
miR-16-5p Suppression Protects Human Cardiomyocytes against Endoplasmic Reticulum and Oxidative Stress-Induced Injury

 ,
,  , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Patients and Control Subjects
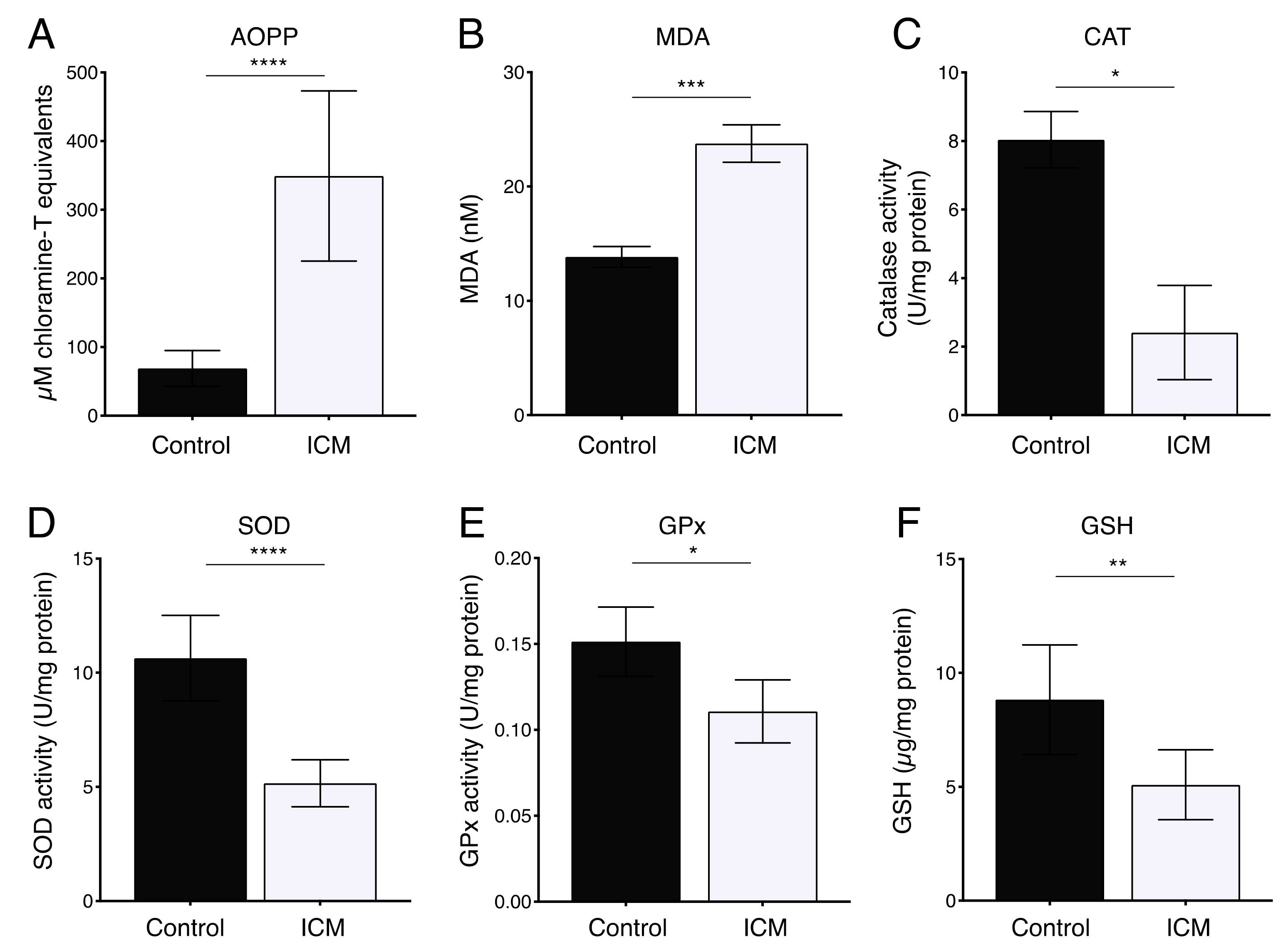
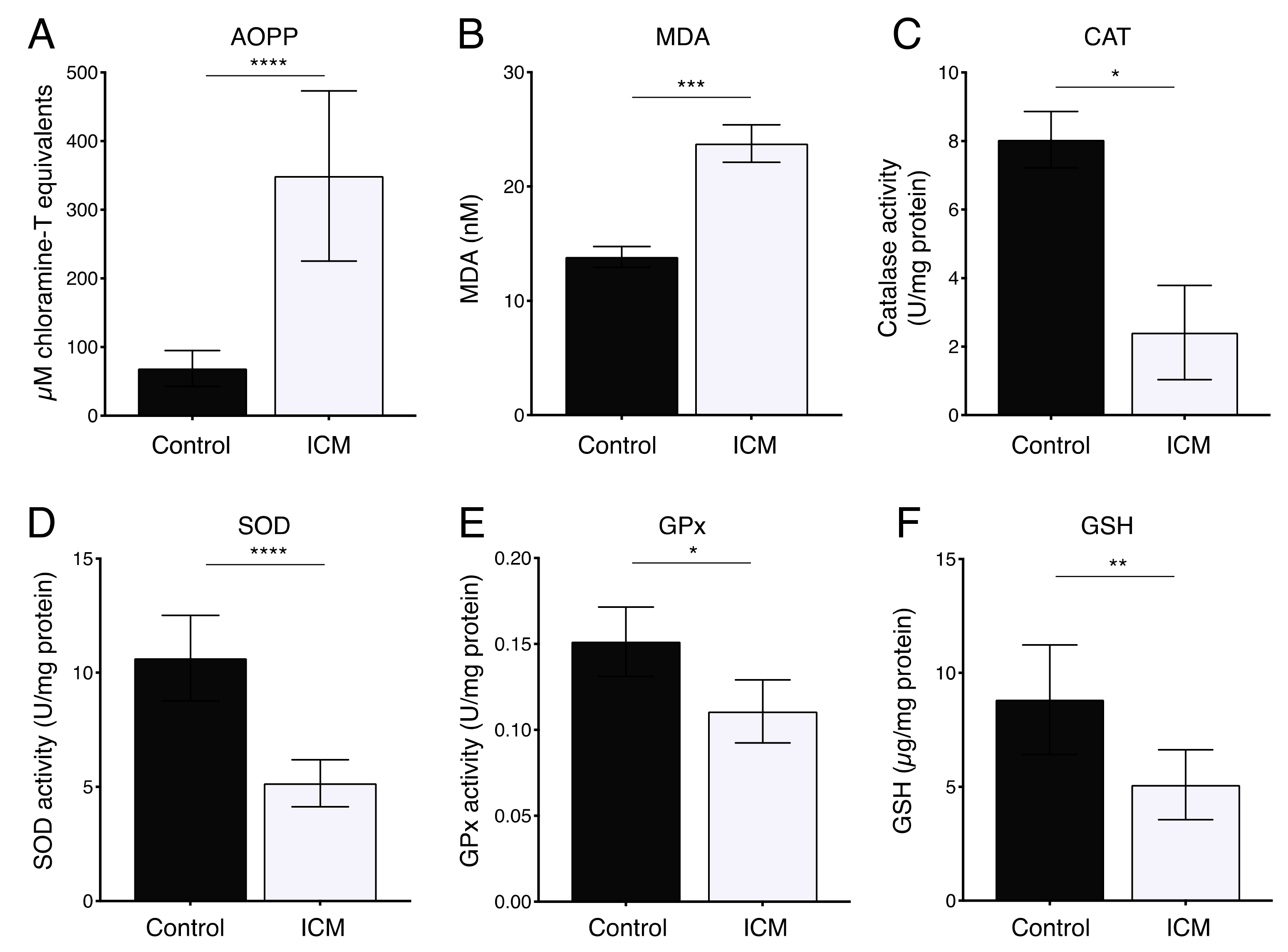
2.2. Increased Oxidative Stress in Plasma from ICM Patients
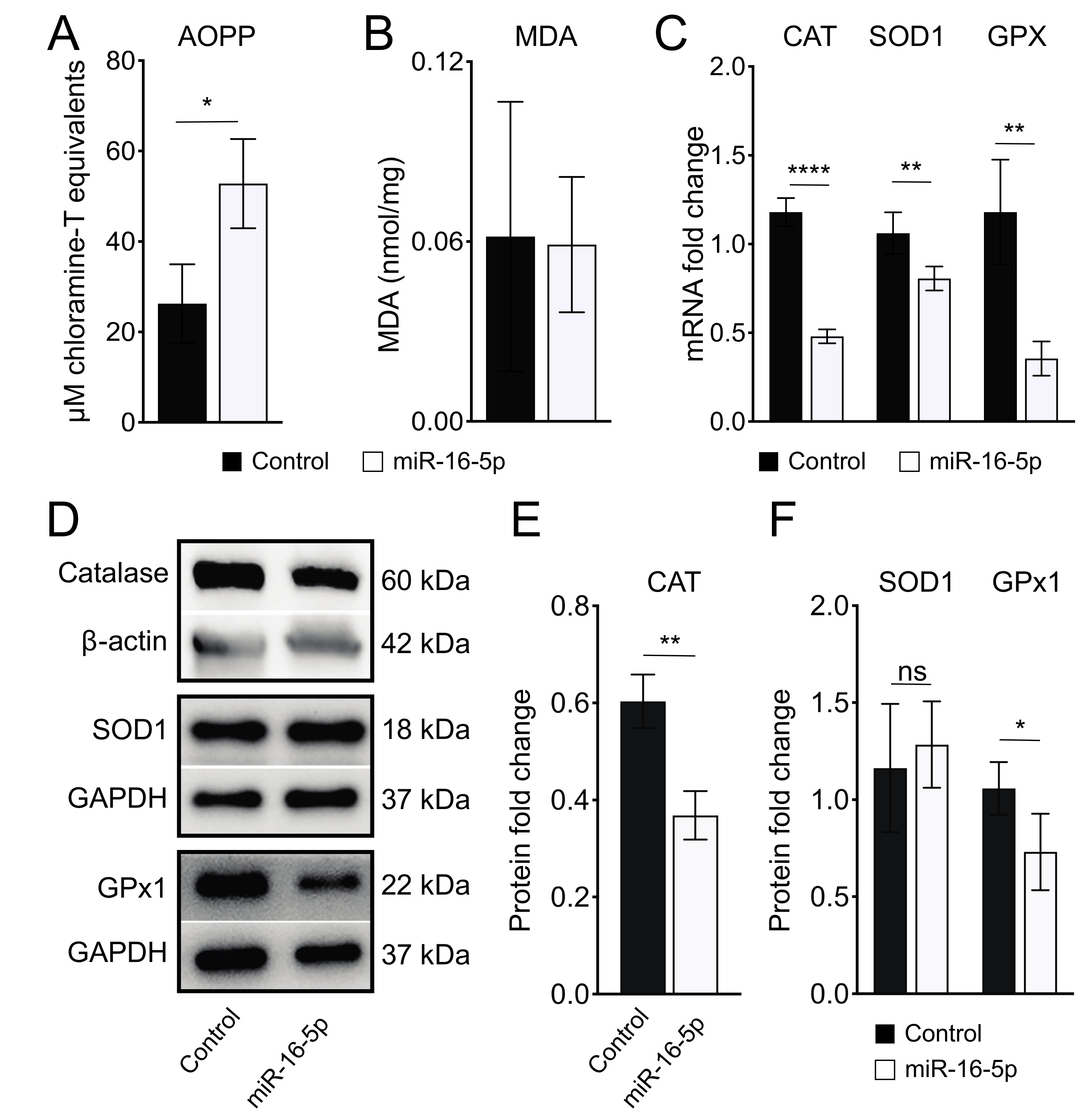
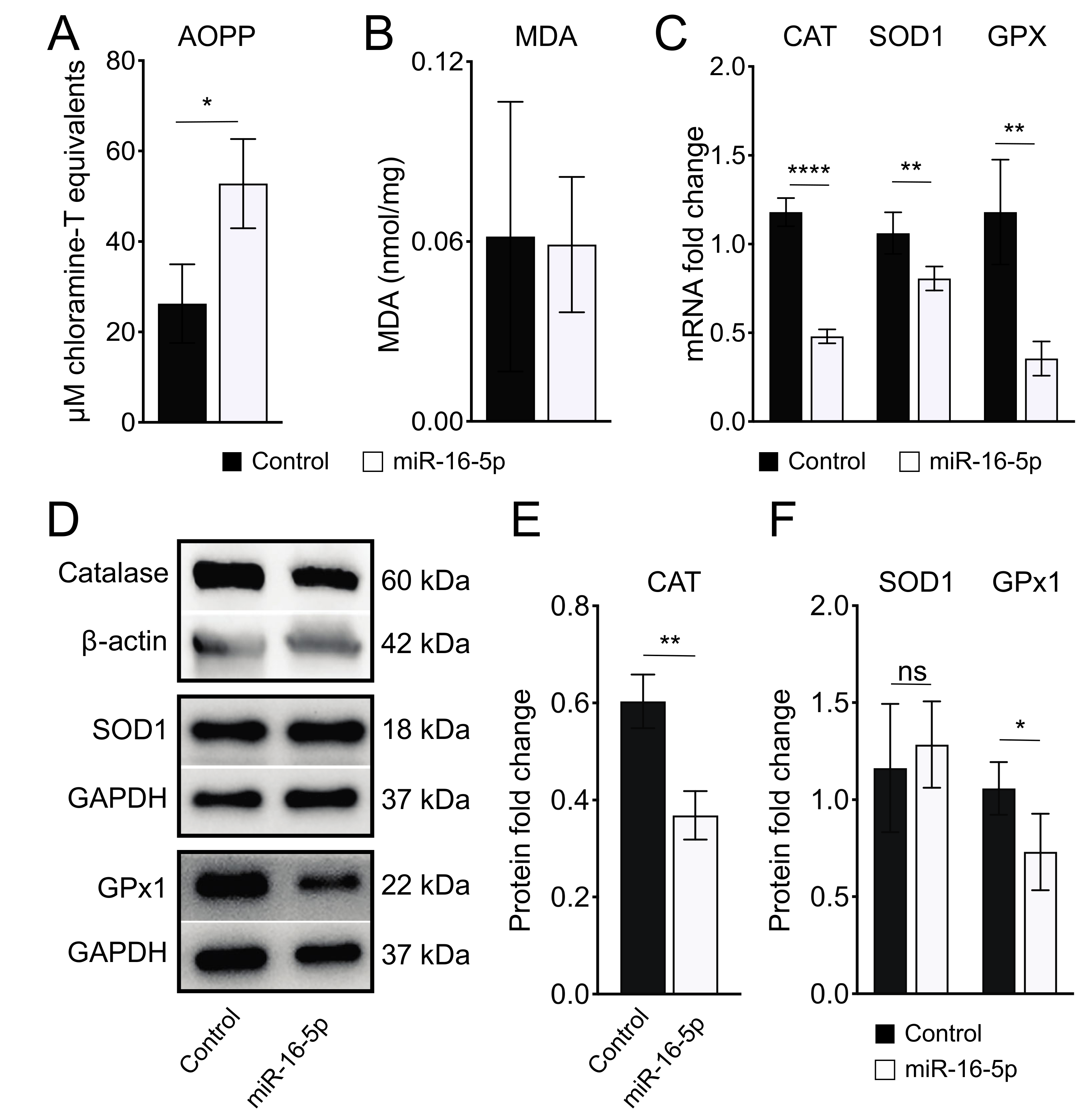
2.3. miR-16-5p Overexpression Induces Oxidative Stress in Human Cardiomyoblas
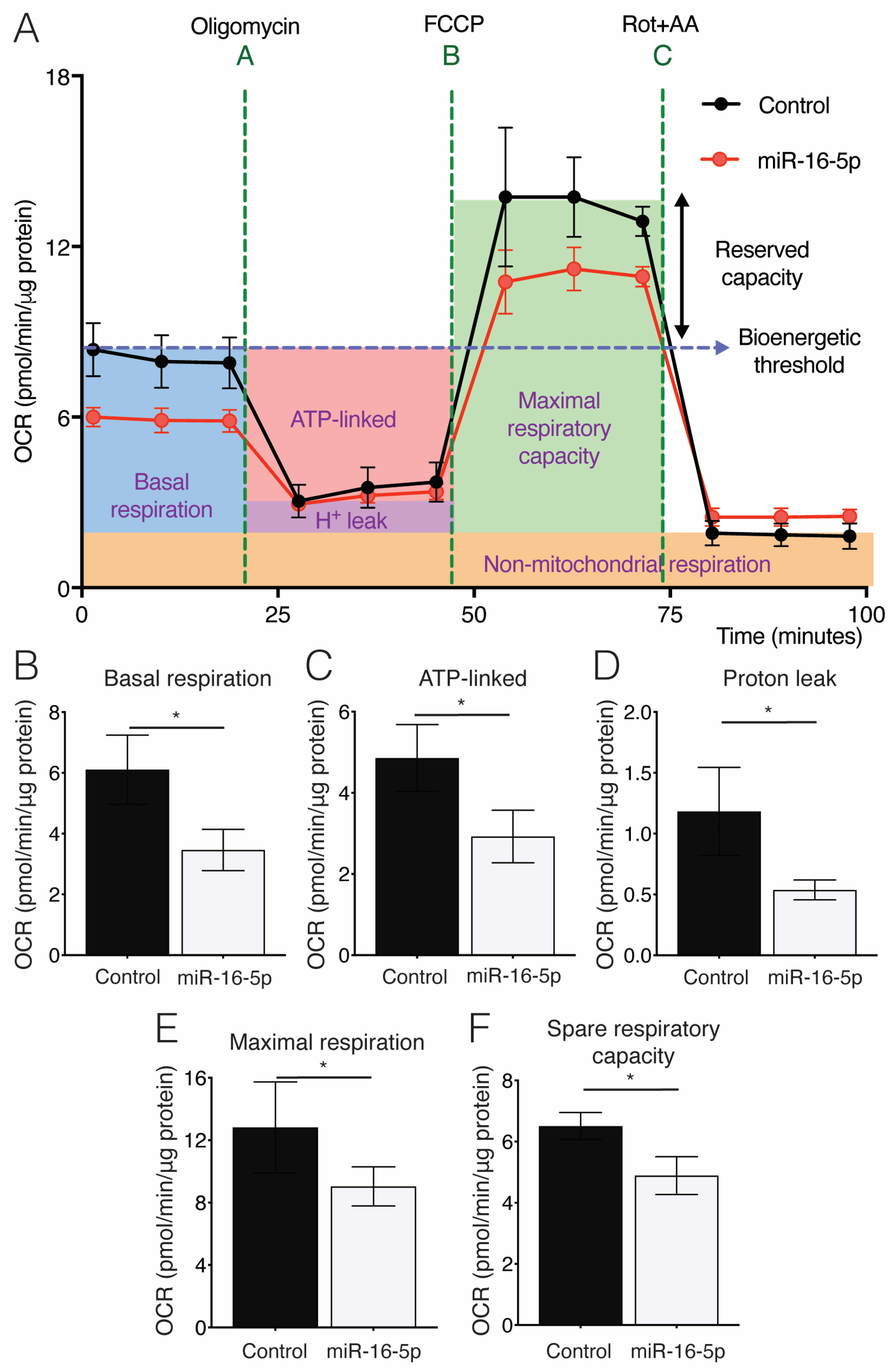
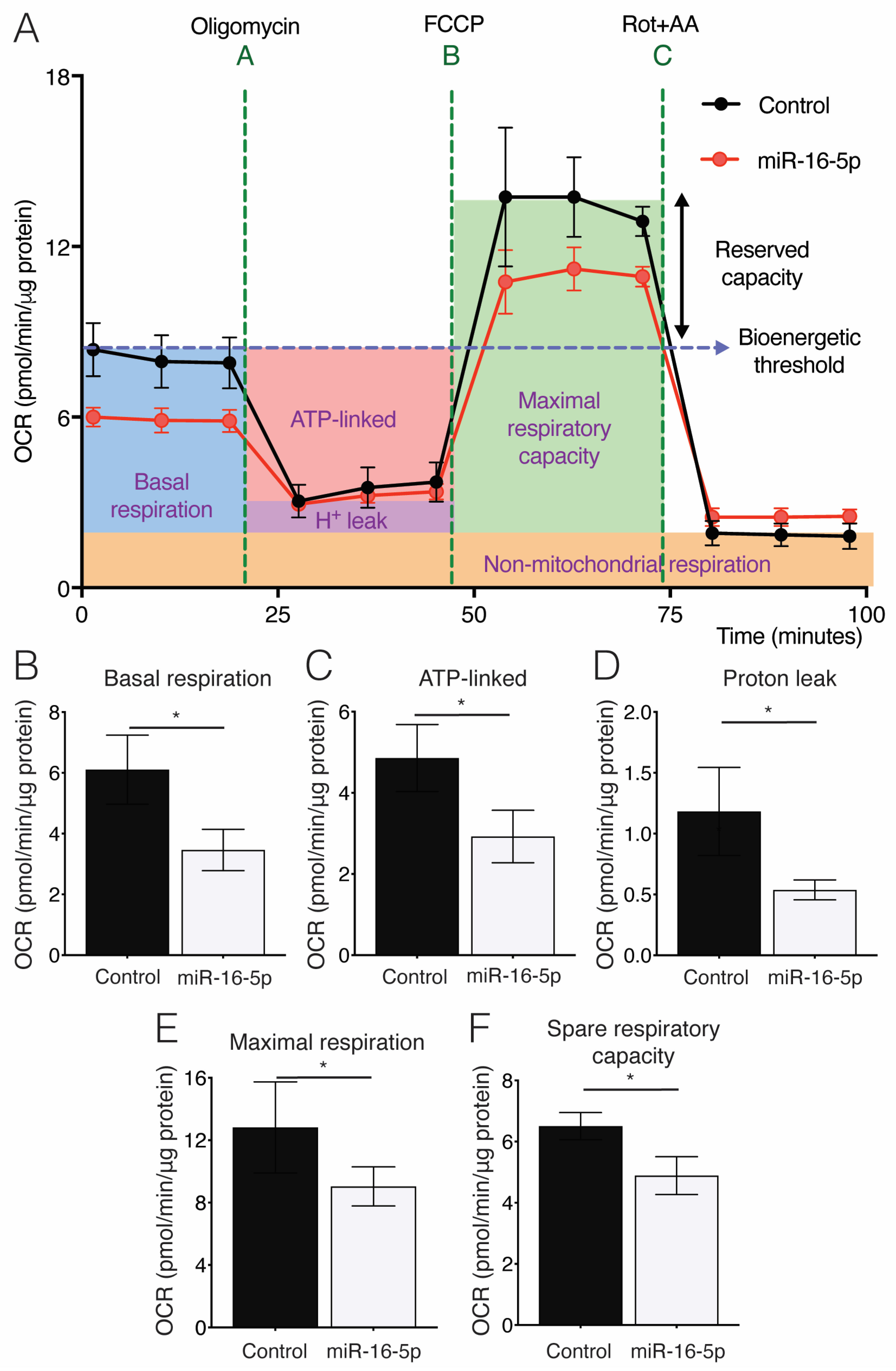
2.4. Effect of miR-16-5p on Mitochondrial Respiration in Cultured Human Cardiomyoblasts
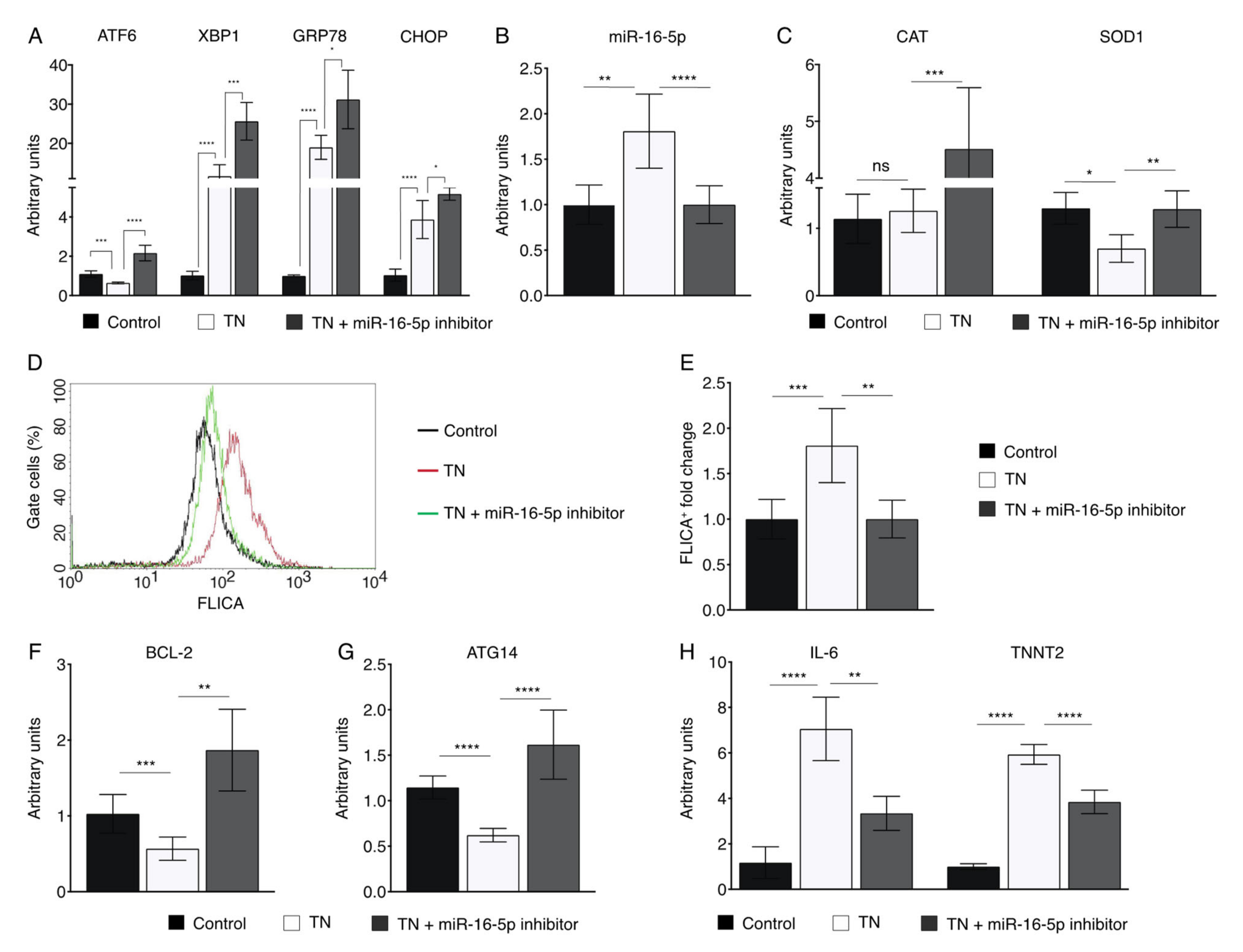
2.5. miR-16-5p Suppression Promotes the Cytoprotective Role of ER Stress in Human Cardiomyoblasts
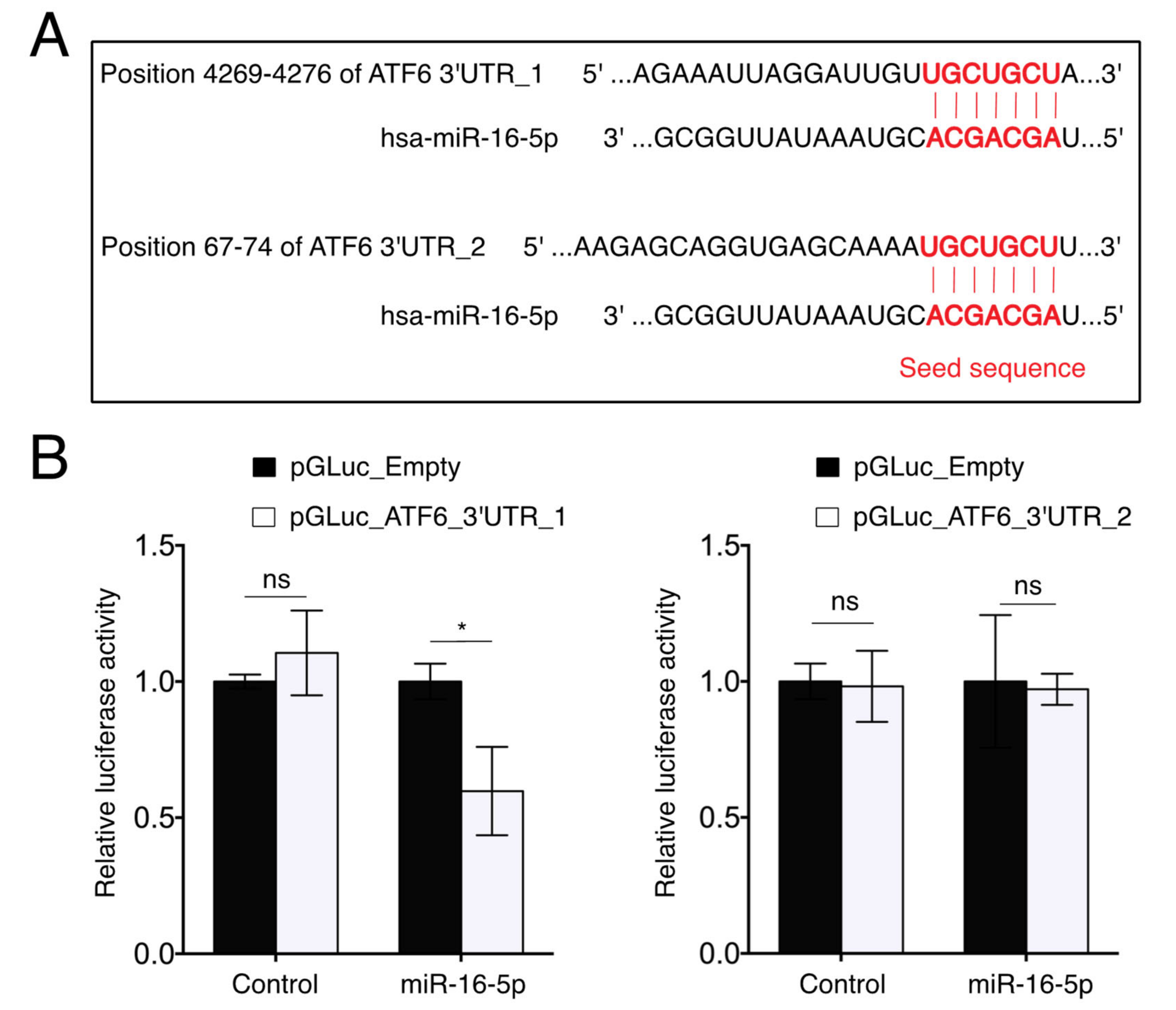
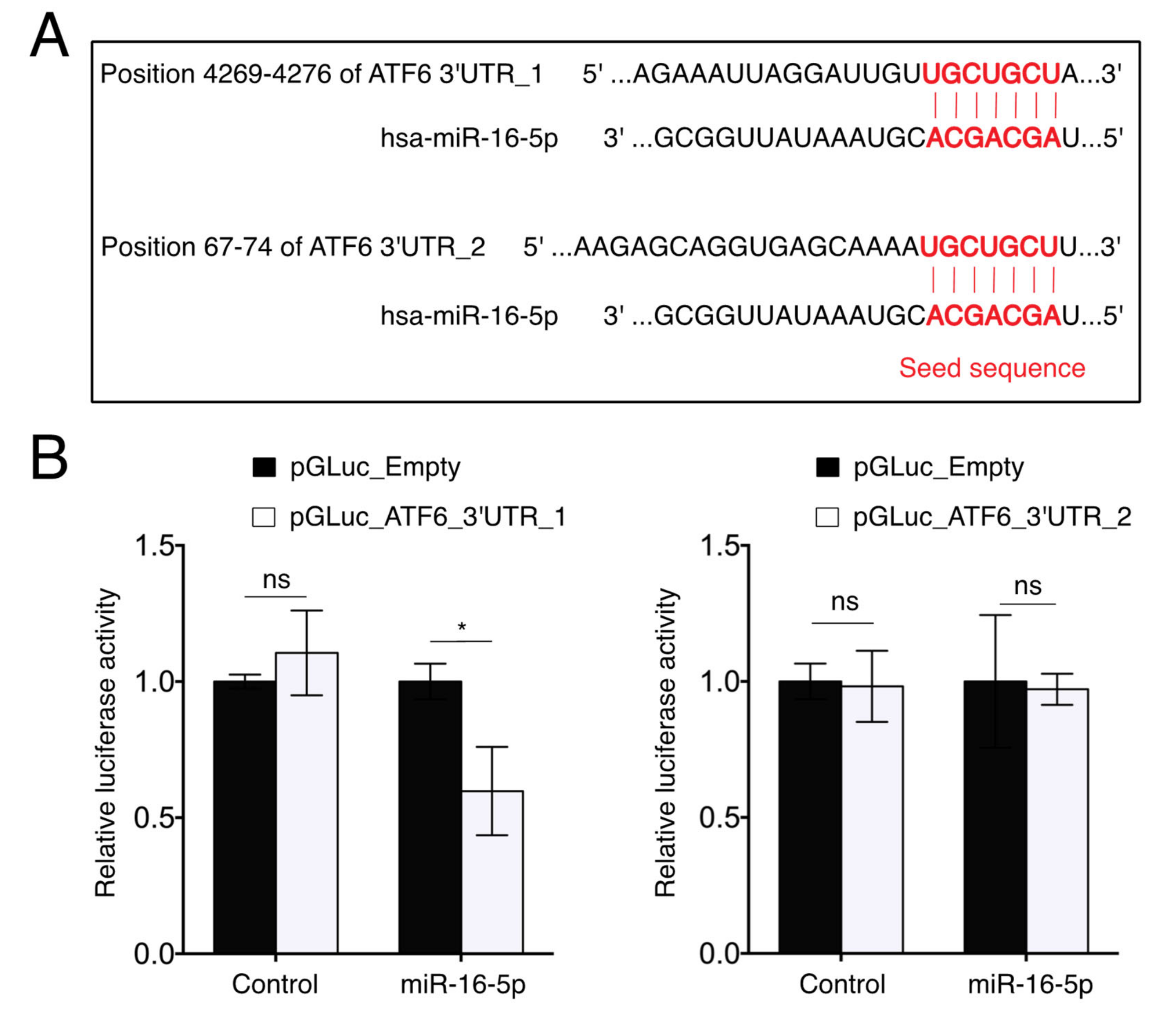
2.6. miR-16-5p Directly Targets ATF6
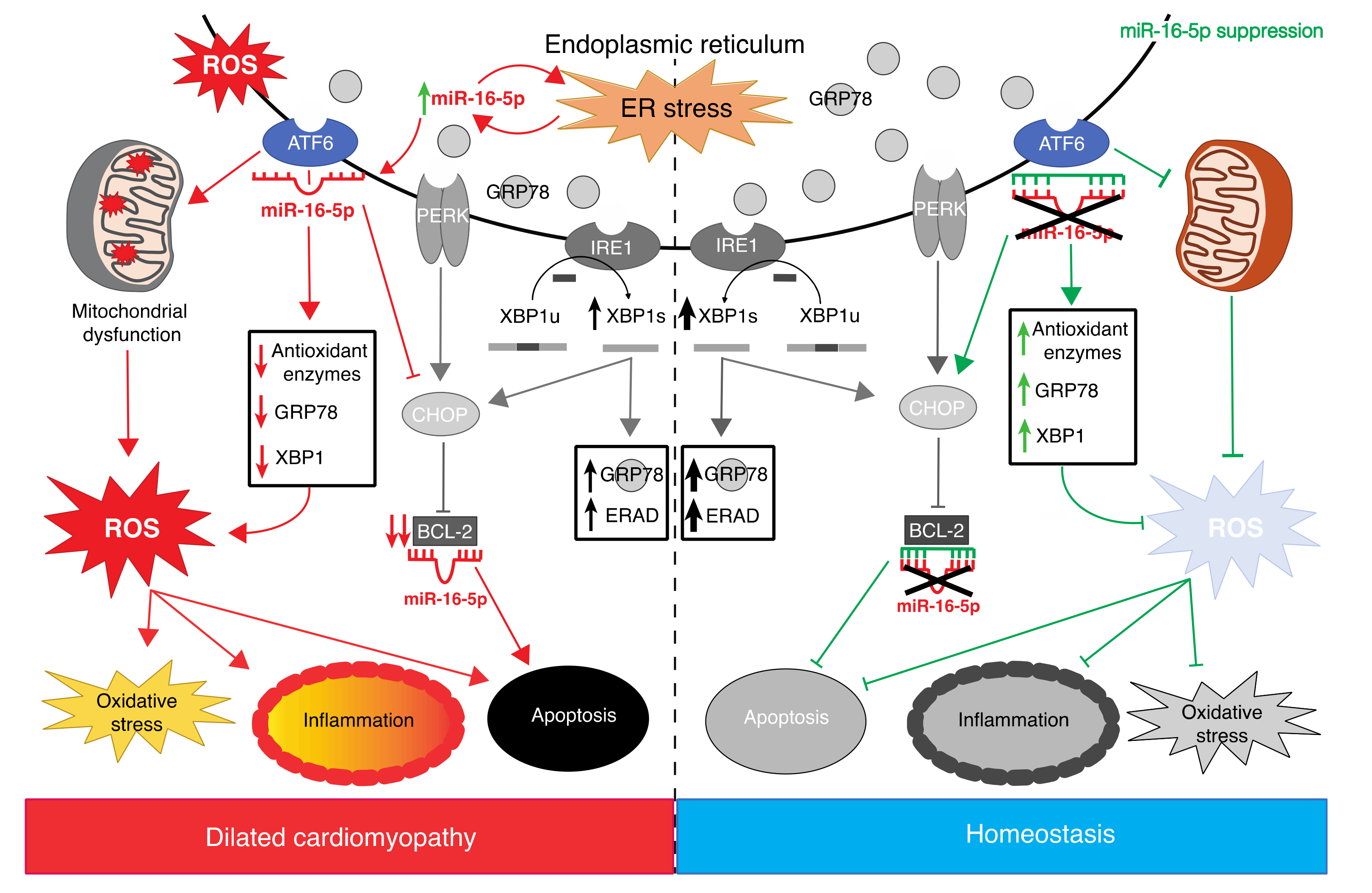
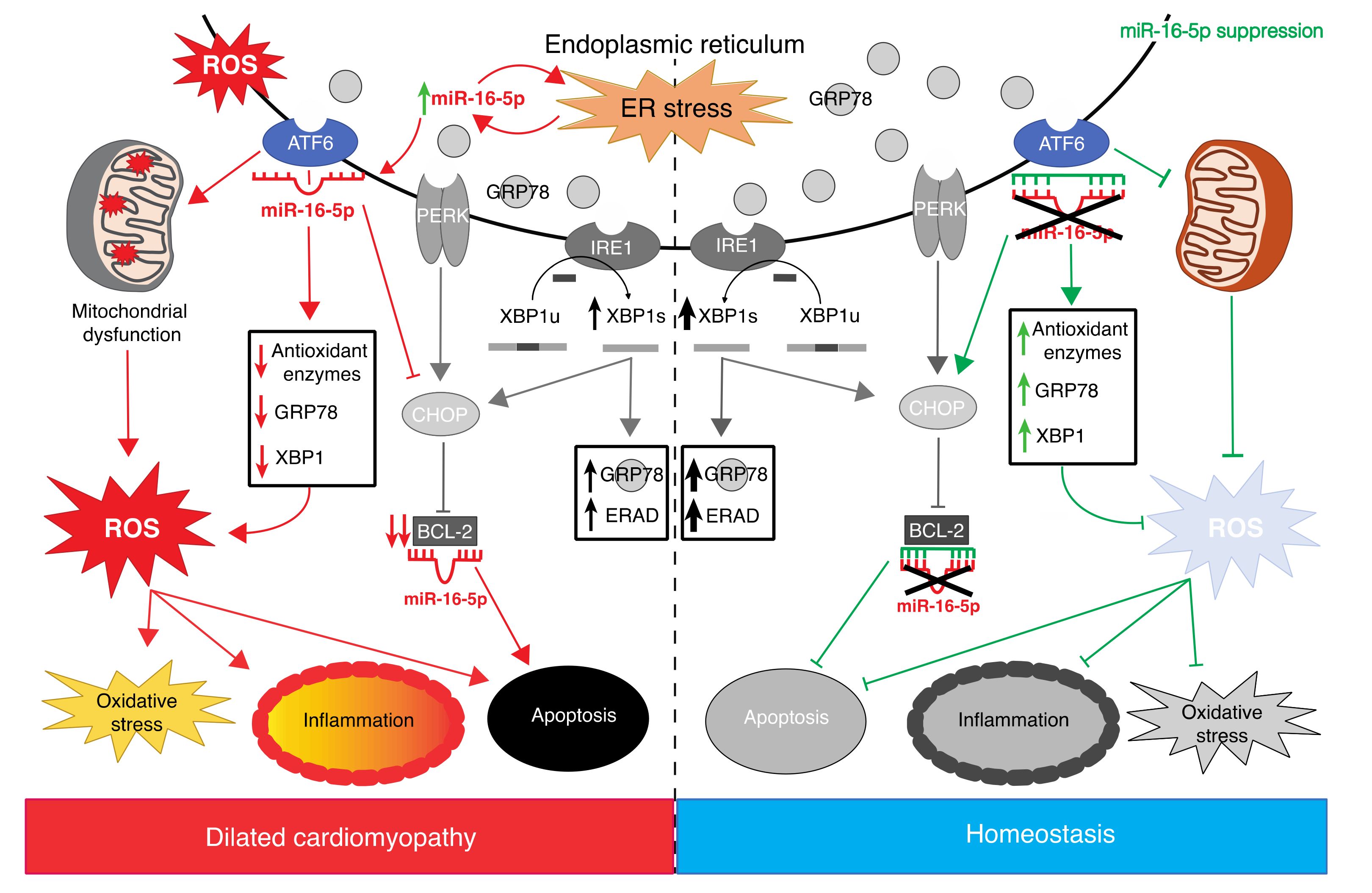
3. Discussion
4. Materials and Methods
4.1. Study Population and Blood Sampling
4.2. Oxidative Damage Determination
4.3. Enzymatic and Non-Enzymatic Antioxidant Systems
4.4. Cell Culture and Transfection
4.5. RNA Isolation and Real-Time Quantitative RT-qPCR
4.6. Western Blot Analysis
4.7. Oxygen Consumption Rate (OCR) Measurement
4.8. Luciferase Reporter Assay
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a Revised Definition of Dilated Cardiomyopathy, Hypokinetic Non-Dilated Cardiomyopathy, and Its Implications for Clinical Practice: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehlik, J.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Christie, J.D.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Hertz, M.I. The Registry of the International Society for Heart and Lung Transplantation: Twenty-Eighth Adult Heart Transplant Report—2011. J. Heart Lung Transplant. 2011, 30, 1078–1094. [Google Scholar] [CrossRef]
- Merlo, M.; Stolfo, D.; Anzini, M.; Negri, F.; Pinamonti, B.; Barbati, G.; Ramani, F.; Lenarda, A.D.; Sinagra, G. Persistent Recovery of Normal Left Ventricular Function and Dimension in Idiopathic Dilated Cardiomyopathy during Long-term Follow-up: Does Real Healing Exist? J. Am. Heart Assoc. 2015, 4, e001504. [Google Scholar] [CrossRef] [Green Version]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen Free Radicals and Congestive Heart Failure. Heart 1991, 65, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.F.; Singal, P.K. Antioxidant and Oxidative Stress Changes during Heart Failure Subsequent to Myocardial Infarction in Rats. Am. J. Pathol. 1996, 148, 291–300. [Google Scholar]
- Hill, M.F.; Singal, P.K. Right and Left Myocardial Antioxidant Responses During Heart Failure Subsequent to Myocardial Infarction. Circulation 1997, 96, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated Levels of 8-Iso-Prostaglandin F 2α in Pericardial Fluid of Patients with Heart Failure: A Potential Role for In Vivo Oxidant Stress in Ventricular Dilatation and Progression to Heart Failure. Circulation 1998, 97, 1536–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA Damage and Dysfunction Associated with Oxidative Stress in Failing Hearts After Myocardial Infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.; et al. Oxidant Stress from Nitric Oxide Synthase–3 Uncoupling Stimulates Cardiac Pathologic Remodeling from Chronic Pressure Load. J. Clin. Invest. 2005, 115, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of Mitochondrial Peroxiredoxin-3 Prevents Left Ventricular Remodeling and Failure After Myocardial Infarction in Mice. Circulation 2006, 113, 1779–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesselli, D.; Jakoniuk, I.; Barlucchi, L.; Beltrami, A.P.; Hintze, T.H.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Oxidative Stress–Mediated Cardiac Cell Death Is a Major Determinant of Ventricular Dysfunction and Failure in Dog Dilated Cardiomyopathy. Circ. Res. 2001, 89, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Mollnau, H.; Oelze, M.; August, M.; Wendt, M.; Daiber, A.; Schulz, E.; Baldus, S.; Kleschyov, A.L.; Materne, A.; Wenzel, P.; et al. Mechanisms of Increased Vascular Superoxide Production in an Experimental Model of Idiopathic Dilated Cardiomyopathy. ATVB 2005, 25, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minhas, K.M.; Saraiva, R.M.; Schuleri, K.H.; Lehrke, S.; Zheng, M.; Saliaris, A.P.; Berry, C.E.; Vandegaer, K.M.; Li, D.; Hare, J.M. Xanthine Oxidoreductase Inhibition Causes Reverse Remodeling in Rats with Dilated Cardiomyopathy. Circ. Res. 2006, 98, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Ma, Y.; Zhang, W.; Bao, D.; Dong, W.; Lian, H.; Huang, L.; Zhang, L. Knockdown of Cytochrome P450 2E1 Inhibits Oxidative Stress and Apoptosis in the CTnT R141W Dilated Cardiomyopathy Transgenic Mice. Hypertension 2012, 60, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, J.; Arnér, E.S.J. Reactive Oxygen Species, Antioxidants, and the Mammalian Thioredoxin. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Giordano, F.J. Oxygen, Oxidative Stress, Hypoxia, and Heart Failure. J. Clin. Invest. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; Yang, L.; Zhang, M.; Gu, Y. Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Contributes to Aldosterone-Induced Apoptosis in Tubular Epithelial Cells. Biochem. Biophys. Res. Commun. 2012, 418, 451–456. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Gu, S.; Chen, C.; Jiang, X.; Zhang, Z. ROS-Mediated Endoplasmic Reticulum Stress and Mitochondrial Dysfunction Underlie Apoptosis Induced by Resveratrol and Arsenic Trioxide in A549 Cells. Chem. Biol. Interact. 2016, 245, 100–109. [Google Scholar] [CrossRef] [PubMed]
- McLendon, P.M.; Robbins, J. Proteotoxicity and Cardiac Dysfunction. Circ. Res. 2015, 116, 1863–1882. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, A.; Blackwood, E.A.; Glembotski, C.C. ER Protein Quality Control and the Unfolded Protein Response in the Heart. In Coordinating Organismal Physiology Through the Unfolded Protein Response; Current Topics in Microbiology and Immunology; Wiseman, R.L., Haynes, C.M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 414, pp. 193–213. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Jin, J.K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, T.; Kitakaze, M. ER Stress in Cardiovascular Disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic Reticulum Stress in the Heart: Insights into Mechanisms and Drug Targets: ER Stress in the Heart. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callis, T.E.; Chen, J.-F.; Wang, D.-Z. MicroRNAs in Skeletal and Cardiac Muscle Development. DNA Cell Biol. 2007, 26, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Wang, D.-Z. Taking MicroRNAs to Heart. Trends Mol. Med. 2008, 14, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Jiao, K. Functions of MiRNAs during Mammalian Heart Development. Int. J. Mol. Sci. 2016, 17, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-Specific MiRNA in Cardiogenesis, Heart Function, and Cardiac Pathology (with Focus on Myocardial Infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Duygu, B.; de Windt, L.J.; da Costa Martins, P.A. Targeting MicroRNAs in Heart Failure. Trends Cardiovasc. Med. 2016, 26, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Colpaert, R.M.W.; Calore, M. MicroRNAs in Cardiac Diseases. Cells 2019, 8, 737. [Google Scholar] [CrossRef] [Green Version]
- Kreutzer, F.P.; Fiedler, J.; Thum, T. Non-coding RNAs: Key Players in Cardiac Disease. J. Physiol. 2020, 598, 2995–3003. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Duan, L.; Li, Y.; Liu, B. Long Noncoding RNA/Circular Noncoding RNA–MiRNA–MRNA Axes in Cardiovascular Diseases. Life Sci. 2019, 233, 116440. [Google Scholar] [CrossRef]
- Omidkhoda, N.; Wallace Hayes, A.; Reiter, R.J.; Karimi, G. The Role of MicroRNAs on Endoplasmic Reticulum Stress in Myocardial. Ischemia Card. Hypertrophy. Pharmacol. Res. 2019, 150, 104516. [Google Scholar] [CrossRef]
- Calderon-Dominguez, M.; Belmonte, T.; Quezada-Feijoo, M.; Ramos-Sánchez, M.; Fernández-Armenta, J.; Pérez-Navarro, A.; Cesar, S.; Peña-Peña, L.; Vea, À.; Llorente-Cortés, V.; et al. Emerging Role of MicroRNAs in Dilated Cardiomyopathy: Evidence Regarding Etiology. Transl. Res. 2020, 215, 86–101. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, C.K.V.; Macêdo, C.T.; Cavalcante, B.R.R.; de Alcântara, A.C.; Silva, D.N.; Bezerra, M.d.R.; Caria, A.C.I.; Tavora, F.R.F.; Neto, J.D.d.S.; Noya-Rabelo, M.M.; et al. Circulating MiRNAs as Potential Biomarkers Associated with Cardiac Remodeling and Fibrosis in Chagas Disease Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-Dominguez, M.; Mangas, A.; Belmonte, T.; Quezada-Feijoo, M.; Ramos, M.; Toro, R. Ischemic Dilated Cardiomyopathy Pathophysiology through MicroRNA-16-5p. Rev. Española Cardiol. (Engl. Ed.) 2021, 74, 740–749. [Google Scholar] [CrossRef]
- Kaneda, H.; Taguchi, J.; Ogasawara, K.; Aizawa, T.; Ohno, M. Increased Level of Advanced Oxidation Protein Products in Patients with Coronary Artery Disease. Atherosclerosis 2002, 162, 221–225. [Google Scholar] [CrossRef]
- Lynch, T.L.; Sivaguru, M.; Velayutham, M.; Cardounel, A.J.; Michels, M.; Barefield, D.; Govindan, S.; dos Remedios, C.; van der Velden, J.; Sadayappan, S. Oxidative Stress in Dilated Cardiomyopathy Caused by MYBPC3 Mutation. Oxidative Med. Cell. Longev. 2015, 2015, 424751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial Dysfunction and Oxidative Stress in Heart Disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Metformin Attenuates ER Stress–Induced Mitochondrial Dysfunction. Transl. Res. 2017, 190, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Rasool, M.; Malik, A.; Butt, T.T.; Ashraf, M.A.B.; Rasool, R.; Zahid, A.; Waquar, S.; Asif, M.; Zaheer, A.; Jabbar, A.; et al. Implications of Advanced Oxidation Protein Products (AOPPs), Advanced Glycation End Products (AGEs) and Other Biomarkers in the Development of Cardiovascular Diseases. Saudi J. Biol. Sci. 2019, 26, 334–339. [Google Scholar] [CrossRef]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of MiR-15 Protects Against Cardiac Ischemic Injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Y.; Teng, Z. Downregulation of MiR-16 Protects H9c2(2-1) Cells against Hypoxia/Reoxygenation Damage by Targeting CIAPIN1 and Regulating the NF-κB Pathway. Mol. Med. Rep. 2019, 20, 3113–3122. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shang, Y.; Dai, S.; Wu, W.; Yi, F.; Cheng, L. MicroRNA-16-5p Aggravates Myocardial Infarction Injury by Targeting the Expression of Insulin Receptor Substrates 1 and Mediating Myocardial Apoptosis and Angiogenesis. CNR 2020, 17, 11–17. [Google Scholar] [CrossRef]
- Liu, J.; Sun, F.; Wang, Y.; Yang, W.; Xiao, H.; Zhang, Y.; Lu, R.; Zhu, H.; Zhuang, Y.; Pan, Z.; et al. Suppression of MicroRNA-16 Protects against Acute Myocardial Infarction by Reversing Beta2-Adrenergic Receptor down-Regulation in Rats. Oncotarget 2017, 8, 20122–20132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Z.P.; Xu, P.; Shi, Y.; Gao, A.-M. MicroRNA-93 Inhibits Ischemia-Reperfusion Induced Cardiomyocyte Apoptosis by Targeting PTEN. Oncotarget 2016, 7, 28796–28805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prola, A.; Nichtova, Z.; Pires Da Silva, J.; Piquereau, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Ventura-Clapier, R.; Garnier, A.; Zahradnik, I.; et al. Endoplasmic Reticulum Stress Induces Cardiac Dysfunction through Architectural Modifications and Alteration of Mitochondrial Function in Cardiomyocytes. Cardiovasc. Res. 2019, 115, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Samidurai, A.; Thompson, J.; Hu, Y.; Das, A.; Willard, B.; Lesnefsky, E.J. Endoplasmic Reticulum Stress-Mediated Mitochondrial Dysfunction in Aged Hearts. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165899. [Google Scholar] [CrossRef]
- Wang, S.; Hu, B.; Ding, Z.; Dang, Y.; Wu, J.; Li, D.; Liu, X.; Xiao, B.; Zhang, W.; Ren, R.; et al. ATF6 Safeguards Organelle Homeostasis and Cellular Aging in Human Mesenchymal Stem Cells. Cell Discov. 2018, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Paxman, R.J.; Plate, L.; Kelly, J.W.; Wiseman, R.L.; Glembotski, C.C. Pharmacologic ATF6 Activation Confers Global Protection in Widespread Disease Models by Reprograming Cellular Proteostasis. Nat. Commun. 2019, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.-J.; Xiong, Z.; Lu, X.; Ye, M.; Han, X.; Jiang, R. ATF6 Upregulates XBP1S and Inhibits ER Stress-Mediated Apoptosis in Osteoarthritis Cartilage. Cell. Signal. 2014, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Hegde, R.S. Regulation of Basal Cellular Physiology by the Homeostatic Unfolded Protein Response. Journal of Cell Biology 2010, 189, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Liu, Y.; Adachi, M.; Zhao, S.; Hareyama, M.; Koong, A.C.; Luo, D.; Rando, T.A.; Imai, K.; Shinomura, Y. Preventing Oxidative Stress: A New Role for XBP1. Cell Death Differ. 2009, 16, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. MiR-15 and MiR-16 Induce Apoptosis by Targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [Green Version]
- Slomp, A.; Peperzak, V. Role and Regulation of Pro-Survival BCL-2 Proteins in Multiple Myeloma. Front. Oncol. 2018, 8, 533. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the Cardiomyopathies: A Position Statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2007, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belmonte, T.; Mangas, A.; Calderon-Dominguez, M.; Quezada-Feijoo, M.; Ramos, M.; Campuzano, O.; Gomez, S.; Peña, M.L.; Cubillos-Arango, A.M.; Dominguez, F.; et al. Peripheral MicroRNA Panels to Guide the Diagnosis of Familial Cardiomyopathy. Transl. Res. 2020, 218, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Roberts, L.J. Measurement of Lipid Peroxidation. Free Radic. Res. 1998, 28, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Friedlander, M.; Capeillère-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced Oxidation Protein Products as a Novel Marker of Oxidative Stress in Uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Aebi, H. Catalase in Vitro. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1984; Volume 105, pp. 121–126. [Google Scholar] [CrossRef]
- Bamforth, C.W. Superoxide dismutase in barley. J. Inst. Brew. 1983, 89, 420–423. [Google Scholar] [CrossRef]
- Günzler, W.A.; Kremers, H.; Flohé, L. An Improved Coupled Test Procedure for Glutathione Peroxidase (EC 1.11.1.9.) in Blood. Clin. Chem. Lab. Med. 1974, 12, 444–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Nuebel, E.; Wisidagama, D.R.R.; Setoguchi, K.; Hong, J.S.; Van Horn, C.M.; Imam, S.S.; Vergnes, L.; Malone, C.S.; Koehler, C.M.; et al. Measuring Energy Metabolism in Cultured Cells, Including Human Pluripotent Stem Cells and Differentiated Cells. Nat. Protoc. 2012, 7, 1068–1085. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Healthy Control (n = 12) | Ischemic DCM (n = 12) | p-Value |
|---|---|---|---|
| Age (years) a, means ± SD | 45.2 ± 14.4 | 63.0 ± 8.8 | 0.006 |
| Sex (male) | 62.5% | 66.7% | ns |
| BMI (Kg/m2) a, means ± SD | 22.9 ± 3.51 | 29.0 ± 2.9 | 0.003 |
| LVEF (%) a, means ± SD | 68.5 ± 6.05 | 34.0 ± 5.8 | <0.001 |
| LVEDD (mm) b, means ± SD | 45.7 ± 5.3 | 61.7 ± 6.5 | <0.001 |
| LVESD (mm) a, means ± SD | 28.0 ± 4.2 | 46.2 ± 15.5 | 0.004 |
| LA (mm) a, means ± SD | 36.6 ± 6.9 | 47.2 ± 5.9 | 0.002 |
| Sphericity index a, means ± SD | 0.6 ± 0,03 | 0.7 ± 0.04 | 0.03 |
| TAPSE (mm) a, means ± SD | 19.09 ± 4.37 | 21.44 ± 4.21 | ns |
| MAPSE (mm) a, means ± SD | 18.77 ± 2.88 | 10.98 ± 2.24 | <0.001 |
| MI | - | 10 (83.4%) | 0.001 |
| TI | - | 6 (50%) | 0.02 |
| E/e′ ratio b, means ± SD | 7.59 ± 1.59 | 18.12 ± 7.62 | <0.001 |
| NYHA class | I | II | <0.001 |
| miR-16-5p expression levels (log2) a | 7.522 ± 0.43 | 8.232 ± 0.52 | 0.005 |
| Gene | Forward | Reverse |
|---|---|---|
| ATF6 | 5′-AATACTGAACTATGGACCTATGAGCA-3′ | 5′-TTGCAGGGCTCACACTAGG-3′ |
| ATG14 | 5′-TGGGGACTACTCTGCCTACTACA-3′ | 5′-GGGTTACTCTGCTCCATGTCA-3′ |
| BCL-2 | 5′-AGCACGTGCACAGCTTCA-3′ | 5′-GTCCACGGGTGAAACAGC-3′ |
| CATALASE | 5′-TCTGGACAAGTACAATGCTGAGA-3′ | 5′-TAAGCTTCGCTGCACAGGT-3′ |
| CHOP | 5′-TCACCACACCTGAAAGCAGA-3′ | 5′-TCTTGCAGGTCCTCATACCA-3′ |
| GAPDH | 5′-AGCCACATCGCTCAGACAC-3′ | 5′-AATACGACCAAATCCGTTGACT-3′ |
| GRP78 | 5′-AATGACCAGAATCGCCTGAC-3′ | 5′-ATGCGCTCCTTGAGCTTTT-3′ |
| GSH | 5′-CCTGCTAGTGGATGCTGTCA-3′ | 5′-TCATCCTGTTTGATGGTGCT-3′ |
| IL-6 | 5′-GATGAGTACAAAAGTCCTGATCCA-3′ | 5′-CTGCAGCCACTGGTTCTGT-3′ |
| SOD1 | 5′-TCCATGTTCATGAGTTTGGAGAT-3′ | 5′-CCCACCGTGTTTTCTGGATA-3′ |
| TNNT2 | 5′-GGCTGCAGTGGCTACAGG-3′ | 5′-CTGTCACCAGGCAATACAGC-3′ |
| XBP1 | 5′-TGCGTAGTCTGGAGCTATGGT-3′ | 5′-CCCGACAGAAGCAGAACTTT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toro, R.; Pérez-Serra, A.; Mangas, A.; Campuzano, O.; Sarquella-Brugada, G.; Quezada-Feijoo, M.; Ramos, M.; Alcalá, M.; Carrera, E.; García-Padilla, C.; et al. miR-16-5p Suppression Protects Human Cardiomyocytes against Endoplasmic Reticulum and Oxidative Stress-Induced Injury. Int. J. Mol. Sci. 2022, 23, 1036. https://doi.org/10.3390/ijms23031036
Toro R, Pérez-Serra A, Mangas A, Campuzano O, Sarquella-Brugada G, Quezada-Feijoo M, Ramos M, Alcalá M, Carrera E, García-Padilla C, et al. miR-16-5p Suppression Protects Human Cardiomyocytes against Endoplasmic Reticulum and Oxidative Stress-Induced Injury. International Journal of Molecular Sciences. 2022; 23(3):1036. https://doi.org/10.3390/ijms23031036
Chicago/Turabian StyleToro, Rocío, Alexandra Pérez-Serra, Alipio Mangas, Oscar Campuzano, Georgia Sarquella-Brugada, Maribel Quezada-Feijoo, Mónica Ramos, Martin Alcalá, Esther Carrera, Carlos García-Padilla, and et al. 2022. "miR-16-5p Suppression Protects Human Cardiomyocytes against Endoplasmic Reticulum and Oxidative Stress-Induced Injury" International Journal of Molecular Sciences 23, no. 3: 1036. https://doi.org/10.3390/ijms23031036
APA StyleToro, R., Pérez-Serra, A., Mangas, A., Campuzano, O., Sarquella-Brugada, G., Quezada-Feijoo, M., Ramos, M., Alcalá, M., Carrera, E., García-Padilla, C., Franco, D., & Bonet, F. (2022). miR-16-5p Suppression Protects Human Cardiomyocytes against Endoplasmic Reticulum and Oxidative Stress-Induced Injury. International Journal of Molecular Sciences, 23(3), 1036. https://doi.org/10.3390/ijms23031036