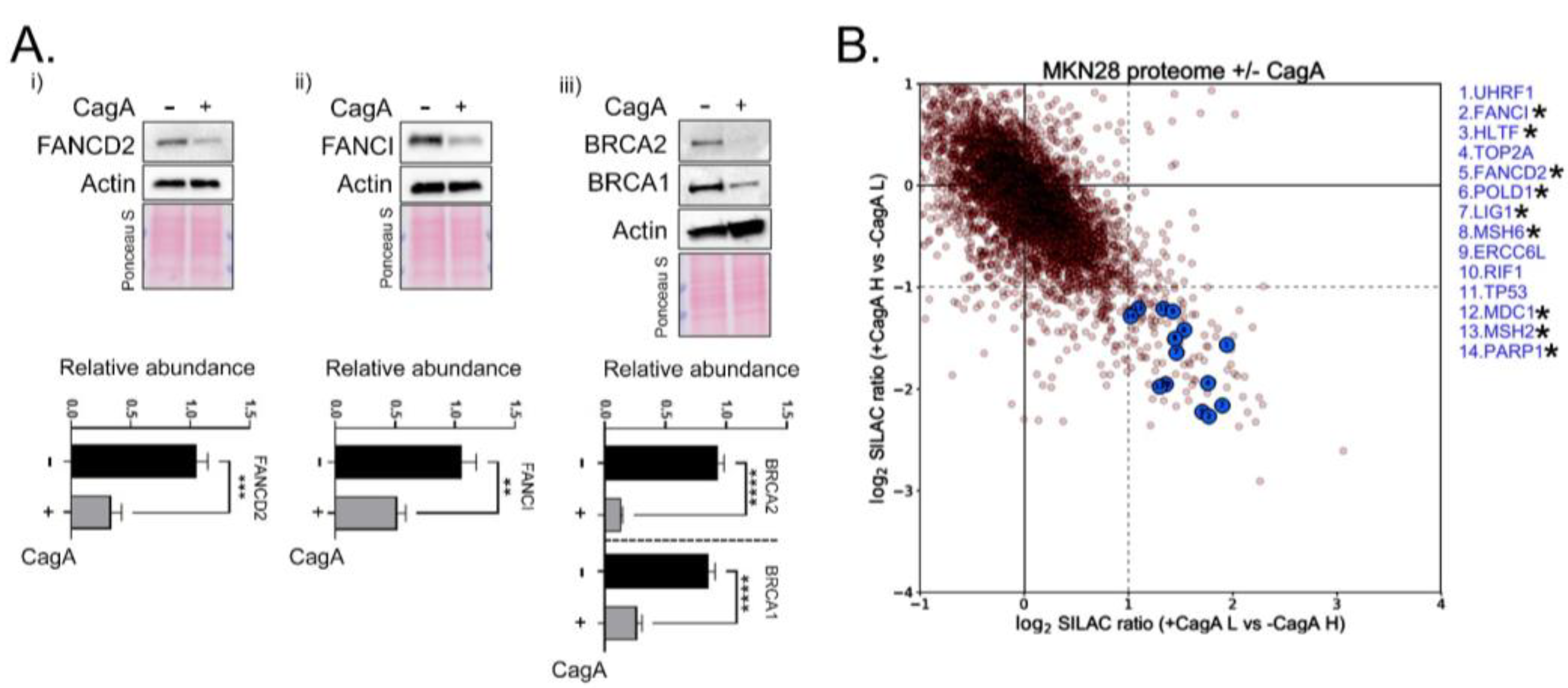
2.2. Proteomic Approach Further Revealed a Reduction in Fanconi Anemia Factors
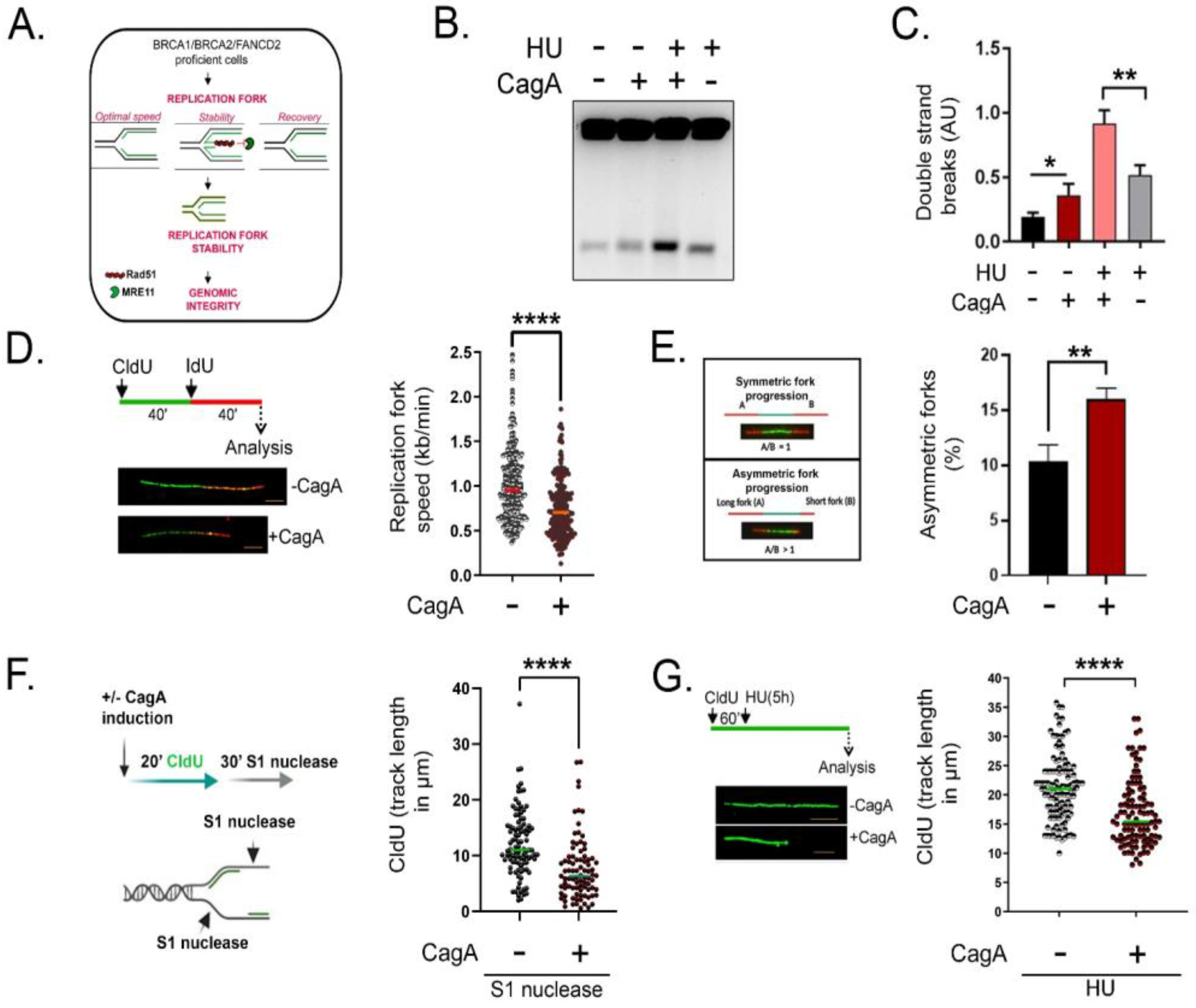
2.3. Impairment in Fanconi Anemia Gene Expression Compromises Replication Fork Stability and Induces DSBs in CagA-Expressing Cells
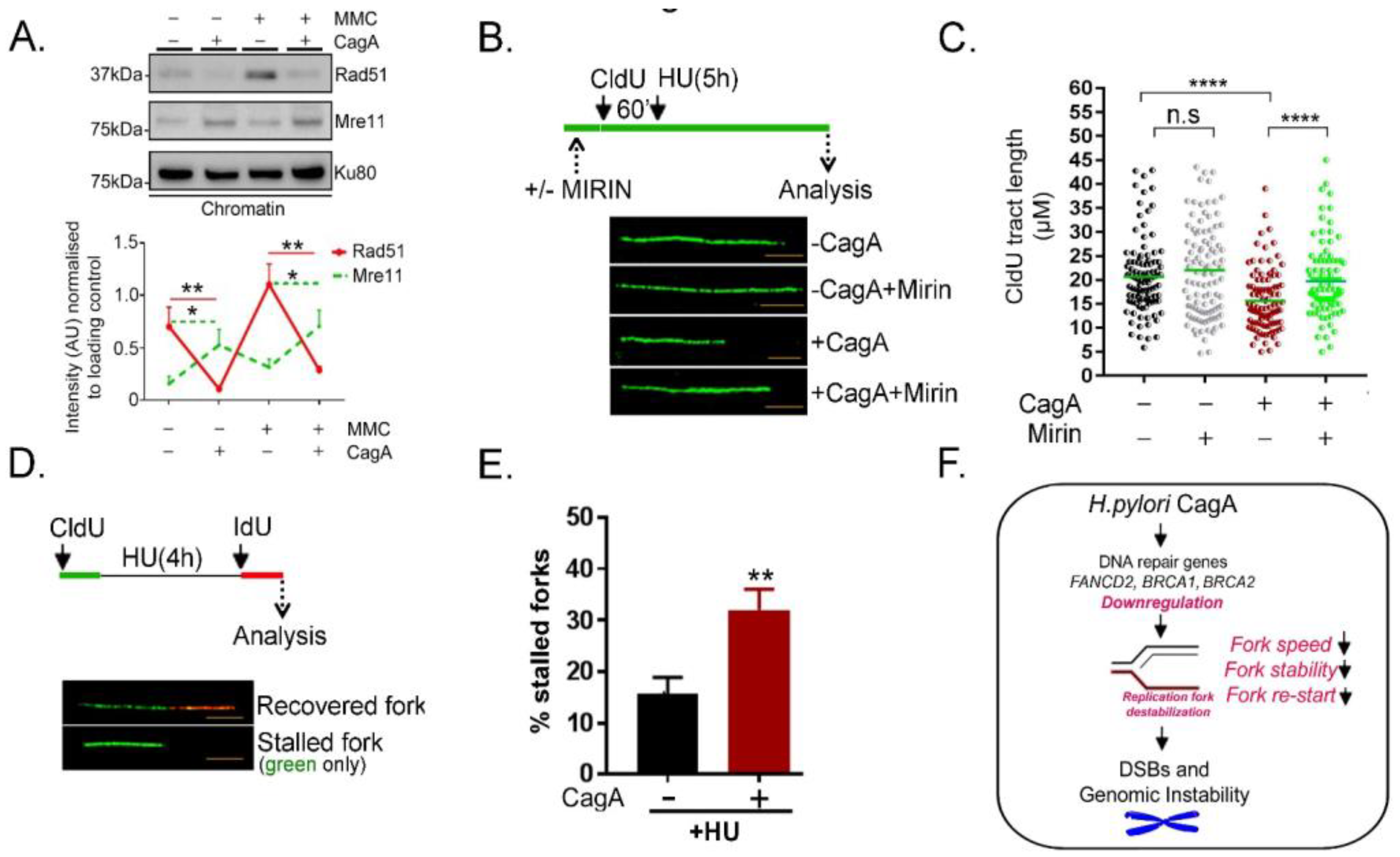
2.4. Poor Rad51 Chromatin Association, Increased MRE11 Nucleolytic Attack Triggers Replication Fork Instability and Stalled Replication Fork Formation in CagA Expressing Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Drugs, and Antibodies
4.2. H. pylori Infection
4.3. RNA Extraction and Q-PCR
4.4. DNA Fibre Assay
4.5. Pulsed-Field Gel Electrophoresis
4.6. Mass Spectrometry Data Acquisition and Analysis
4.7. Chromatin Fractionation
4.8. Immunofluorescence
4.9. Cell Cycle Analysis (FACS)
4.10. RNA-Sequencing and Raw Data Processing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Noto, J.M.; Peek, R.M., Jr. The Helicobacter pylori cag Pathogenicity Island. Methods Mol. Biol. 2012, 921, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microb. 2014, 15, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsonnet, J.; Friedman, G.D.; Orentreich, N.; Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997, 40, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Tada, M.; Nagai, H.; Sasaki, S.; Nakao, M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 1998, 115, 642–648. [Google Scholar] [CrossRef]
- Hanada, K.; Uchida, T.; Tsukamoto, Y.; Watada, M.; Yamaguchi, N.; Yamamoto, K.; Shiota, S.; Moriyama, M.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect Immun. 2014, 82, 4182–4189. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Au, B.; Kulkarni, M.; Shen, Y.; Lim, K.J.; Maimaiti, J.; Wong, C.K.; Luijten, M.N.H.; Chong, H.C.; Lim, E.H.; et al. Chromosomal instability-induced senescence potentiates cell non-autonomous tumourigenic effects. Oncogenesis 2018, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Ooki, T.; Murata-Kamiya, N.; Komura, D.; Tahmina, K.; Wu, W.; Takahashi-Kanemitsu, A.; Knight, C.T.; Kunita, A.; Suzuki, N.; et al. Helicobacter pylori CagA elicits BRCAness to induce genome instability that may underlie bacterial gastric carcinogenesis. Cell Host Microb. 2021, 29, 941–958. [Google Scholar] [CrossRef]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori Infection Causes Characteristic DNA Damage Patterns in Human Cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef] [Green Version]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.O.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef] [Green Version]
- Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Ohba, Y.; Takahashi, M.; Hatakeyama, M. Helicobacter pylori CagA causes mitotic impairment and induces chromosomal instability. J. Biol. Chem. 2009, 284, 22166–22172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Gruter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Muller, A.H. pylori-Induced DNA Strand Breaks Are Introduced by Nucleotide Excision Repair Endonucleases and Promote NF-kappaB Target Gene Expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Murata-Kamiya, N.; Hirayama, T.; Ohba, Y.; Hatakeyama, M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. J. Exp. Med. 2010, 207, 2157–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Noto, J.M.; Zaika, E.; Romero-Gallo, J.; Piazuelo, M.B.; Schneider, B.; El-Rifai, W.; Correa, P.; Peek, R.M.; Zaika, A.I. Bacterial CagA protein induces degradation of p53 protein in a p14ARF-dependent manner. Gut 2015, 64, 1040–1048. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Techer, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Techer, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016, 12, e1006007. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghunandan, M.; Chaudhury, I.; Kelich, S.L.; Hanenberg, H.; Sobeck, A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi Anemia core complex. Cell Cycle 2015, 14, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144. [Google Scholar] [CrossRef] [PubMed]
- Somyajit, K.; Spies, J.; Coscia, F.; Kirik, U.; Rask, M.B.; Lee, J.H.; Neelsen, K.J.; Mund, A.; Jensen, L.J.; Paull, T.T.; et al. Homology-directed repair protects the replicating genome from metabolic assaults. Dev. Cell 2021, 56, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Tsujii, M.; Wang, J.; Kondo, J.; Akasaka, T.; Jin, Y.; Li, W.; Nakamura, T.; Nishida, T.; Iijima, H.; et al. CagA mediates epigenetic regulation to attenuate let-7 expression in Helicobacter pylori-related carcinogenesis. Gut 2013, 62, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Zamperone, A.; Cohen, D.; Stein, M.; Viard, C.; Musch, A. Inhibition of polarity-regulating kinase PAR1b contributes to Helicobacter pylori inflicted DNA Double Strand Breaks in gastric cells. Cell Cycle 2019, 18, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef]
- Chen, H.N.; Wang, Z.; Li, X.; Zhou, Z.G. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: Evidence from a meta-analysis. Gastric Cancer. 2016, 19, 166–175. [Google Scholar] [CrossRef]
- Fukase, K.; Kato, M.; Kikuchi, S.; Inoue, K.; Uemura, N.; Okamoto, S.; Terao, S.; Amagai, K.; Hayashi, S.; Asaka, M.; et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet 2008, 372, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, Y.; Yamaguchi, N.; Teshima, R.; Narahara, H.; Yamaoka, Y.; Anai, H.; Nishida, Y.; Hanada, K. Detection of DNA double-strand breaks by pulsed-field gel electrophoresis. Genes Cells 2017, 22, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucl. Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Tay, L.S.; Krishnan, V.; Sankar, H.; Chong, Y.L.; Chuang, L.S.H.; Tan, T.Z.; Kolinjivadi, A.M.; Kappei, D.; Ito, Y. RUNX Poly(ADP-Ribosyl)ation and BLM Interaction Facilitate the Fanconi Anemia Pathway of DNA Repair. Cell Rep. 2018, 24, 1747–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolinjivadi, A.M.; Sankar, H.; Choudhary, R.; Tay, L.S.; Tan, T.Z.; Murata-Kamiya, N.; Voon, D.C.-C.; Kappei, D.; Hatakeyama, M.; Krishnan, V.; et al. The H. pylori CagA Oncoprotein Induces DNA Double Strand Breaks through Fanconi Anemia Pathway Downregulation and Replication Fork Collapse. Int. J. Mol. Sci. 2022, 23, 1661. https://doi.org/10.3390/ijms23031661
Kolinjivadi AM, Sankar H, Choudhary R, Tay LS, Tan TZ, Murata-Kamiya N, Voon DC-C, Kappei D, Hatakeyama M, Krishnan V, et al. The H. pylori CagA Oncoprotein Induces DNA Double Strand Breaks through Fanconi Anemia Pathway Downregulation and Replication Fork Collapse. International Journal of Molecular Sciences. 2022; 23(3):1661. https://doi.org/10.3390/ijms23031661
Chicago/Turabian StyleKolinjivadi, Arun Mouli, Haresh Sankar, Ramveer Choudhary, Lavina Sierra Tay, Tuan Zea Tan, Naoko Murata-Kamiya, Dominic Chih-Cheng Voon, Dennis Kappei, Masanori Hatakeyama, Vaidehi Krishnan, and et al. 2022. "The H. pylori CagA Oncoprotein Induces DNA Double Strand Breaks through Fanconi Anemia Pathway Downregulation and Replication Fork Collapse" International Journal of Molecular Sciences 23, no. 3: 1661. https://doi.org/10.3390/ijms23031661
APA StyleKolinjivadi, A. M., Sankar, H., Choudhary, R., Tay, L. S., Tan, T. Z., Murata-Kamiya, N., Voon, D. C.-C., Kappei, D., Hatakeyama, M., Krishnan, V., & Ito, Y. (2022). The H. pylori CagA Oncoprotein Induces DNA Double Strand Breaks through Fanconi Anemia Pathway Downregulation and Replication Fork Collapse. International Journal of Molecular Sciences, 23(3), 1661. https://doi.org/10.3390/ijms23031661