
Analogs of the Heat Shock Protein 70 Inhibitor MKT-077 Suppress Medullary Thyroid Carcinoma Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
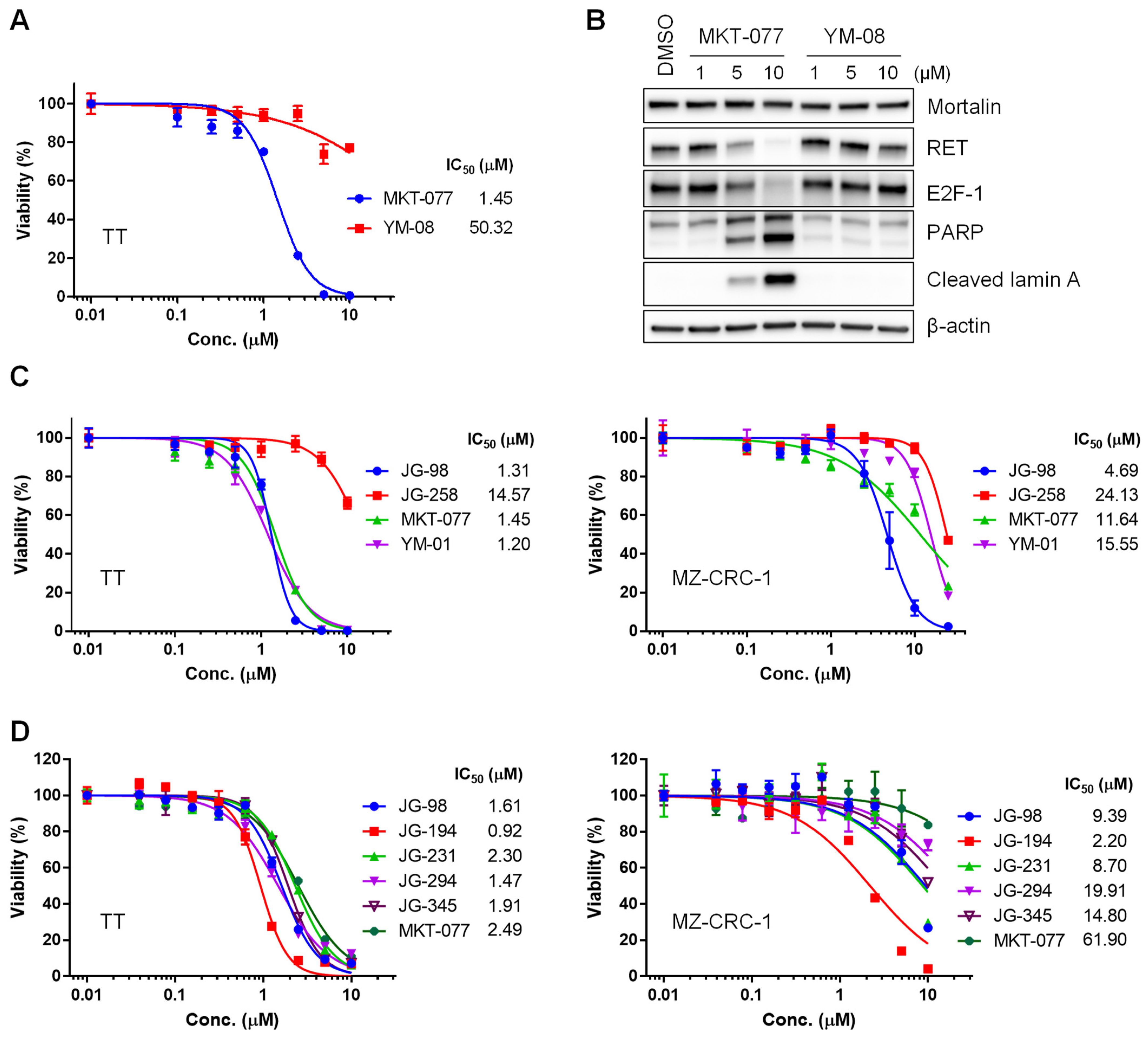
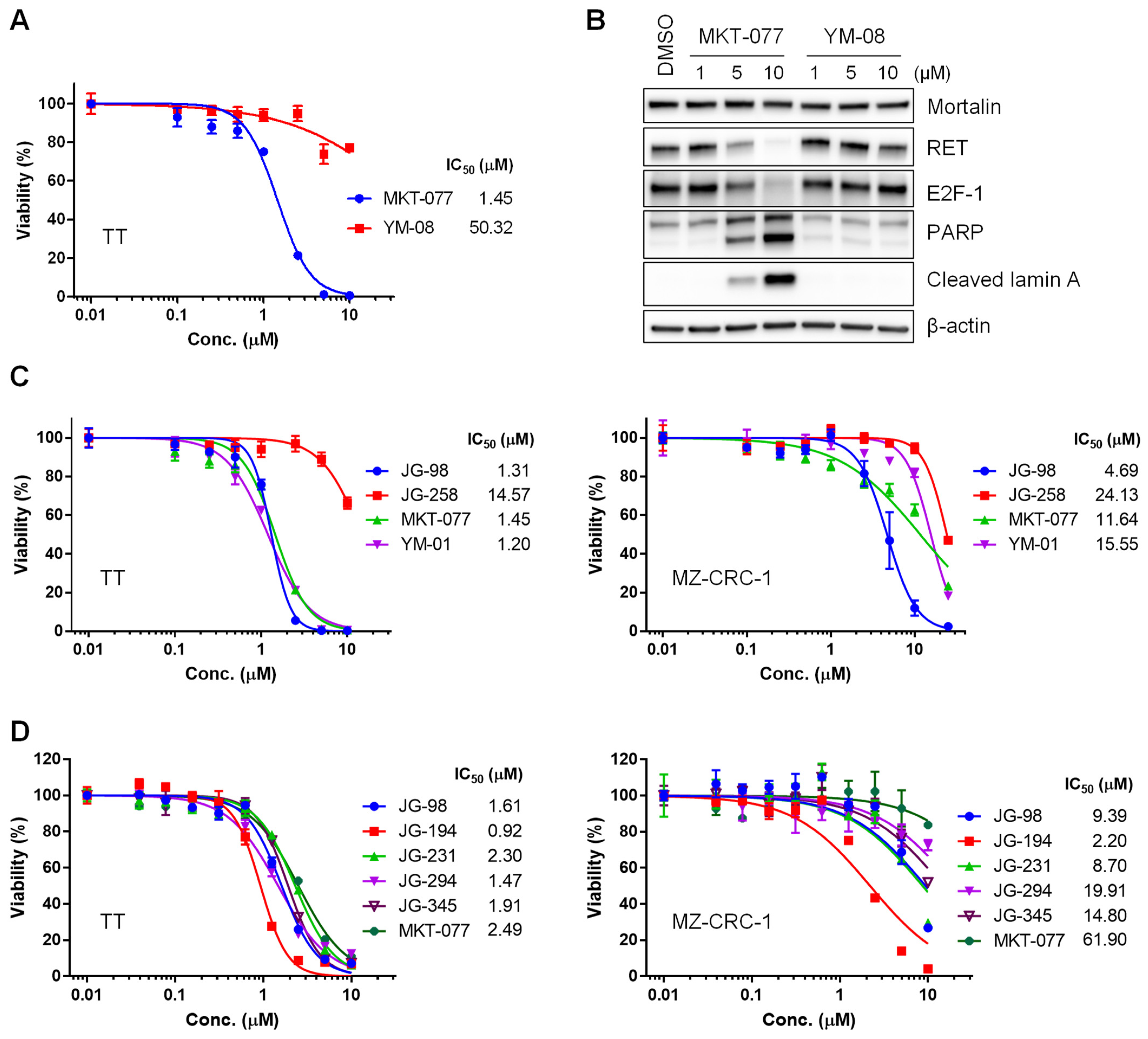
2.1. Benzothiazole Rhodacyanine Derivatives of MKT-077 Effectively Suppress the Viability of Human MTC Cell Lines, TT and MZ-CRC-1, in Cultures
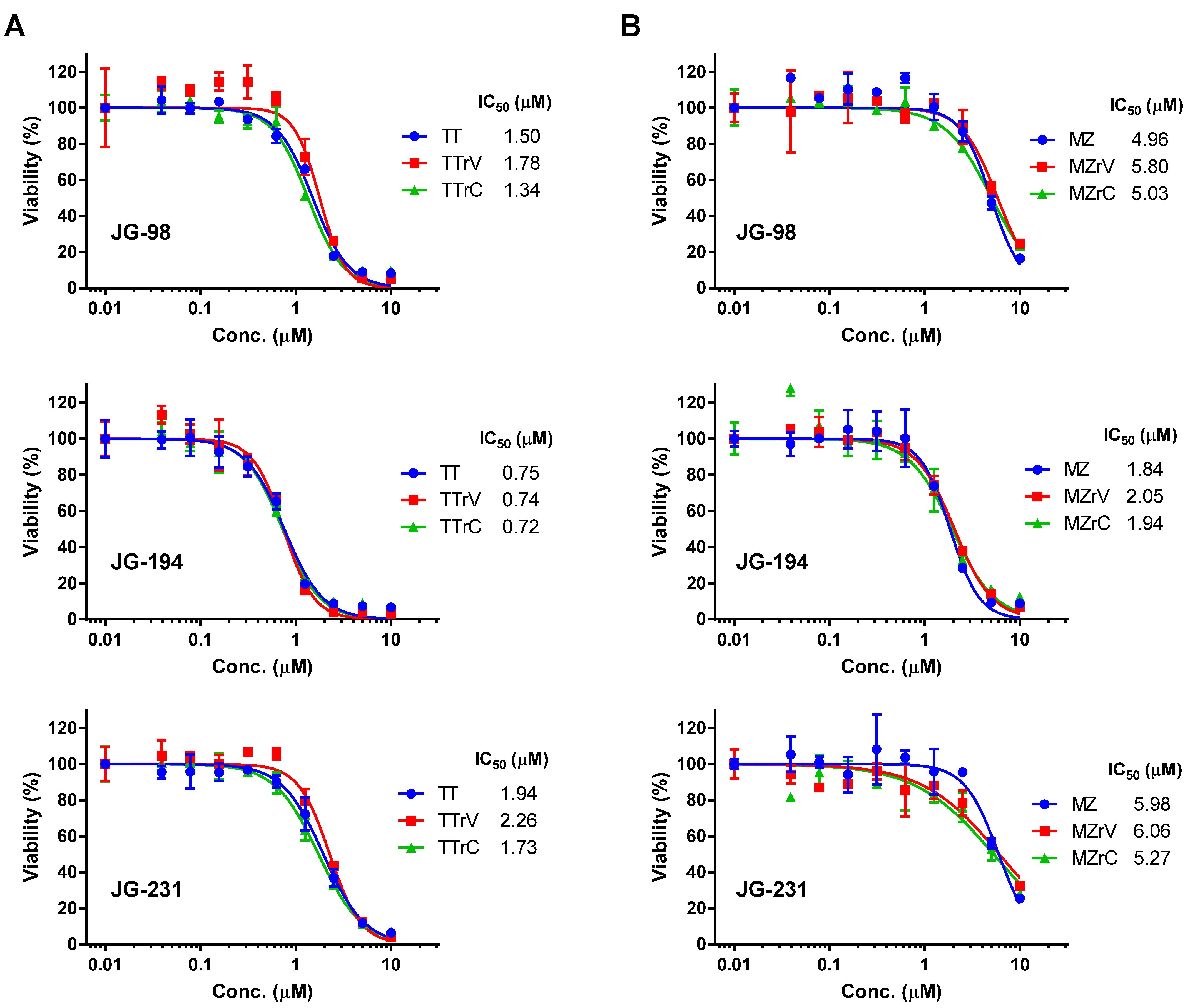
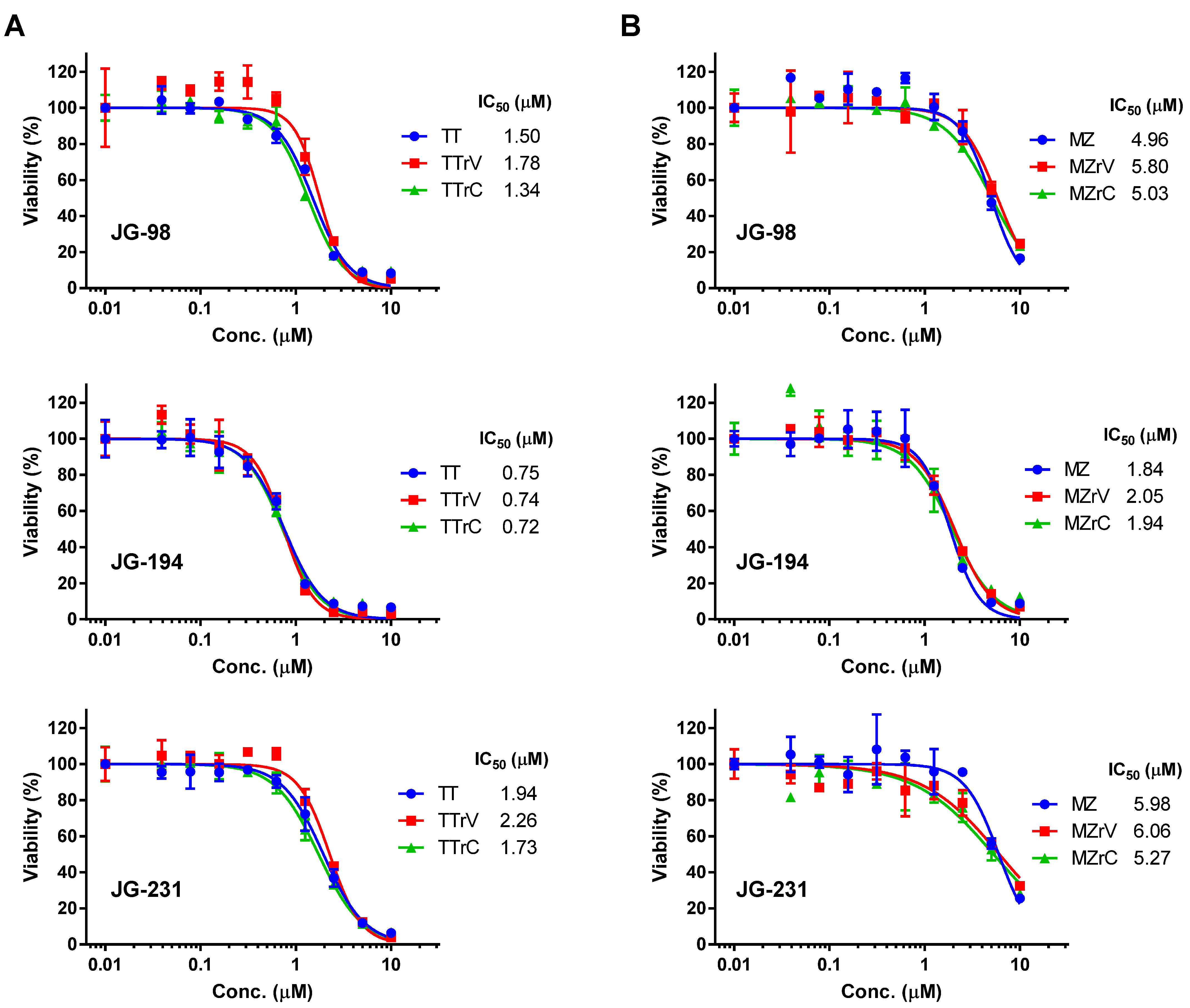
2.2. JG-98 Analogs Effectively Suppress the Viability of Vandetanib- and Cabozantinib-Resistant MTC Cells
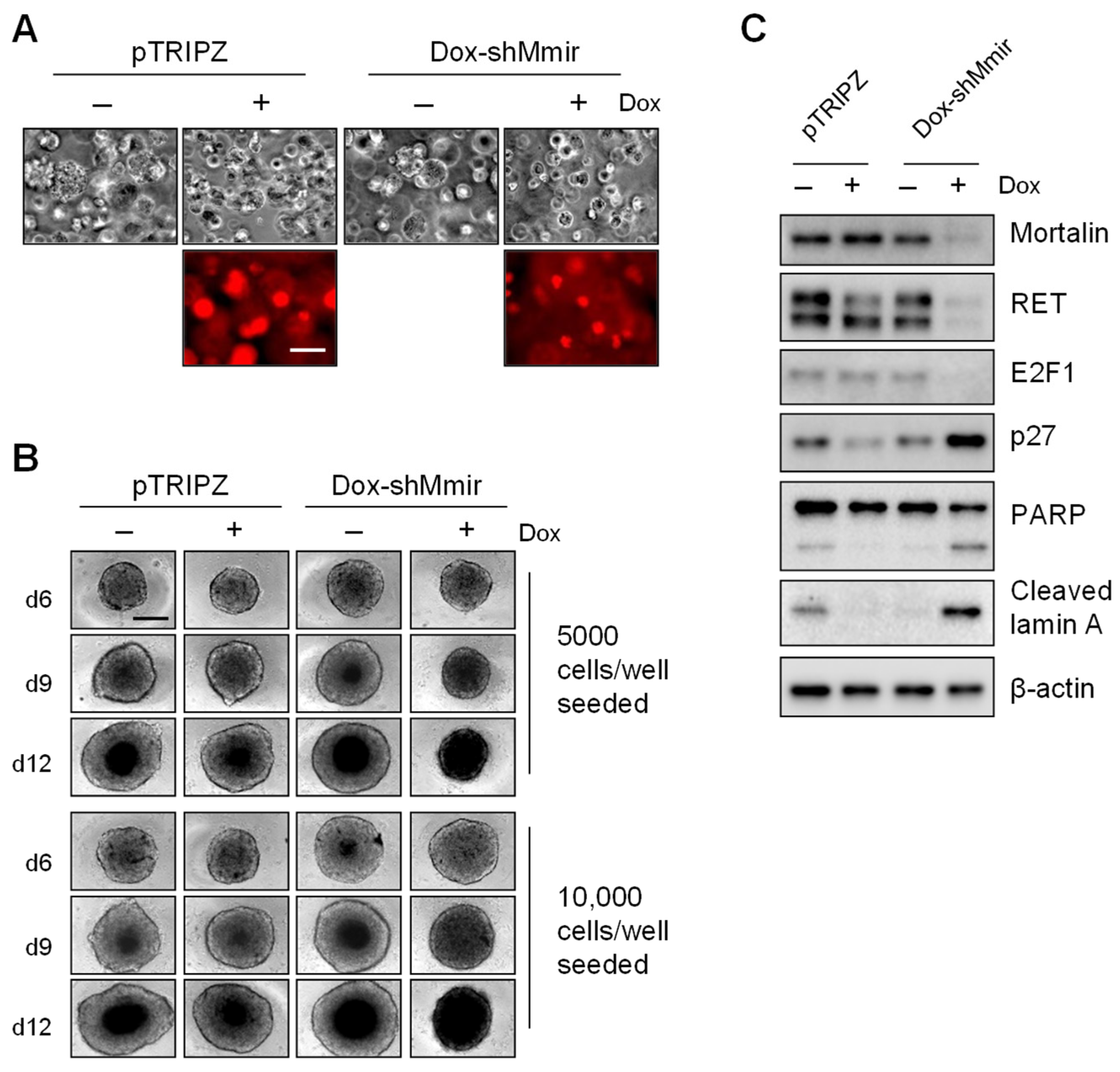
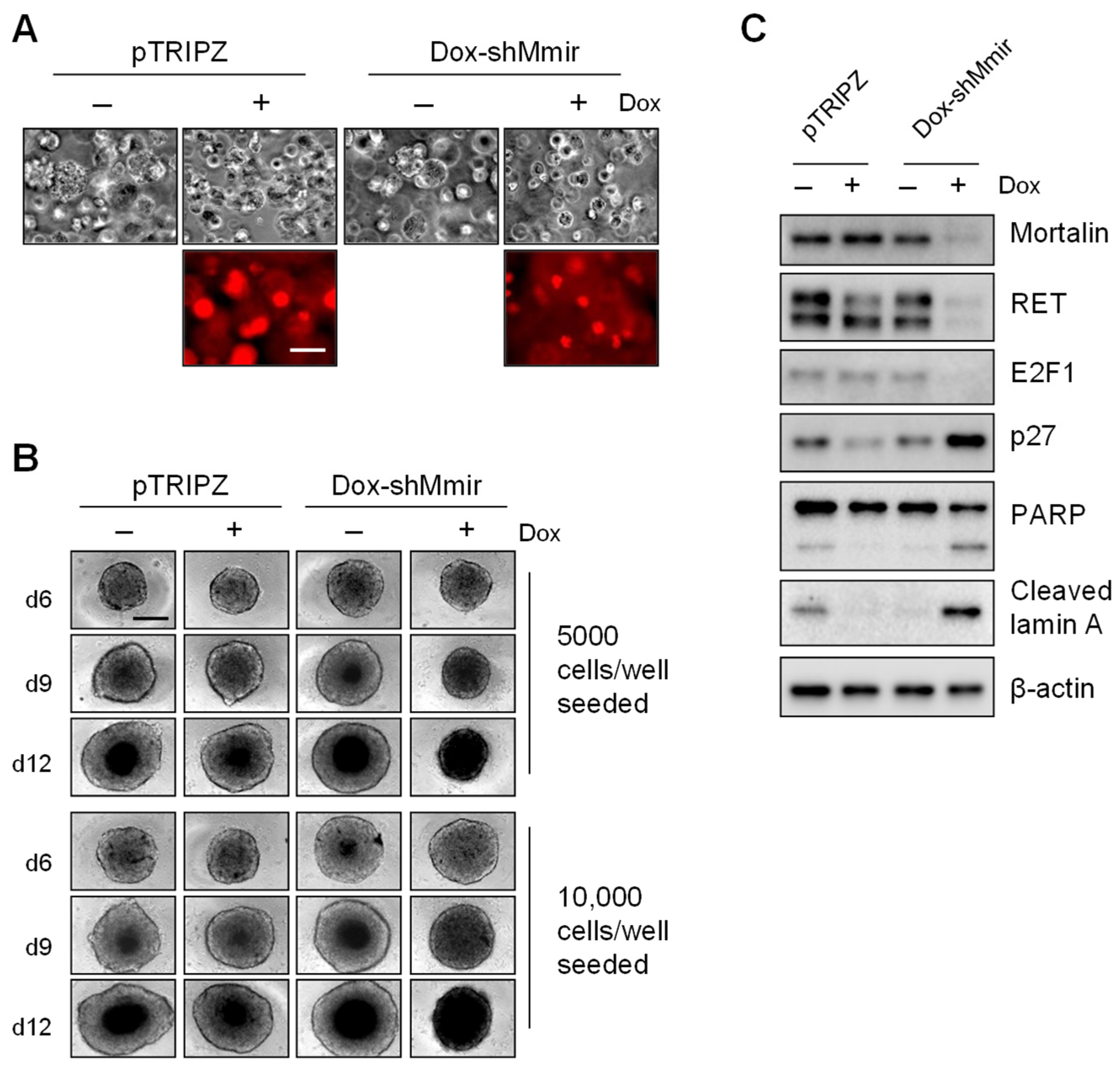
2.3. Mortalin Depletion Induces Growth Inhibition in TT Cells Grown in Three-Dimensional (3D) Cultures
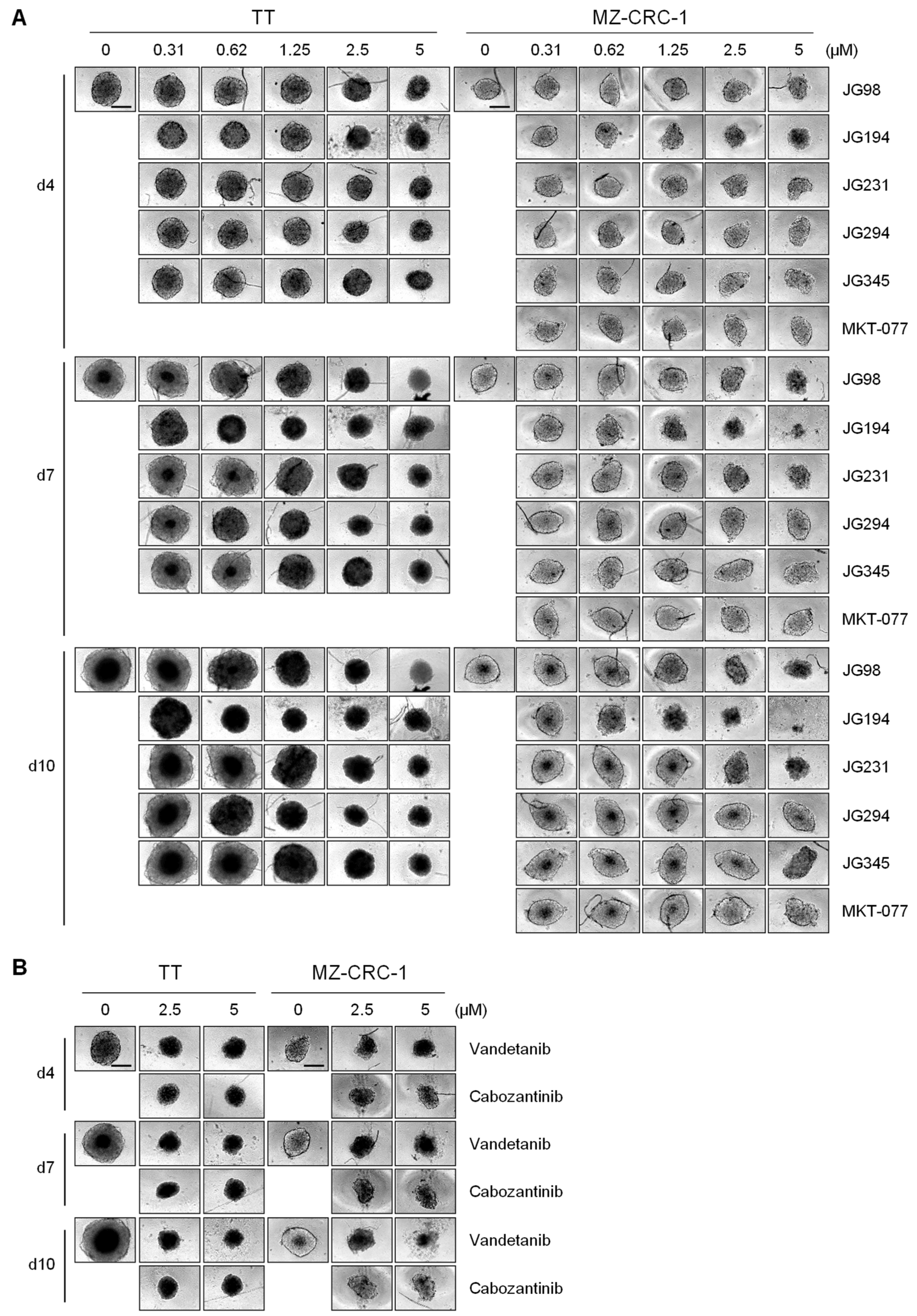
2.4. JG-98 Analogs Effectively Suppress MTC Cell Growth in 3D Cultures
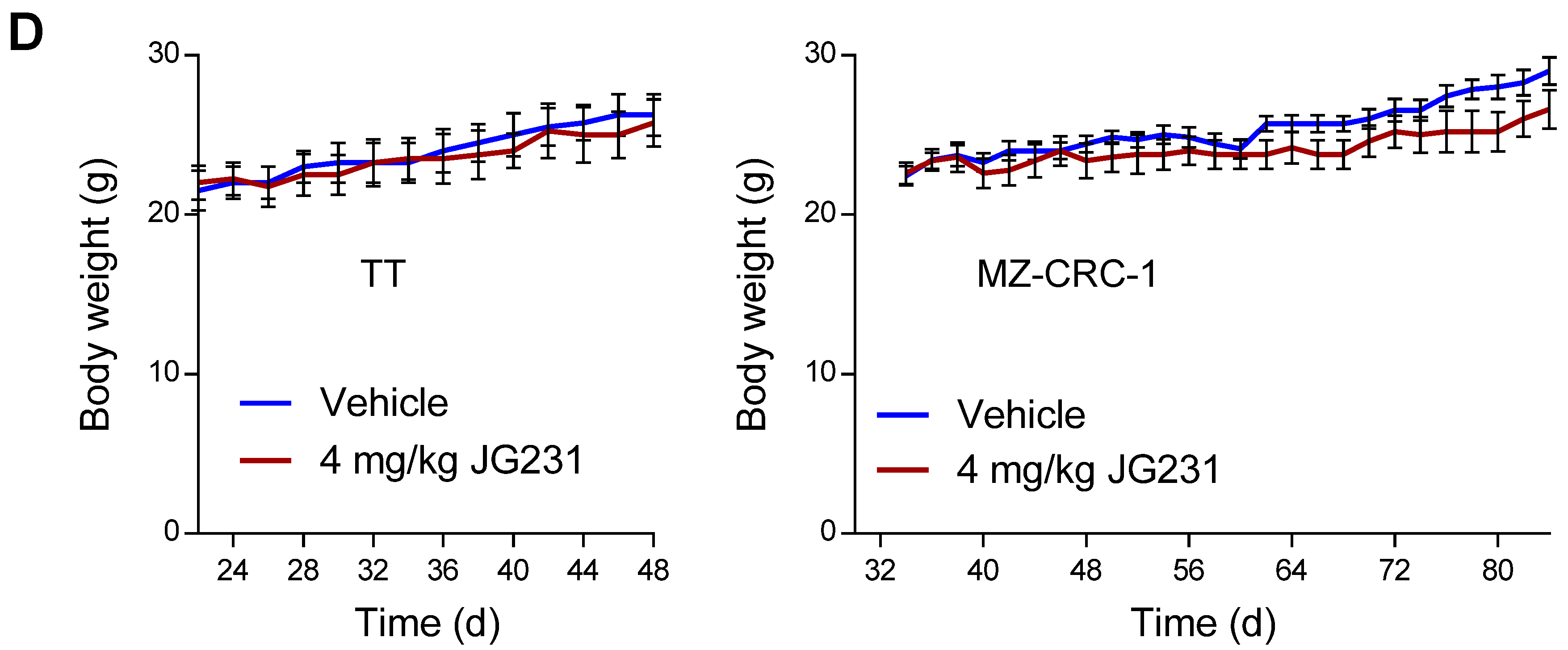
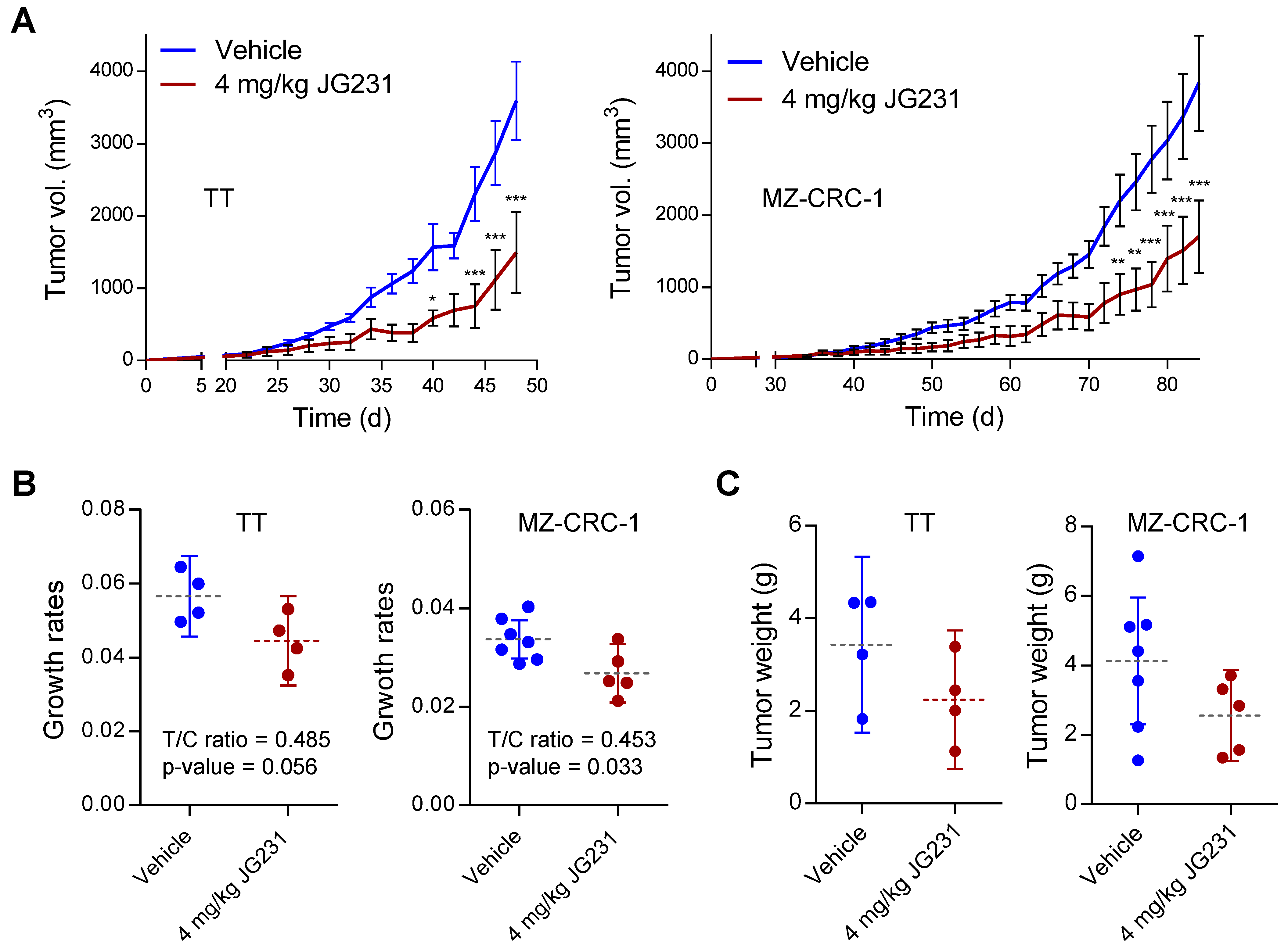
2.5. The Effects of JG-231 on TT and MZ-CRC-1 Xenografts in Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Analysis of Cell Viability
4.3. Immunoblotting
4.4. Tumor Xenograft Studies
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tuttle, R.M.; Ball, D.W.; Byrd, D.; Daniels, G.H.; Dilawari, R.A.; Doherty, G.M.; Duh, Q.Y.; Ehya, H.; Farrar, W.B.; Haddad, R.I.; et al. Medullary carcinoma. J. Natl. Compr. Cancer Netw. 2010, 8, 512–530. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J.; et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 2013, 98, E364–E369. [Google Scholar] [CrossRef] [Green Version]
- Boichard, A.; Croux, L.; Al Ghuzlan, A.; Broutin, S.; Dupuy, C.; Leboulleux, S.; Schlumberger, M.; Bidart, J.M.; Lacroix, L. Somatic RAS mutations occur in a large proportion of sporadic RET-negative medullary thyroid carcinomas and extend to a previously unidentified exon. J. Clin. Endocrinol. Metab. 2012, 97, E2031–E2035. [Google Scholar] [CrossRef] [Green Version]
- Ciampi, R.; Mian, C.; Fugazzola, L.; Cosci, B.; Romei, C.; Barollo, S.; Cirello, V.; Bottici, V.; Marconcini, G.; Rosa, P.M.; et al. Evidence of a Low Prevalence of RAS Mutations in a Large Medullary Thyroid Cancer Series. Thyroid 2013, 23, 50–57. [Google Scholar] [CrossRef]
- Ichihara, M.; Murakumo, Y.; Takahashi, M. RET and neuroendocrine tumors. Cancer Lett. 2004, 204, 197–211. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, D.; Santoro, M.; Schlumberger, M. The importance of the RET gene in thyroid cancer and therapeutic implications. Nat. Rev. Endocrinol. 2021, 17, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.G.; Ghiraldeli, L.P.; Pardee, T.S. Mitochondria in cancer metabolism, an organelle whose time has come? Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Don, A.S.; Hogg, P.J. Mitochondria as cancer drug targets. Trends Mol. Med. 2004, 10, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Park, J.I. Mitochondria-targeted nitroxide, Mito-CP, suppresses medullary thyroid carcinoma cell survival in vitro and in vivo. J. Clin. Endocrinol. Metab. 2013, 98, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Starenki, D.; Hong, S.K.; Wu, P.K.; Park, J.I. Vandetanib and cabozantinib potentiate mitochondria-targeted agents to suppress medullary thyroid carcinoma cells. Cancer Biol. Ther. 2017, 18, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Hong, S.K.; Lloyd, R.V.; Park, J.I. Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 2015, 34, 4624–4634. [Google Scholar] [CrossRef] [Green Version]
- Koya, K.; Li, Y.; Wang, H.; Ukai, T.; Tatsuta, N.; Kawakami, M.; Shishido, T.; Chen, L.B. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996, 56, 538–543. [Google Scholar]
- Rousaki, A.; Miyata, Y.; Jinwal, U.K.; Dickey, C.A.; Gestwicki, J.E.; Zuiderweg, E.R. Allosteric drugs: The interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J. Mol. Biol 2011, 411, 614–632. [Google Scholar] [CrossRef] [Green Version]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef]
- Miyata, Y.; Li, X.; Lee, H.F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Li, X.; Moses, M.A.; Gilbert, L.A.; Kalyanaraman, C.; Young, Z.T.; Chernova, M.; Journey, S.N.; Weissman, J.S.; Hann, B.; et al. Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J. Med. Chem. 2018, 61, 6163–6177. [Google Scholar] [CrossRef]
- Li, X.; Srinivasan, S.R.; Connarn, J.; Ahmad, A.; Young, Z.T.; Kabza, A.M.; Zuiderweg, E.R.; Sun, D.; Gestwicki, J.E. Analogs of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, as Anti-Cancer Agents. ACS Med. Chem. Lett. 2013, 4, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Park, J.I. Selective Mitochondrial Uptake of MKT-077 Can Suppress Medullary Thyroid Carcinoma Cell Survival In Vitro and In Vivo. Endocrinol. Metab. 2015, 30, 593–603. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Hather, G.; Liu, R.; Bandi, S.; Mettetal, J.; Manfredi, M.; Shyu, W.C.; Donelan, J.; Chakravarty, A. Growth rate analysis and efficient experimental design for tumor xenograft studies. Cancer Inform. 2014, 13, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister-Broekema, M.; Freilich, R.; Jagadeesan, C.; Rauch, J.N.; Bengoechea, R.; Motley, W.W.; Kuiper, E.F.E.; Minoia, M.; Furtado, G.V.; van Waarde, M.; et al. Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat. Commun. 2018, 9, 5342. [Google Scholar] [CrossRef] [Green Version]
- Colvin, T.A.; Gabai, V.L.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, I.A.; et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.K.; Hong, S.K.; Chen, W.; Becker, A.E.; Gundry, R.L.; Lin, C.W.; Shao, H.; Gestwicki, J.E.; Park, J.I. Mortalin (HSPA9) facilitates BRAF-mutant tumor cell survival by suppressing ANT3-mediated mitochondrial membrane permeability. Sci. Signal. 2020, 13, eaay1478. [Google Scholar] [CrossRef] [Green Version]
- Soudry, E.; Stern Shavit, S.; Hardy, B.; Morgenstern, S.; Hadar, T.; Feinmesser, R. Heat shock proteins HSP90, HSP70 and GRP78 expression in medullary thyroid carcinoma. Ann. Diagn. Pathol. 2017, 26, 52–56. [Google Scholar] [CrossRef]
- Shao, H.; Li, X.; Hayashi, S.; Bertron, J.L.; Schwarz, D.M.C.; Tang, B.C.; Gestwicki, J.E. Inhibitors of heat shock protein 70 (Hsp70) with enhanced metabolic stability reduce tau levels. Bioorg. Med. Chem. Lett. 2021, 41, 128025. [Google Scholar] [CrossRef]
- Arthan, D.; Hong, S.K.; Park, J.I. Leukemia inhibitory factor can mediate Ras/Raf/MEK/ERK-induced growth inhibitory signaling in medullary thyroid cancer cells. Cancer Lett. 2010, 297, 31–41. [Google Scholar] [CrossRef]
- Park, J.I.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Mol. Cell Biol. 2003, 23, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.K.; Yoon, S.; Moelling, C.; Arthan, D.; Park, J.I. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J. Biol. Chem. 2009, 284, 33006–33018. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.-K.; Starenki, D.; Johnson, O.T.; Gestwicki, J.E.; Park, J.-I. Analogs of the Heat Shock Protein 70 Inhibitor MKT-077 Suppress Medullary Thyroid Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 1063. https://doi.org/10.3390/ijms23031063
Hong S-K, Starenki D, Johnson OT, Gestwicki JE, Park J-I. Analogs of the Heat Shock Protein 70 Inhibitor MKT-077 Suppress Medullary Thyroid Carcinoma Cells. International Journal of Molecular Sciences. 2022; 23(3):1063. https://doi.org/10.3390/ijms23031063
Chicago/Turabian StyleHong, Seung-Keun, Dmytro Starenki, Oleta T. Johnson, Jason E. Gestwicki, and Jong-In Park. 2022. "Analogs of the Heat Shock Protein 70 Inhibitor MKT-077 Suppress Medullary Thyroid Carcinoma Cells" International Journal of Molecular Sciences 23, no. 3: 1063. https://doi.org/10.3390/ijms23031063
APA StyleHong, S.-K., Starenki, D., Johnson, O. T., Gestwicki, J. E., & Park, J.-I. (2022). Analogs of the Heat Shock Protein 70 Inhibitor MKT-077 Suppress Medullary Thyroid Carcinoma Cells. International Journal of Molecular Sciences, 23(3), 1063. https://doi.org/10.3390/ijms23031063