


An Isochroman Analog of CD3254 and Allyl-, Isochroman-Analogs of NEt-TMN Prove to Be More Potent Retinoid-X-Receptor (RXR) Selective Agonists Than Bexarotene

 ,
,  , , and
, , and
Abstract
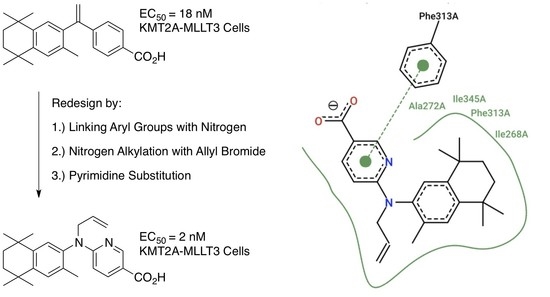
1. Introduction
2. Results: Molecular Modeling
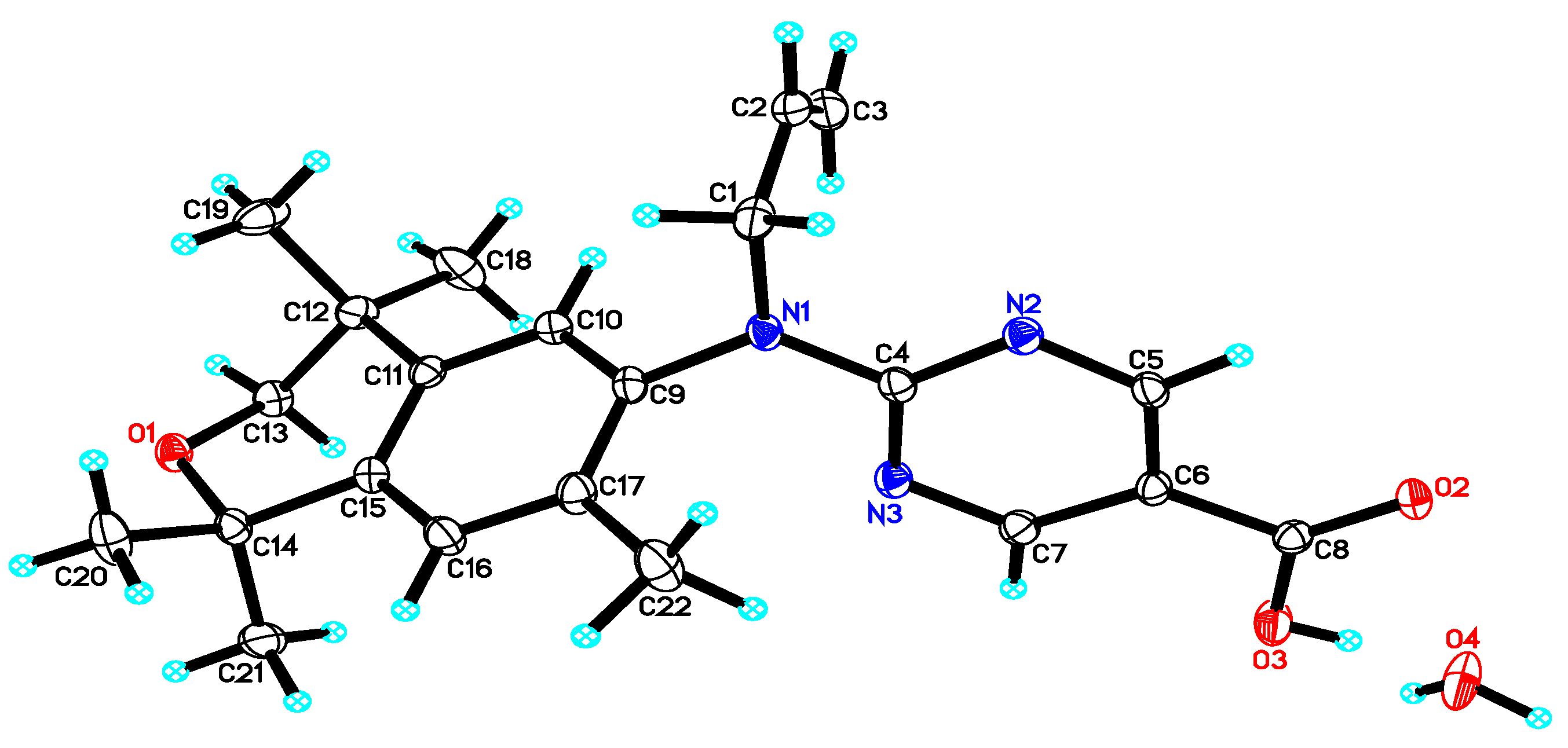
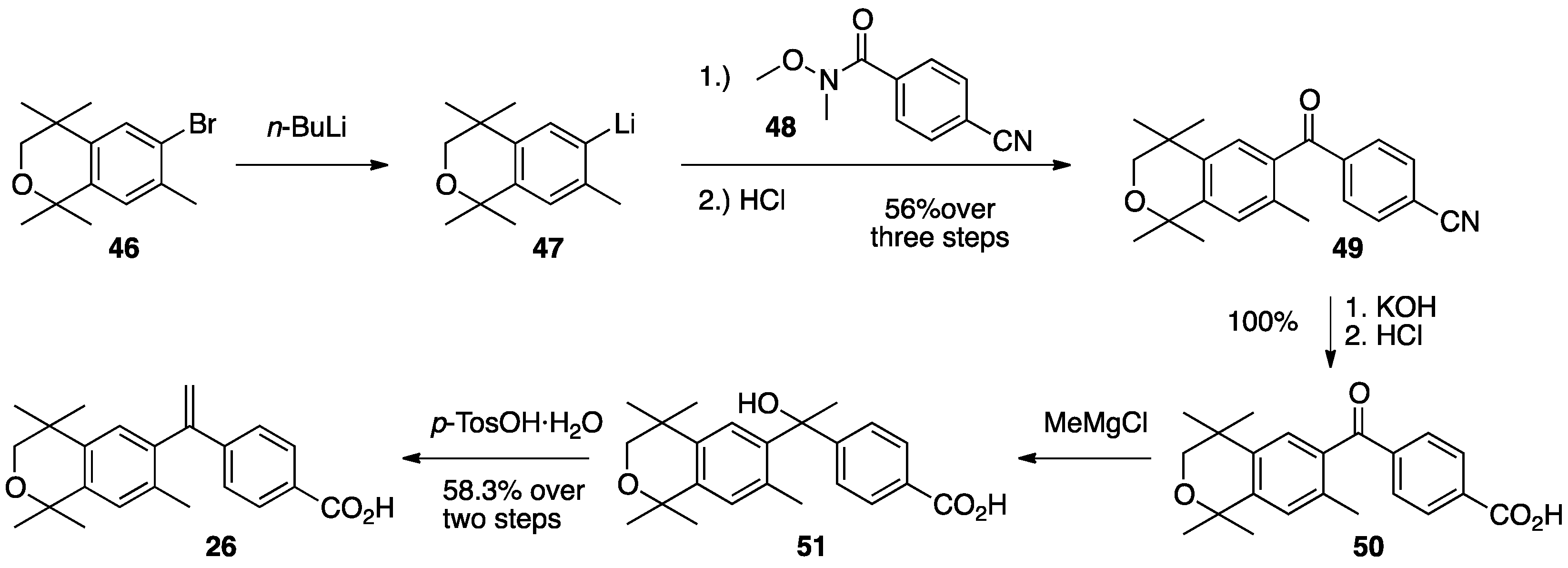
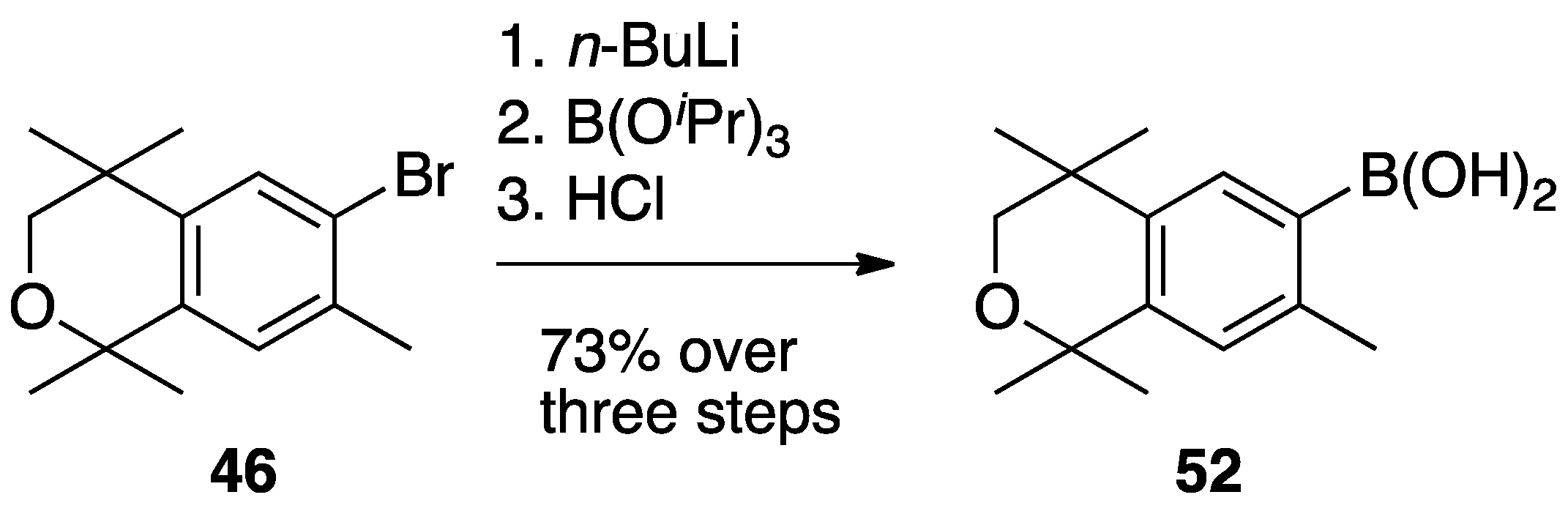
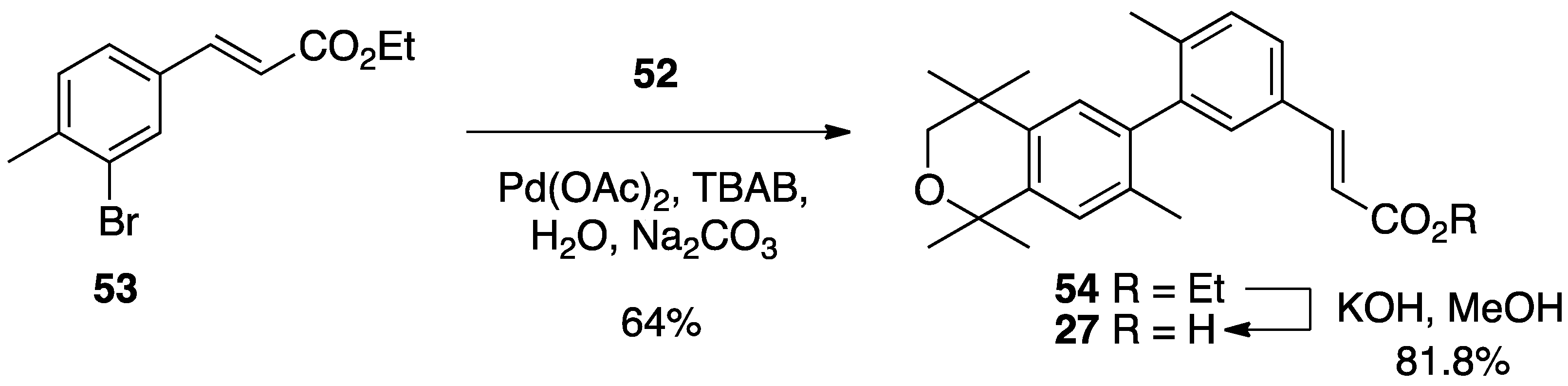
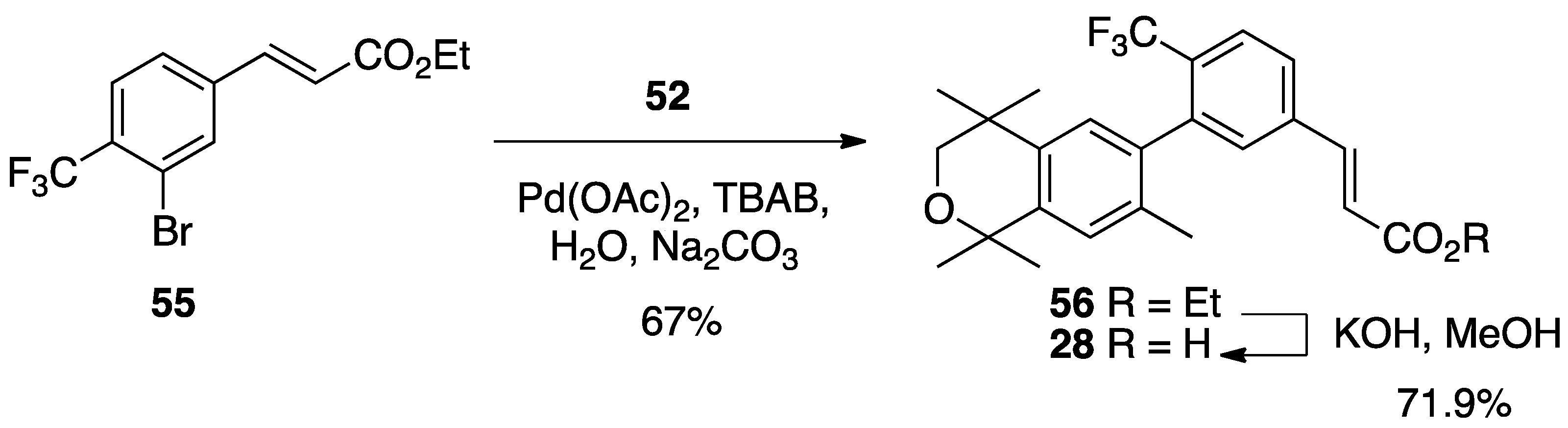
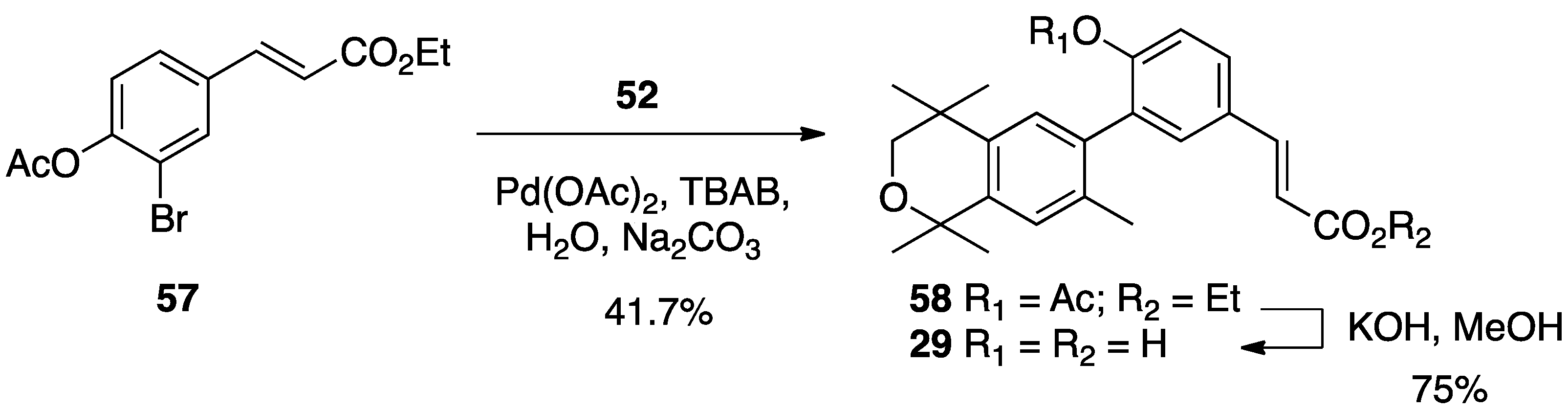
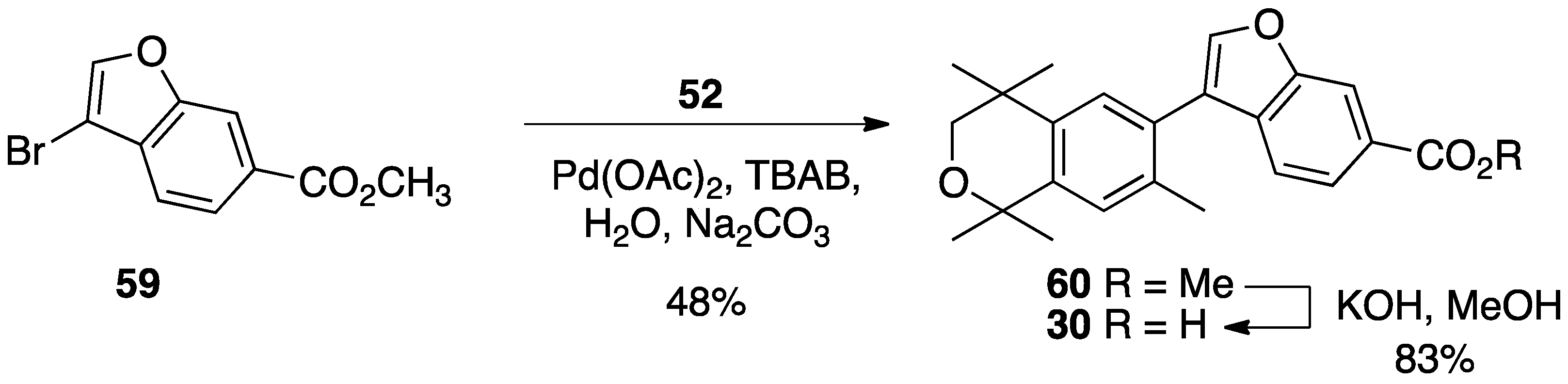
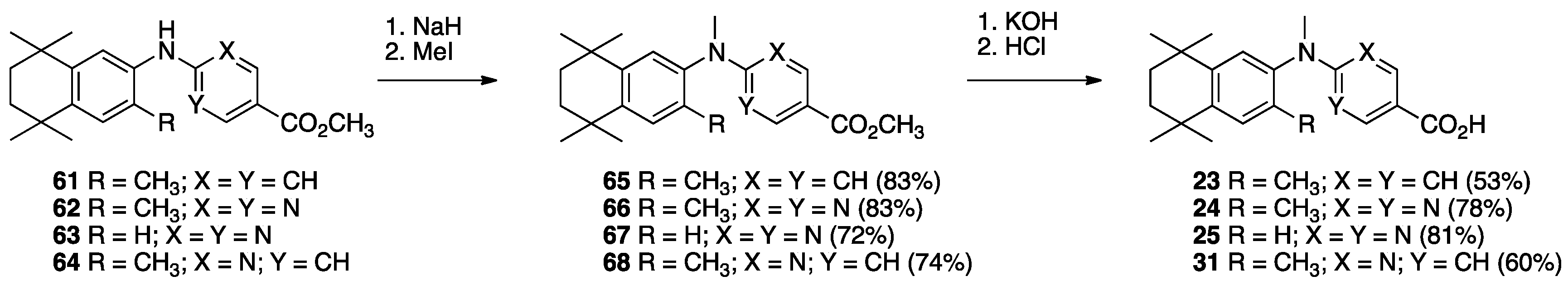
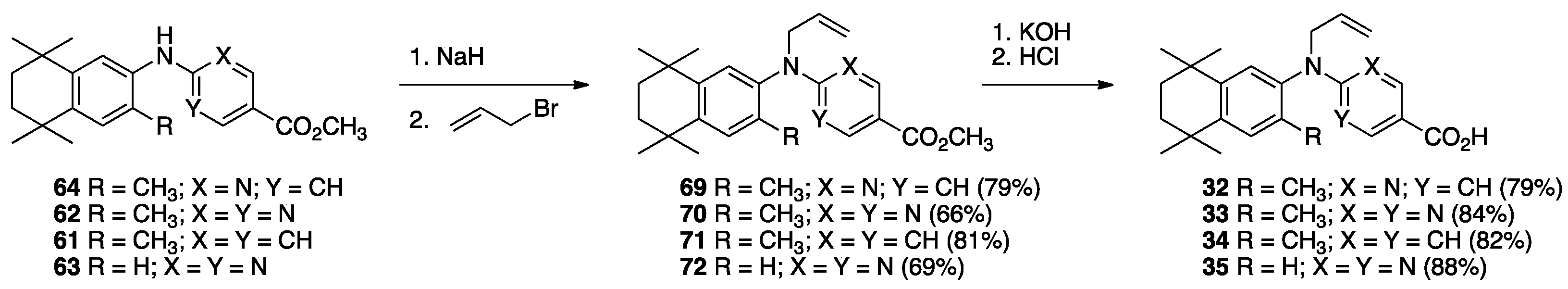
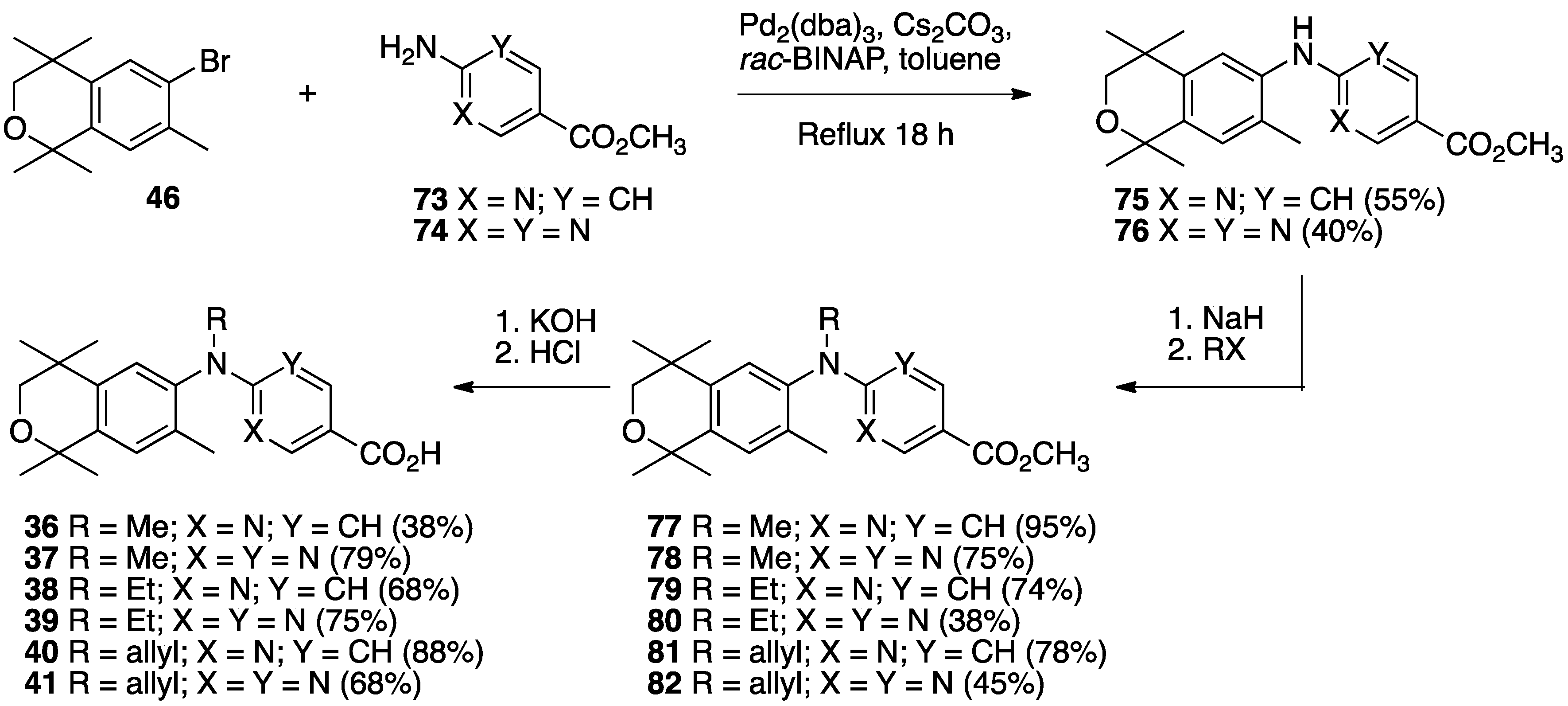
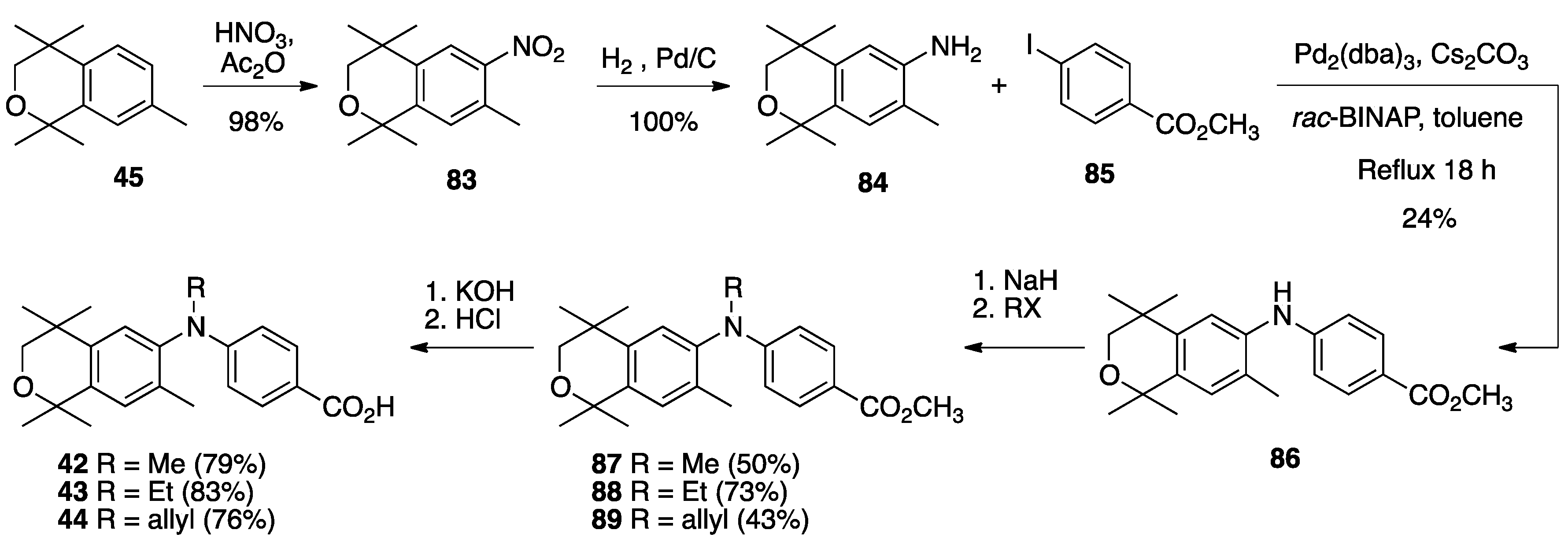
3. Results: Chemistry
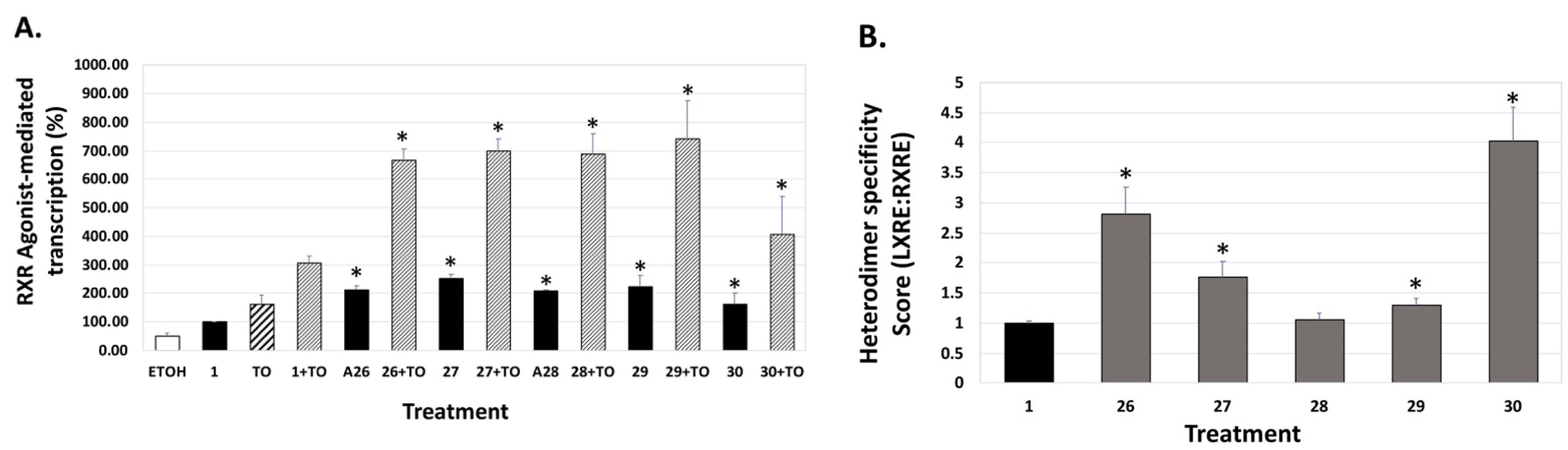
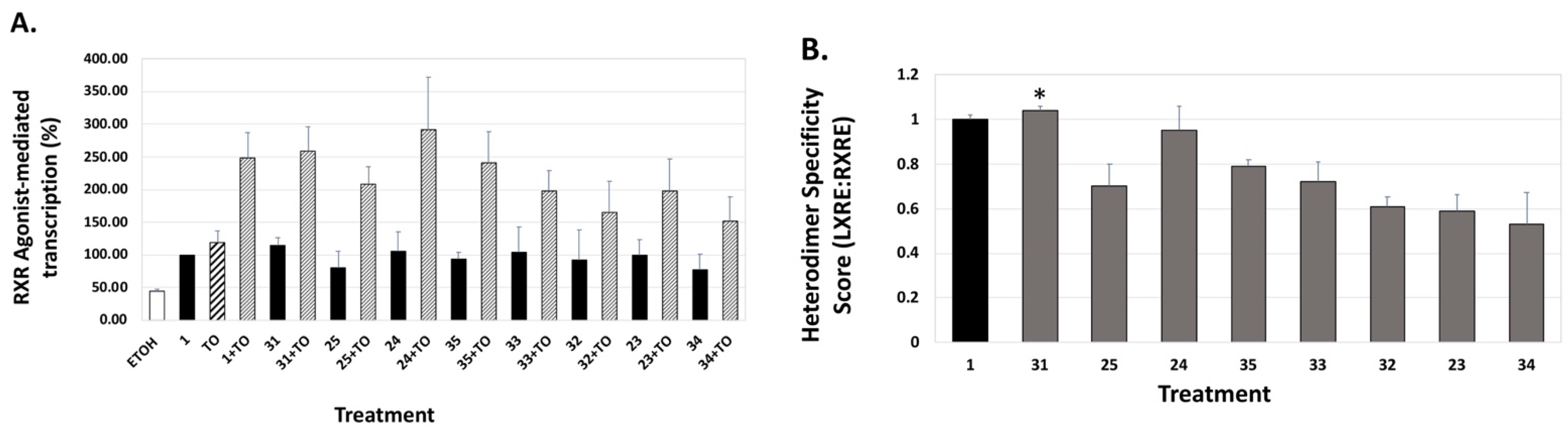
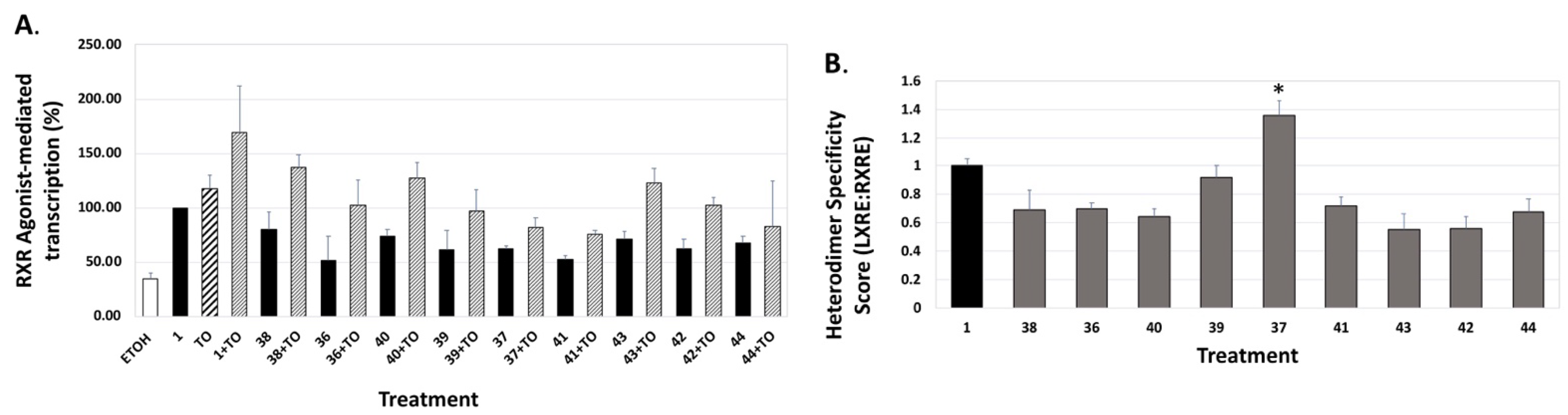
4. Results and Discussion: Biological Assays
5. Conclusions
6. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATRA | all-trans-retinoic acid |
| AD | Alzheimer’s disease |
| 9-cis-RA | 9-cis-retinoic acid |
| CTCL | cutaneous T-cell lymphoma |
| DMF | dimethylformamide |
| DMSO | dimethylsulfoxide |
| DNA | deoxyribonucleic acid |
| FXR | farnesoid-X-receptor |
| HCl | hydrochloric acid |
| HRE | hormone responsive element |
| KOH | potassium hydroxide |
| LBD | ligand binding domain |
| LHS | LXR Heterodimer Specificity |
| LR | lipid risk assessment index |
| LXR | liver-X-receptor |
| LXRE | liver-X-receptor element |
| NaBu | sodium butyrate |
| NR | nuclear receptor |
| POC | proof of concept |
| PPAR | peroxisome proliferator activating receptor |
| RAR | retinoic-acid-receptor |
| RARE | retinoic acid receptor element |
| RXR | retinoid-X-receptor |
| RXRE | retinoid-X-receptor element |
| SNuRMs | specific nuclear receptor modulators |
| SREBP | sterol regulatory element binding protein |
| TR | thyroid hormone receptor |
| VDR | vitamin D receptor |
References
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. The Retinoids; Academic Press: Orlando, FL, USA, 1994; pp. 319–349. [Google Scholar]
- Leid, M.; Kastner, P.; Chambon, P. Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem. Sci. 1992, 17, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. Nuclear Receptor Minireview Series. J. Biol. Chem. 2001, 276, 36863–36864. [Google Scholar] [CrossRef] [PubMed]
- Perlmann, T.; Rangarajan, P.N.; Umesono, K.; Evans, R.M. Determinants for selective RAR and TR recognition of direct repeat HREs. Genes Dev. 1993, 7, 1411–1422. [Google Scholar] [CrossRef]
- Phan, T.Q.; Jow, M.M.; Privalsky, M.L. DNA recognition by thyroid hormone and retinoic acid receptors: 3,4,5 rule modified. Mol. Cell. Endocrinol. 2010, 319, 88–98. [Google Scholar] [CrossRef]
- Forman, B.M.; Yang, C.-R.; Au, M.; Casanova, J.; Ghysdael, J.; Samuels, H.H. A Domain Containing Leucine-Zipper-Like Motifs Mediate Novel in Vivo Interactions between the Thyroid Hormone and Retinoic Acid Receptors. Mol. Endocrinol. 1989, 3, 1610–1626. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Zhang, X.-K.; Lehmann, J.; Hoffmann, B.; Dawson, M.I.; Cameron, J.; Graupner, G.; Hermann, T.; Tran, P.; Pfahl, M. Homodimer formation of retinoid X receptor induced by 9-cis retinoic acid. Nature 1992, 358, 587–591. [Google Scholar] [CrossRef]
- Thompson, P.D.; Remus, L.S.; Hsieh, J.C.; Jurutka, P.W.; Whitfield, G.K.; Galligan, M.A.; Dominguez, C.E.; Haussler, C.A.; Haussler, M.R. Distinct retinoid X receptor activation function-2 residues mediate transactivation in homodimeric and vitamin D receptor heterodimeric contexts. J. Mol. Endocrinol. 2001, 27, 211–227. [Google Scholar] [CrossRef]
- Svensson, S.; Östberg, T.; Jacobsson, M.; Norström, C.; Stefansson, K.; Hallén, D.; Johansson, I.C.; Zachrisson, K.; Ogg, D.; Jendeberg, L. Crystal structure of the heterodimeric complex of LXRα and RXRβ ligand-binding domains in a fully agonistic conformation. EMBO J. 2003, 22, 4625–4633. [Google Scholar] [CrossRef]
- Nahoum, V.; Pérez, E.; Germain, P.; Rodríguez-Barrios, F.; Manzo, F.; Kammerer, S.; Lemaire, G.; Hirsch, O.; Royer, C.A.; Gronemeyer, H.; et al. Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc. Natl. Acad. Sci. USA 2007, 104, 17323–17328. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Umesono, K.; Chen, J.; Evans, R.M. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell 1995, 81, 541–550. [Google Scholar] [CrossRef]
- Lala, D.S.; Mukherjee, R.; Schulman, I.G.; Koch, S.S.C.; Dardashti, L.J.; Nadzan, A.M.; Croston, G.E.; Evans, R.M.; Heyman, R.A. Activation of specific RXR heterodimers by an antagonist of RXR homodimers. Nature 1996, 383, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Lemon, B.D.; Freedman, L.P. Selective effects of ligands on vitamin D3 receptor- and retinoid X receptor-mediated gene activation in vivo. Mol. Cell. Biol. 1996, 16, 1006–1016. [Google Scholar] [CrossRef]
- MacDonald, P.N.; Dowd, D.R.; Nakajima, S.; Galligan, M.A.; Reeder, M.C.; Haussler, C.A.; Ozato, K.; Haussler, M.R. Retinoid X receptors stimulate and 9-cis retinoic acid inhibits 1,25-dihydroxyvitamin D3-activated expression of the rat osteocalcin gene. Mol. Cell. Biol. 1993, 13, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.D.; Jurutka, P.W.; Haussler, C.A.; Whitfield, G.K.; Haussler, M.R. Heterodimeric DNA Binding by the Vitamin D Receptor and Retinoid X Receptors Is Enhanced by 1,25-Dihydroxyvitamin D3 and Inhibited by 9-cis-Retinoic Acid: Evidence for Allosteric Receptor Interactions. J. Biol. Chem. 1998, 273, 8483–8491. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Zhang, X.K.; Graupner, G.; Lee, M.O.; Hermann, T.; Hoffmann, B.; Pfahl, M. Formation of retinoid X receptor homodimers leads to repression of T3 response: Hormonal cross talk by ligand-induced squelching. Mol. Cell. Biol. 1993, 13, 7698–7707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yen, P.M.; Liu, Y.; Sugawara, A.; Chin, W.W. Vitamin D receptors repress basal transcription and exert dominant negative activity on triiodothyronine-mediated transcriptional activity. J. Biol. Chem. 1996, 271, 10910–10916. [Google Scholar] [CrossRef]
- Raval-Pandya, M.; Freedman, L.P.; Li, H.; Christakos, S. Thyroid hormone receptor does not heterodimerize with the vitamin D receptor but represses vitamin D receptor-mediated transactivation. Mol. Endocrinol. 1998, 12, 1367–1379. [Google Scholar] [CrossRef]
- Thompson, P.D.; Hsieh, J.C.; Whitfield, G.K.; Haussler, C.A.; Jurutka, P.W.; Galligan, M.A.; Tillman, J.B.; Spindler, S.R.; Haussler, M.R. Vitamin D receptor displays DNA binding and transactivation as a heterodimer with the retinoid X receptor, but not with the thyroid hormone receptor. J. Cell. Biochem. 1999, 75, 462–480. [Google Scholar] [CrossRef]
- Altucci, L.; Leibowitz, M.D.; Ogilvie, K.M.; de Lera, A.R.; Gronemeyer, H. RAR and RXR modulation in cancer and metabolic disease. Nat. Rev. Drug Discov. 2007, 6, 793–810. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Jong, L.; Fanjul, A.; Cameron, J.F.; Lu, X.P.; Haefner, P.; Dawson, M.I.; Pfahl, M. Retinoids selective for retinoid X receptor response pathways. Science 1992, 258, 1944–1946. [Google Scholar] [CrossRef] [PubMed]
- Jong, L.; Lehmann, J.M.; Hobbs, P.D.; Harlev, E.; Huffman, J.C.; Pfahl, M.; Dawson, M.I. Conformational effects on retinoid receptor selectivity. 1. Effect of 9-double bond geometry on retinoid X receptor activity. J. Med. Chem. 1993, 36, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.I.; Jong, L.; Hobbs, P.D.; Cameron, J.F.; Chao, W.-R.; Pfahl, M.; Lee, M.-O.; Shroot, B.; Pfahl, M. Conformational Effects on Retinoid Receptor Selectivity. 2. Effects of Retinoid Bridging Group on Retinoid X Receptor Activity and Selectivity. J. Med. Chem. 1995, 38, 3368–3383. [Google Scholar] [CrossRef]
- Boehm, M.F.; Zhang, L.; Badea, B.A.; White, S.K.; Mais, D.E.; Berger, E.; Suto, C.M.; Goldman, M.E.; Heyman, R.A. Synthesis and Structure-Activity Relationships of Novel Retinoid X Receptor-Selective Retinoids. J. Med. Chem. 1994, 37, 2930–2941. [Google Scholar] [CrossRef] [PubMed]
- Daiss, J.O.; Burschka, C.; Mills, J.S.; Montana, J.G.; Showell, G.A.; Fleming, I.; Gaudon, C.; Ivanova, D.; Gronemeyer, H.; Tacke, R. Synthesis, Crystal Structure Analysis, and Pharmacological Characterization of Disila-bexarotene, a Disila-Analogue of the RXR-Selective Retinoid Agonist Bexarotene. Organometallics 2005, 24, 3192–3199. [Google Scholar] [CrossRef]
- Zhang, D.; Leal, A.S.; Carapellucci, S.; Shahani, P.H.; Bhogal, J.S.; Ibrahim, S.; Raban, S.; Jurutka, P.W.; Marshall, P.A.; Sporn, M.B.; et al. Testing Novel Pyrimidinyl Rexinoids: A New Paradigm for Evaluating Rexinoids for Cancer Prevention. Cancer Prev. Res. 2019, 12, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.-C.; Prudente, R.Y.; Lamph, W.W. Synergistic effect of a retinoid X receptor-selective ligand bexarotene (LGD1069, Targretin) and paclitaxel (Taxol) in mammary carcinoma. Breast Cancer Res. Treat. 2004, 88, 141–148. [Google Scholar] [CrossRef]
- Cesario, R.M.; Stone, J.; Yen, W.-C.; Bissonnette, R.P.; Lamph, W.W. Differentiation and growth inhibition mediated via the RXR:PPARγ heterodimer in colon cancer. Cancer Lett. 2006, 240, 225–233. [Google Scholar] [CrossRef]
- Yen, W.-C.; Corpuz, M.R.; Prudente, R.Y.; Cooke, T.A.; Bissonnette, R.P.; Negro-Vilar, A.; Lamph, W.W. A Selective Retinoid X Receptor Agonist Bexarotene (Targretin) Prevents and Overcomes Acquired Paclitaxel (Taxol) Resistance in Human Non–Small Cell Lung Cancer. Clin. Cancer Res. 2004, 10, 8656–8664. [Google Scholar] [CrossRef]
- Dragnev, K.H.; Petty, W.J.; Shah, S.J.; Lewis, L.D.; Black, C.C.; Memoli, V.; Nugent, W.C.; Hermann, T.; Negro-Vilar, A.; Rigas, J.R.; et al. A Proof-of-Principle Clinical Trial of Bexarotene in Patients with Non–Small Cell Lung Cancer. Clin. Cancer Res. 2007, 13, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Davies, P.J.A.; Crombie, D.L.; Bischoff, E.D.; Cesario, R.M.; Jow, L.; Hamann, L.G.; Boehm, M.F.; Mondon, C.E.; Nadzan, A.M.; et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 1997, 386, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.I.; Gopal, J.; Haugen, B.R.; Chiu, A.C.; Whaley, K.; Nowlakha, P.; Duvic, M. Central Hypothyroidism Associated with Retinoid X Receptor–Selective Ligands. N. Engl. J. Med. 1999, 340, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, T.; Wang, F.; Tian, H.; Samuels, H.H. Functional Evidence for Retinoid X Receptor (RXR) as a Nonsilent Partner in the Thyroid Hormone Receptor/RXR Heterodimer. Mol. Cell. Biol. 2002, 22, 5782–5792. [Google Scholar] [CrossRef] [PubMed]
- Field, F.J.; Born, E.; Mathur, S.N. LXR/RXR ligand activation enhances basolateral efflux of beta-sitosterol in CaCo-2 cells. J. Lipid Res. 2004, 45, 905–913. [Google Scholar] [CrossRef]
- Murthy, S.; Born, E.; Mathur, S.N.; Field, F.J. LXR/RXR activation enhances basolateral efflux of cholesterol in CaCo-2 cells. J. Lipid Res. 2002, 43, 1054–1064. [Google Scholar] [CrossRef]
- Thacher, S.M.; Standeven, A.M.; Athanikar, J.; Kopper, S.; Castilleja, O.; Escobar, M.; Beard, R.L.; Chandraratna, R.A.S. Receptor Specificity of Retinoid-Induced Epidermal Hyperplasia: Effect of RXR-Selective Agonists and Correlation with Topical Irritation. J. Pharmacol. Exp. Ther. 1997, 282, 528–534. [Google Scholar]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models. Science 2012, 335, 1503. [Google Scholar] [CrossRef]
- McFarland, K.; Spalding, T.A.; Hubbard, D.; Ma, J.-N.; Olsson, R.; Burstein, E.S. Low Dose Bexarotene Treatment Rescues Dopamine Neurons and Restores Behavioral Function in Models of Parkinson’s Disease. ACS Chem. Neurosci. 2013, 4, 1430–1438. [Google Scholar] [CrossRef]
- Cummings, J.L.; Zhong, K.; Kinney, J.W.; Heaney, C.; Moll-Tudla, J.; Joshi, A.; Pontecorvo, M.; Devous, M.; Tang, A.; Bena, J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 4. [Google Scholar] [CrossRef]
- Kabbinavar, F.F.; Zomorodian, N.; Rettig, M.; Khan, F.; Greenwald, D.R.; Davidson, S.J.; DiCarlo, B.A.; Patel, R.; Pandit, L.; Chandraratna, R.; et al. An open-label phase II clinical trial of the RXR agonist IRX4204 in taxane-resistant, castration-resistant metastatic prostate cancer (CRPC). J. Clin. Oncol. 2014, 32, 5073. [Google Scholar] [CrossRef]
- Vuligonda, V.; Thacher, S.M.; Chandraratna, R.A. Enantioselective syntheses of potent retinoid X receptor ligands: Differential biological activities of individual antipodes. J. Med. Chem. 2001, 44, 2298–2303. [Google Scholar] [CrossRef] [PubMed]
- Muccio, D.D.; Brouillette, W.J.; Breitman, T.R.; Taimi, M.; Emanuel, P.D.; Zhang, X.-k.; Chen, G.-q.; Sani, B.P.; Venepally, P.; Reddy, L.; et al. Conformationally Defined Retinoic Acid Analogues. 4. Potential New Agents for Acute Promyelocytic and Juvenile Myelomonocytic Leukemias. J. Med. Chem. 1998, 41, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Atigadda, V.R.; Vines, K.K.; Grubbs, C.J.; Hill, D.L.; Beenken, S.L.; Bland, K.I.; Brouillette, W.J.; Muccio, D.D. Conformationally Defined Retinoic Acid Analogues. 5. Large-Scale Synthesis and Mammary Cancer Chemopreventive Activity for (2E,4E,6Z,8E)-8-(3‘,4‘-Dihydro-1‘(2‘H)-naphthalen-1‘-ylidene)-3,7-dimethyl-2,4,6-octatrienoic Acid (9cUAB30). J. Med. Chem. 2003, 46, 3766–3769. [Google Scholar] [CrossRef]
- Kolesar, J.M.; Hoel, R.; Pomplun, M.; Havighurst, T.; Stublaski, J.; Wollmer, B.; Krontiras, H.; Brouillette, W.; Muccio, D.; Kim, K.; et al. A Pilot, First-in-Human, Pharmacokinetic Study of 9cUAB30 in Healthy Volunteers. Cancer Prev. Res. 2010, 3, 1565–1570. [Google Scholar] [CrossRef]
- Hansen, N.J.; Wylie, R.C.; Phipps, S.M.; Love, W.K.; Andrews, L.G.; Tollefsbol, T.O. The low-toxicity 9-cis UAB30 novel retinoid down-regulates the DNA methyltransferases and has anti-telomerase activity in human breast cancer cells. Int. J. Oncol. 2007, 30, 641–650. [Google Scholar] [CrossRef][Green Version]
- Atigadda, V.R.; Xia, G.; Desphande, A.; Boerma, L.J.; Lobo-Ruppert, S.; Grubbs, C.J.; Smith, C.D.; Brouillette, W.J.; Muccio, D.D. Methyl substitution of a rexinoid agonist improves potency and reveals site of lipid toxicity. J. Med. Chem. 2014, 57, 5370–5380. [Google Scholar] [CrossRef] [PubMed]
- Desphande, A.; Xia, G.; Boerma, L.J.; Vines, K.K.; Atigadda, V.R.; Lobo-Ruppert, S.; Grubbs, C.J.; Moeinpour, F.L.; Smith, C.D.; Christov, K.; et al. Methyl-substituted conformationally constrained rexinoid agonists for the retinoid X receptors demonstrate improved efficacy for cancer therapy and prevention. Bioorganic Med. Chem. 2014, 22, 178–185. [Google Scholar] [CrossRef]
- Michellys, P.Y.; Ardecky, R.J.; Chen, J.H.; Crombie, D.L.; Etgen, G.J.; Faul, M.M.; Faulkner, A.L.; Grese, T.A.; Heyman, R.A.; Karanewsky, D.S.; et al. Novel (2E,4E,6Z)-7-(2-alkoxy-3,5-dialkylbenzene)-3-methylocta-2,4,6-trienoic acid retinoid X receptor modulators are active in models of type 2 diabetes. J. Med. Chem. 2003, 46, 2683–2696. [Google Scholar] [CrossRef]
- Michellys, P.Y.; Ardecky, R.J.; Chen, J.H.; D’Arrigo, J.; Grese, T.A.; Karanewsky, D.S.; Leibowitz, M.D.; Liu, S.; Mais, D.A.; Mapes, C.M.; et al. Design, synthesis, and structure-activity relationship studies of novel 6,7-locked-[7-(2-alkoxy-3,5-dialkylbenzene)-3-methylocta]-2,4,6-trienoic acids. J. Med. Chem. 2003, 46, 4087–4103. [Google Scholar] [CrossRef]
- Michellys, P.Y.; D’Arrigo, J.; Grese, T.A.; Karanewsky, D.S.; Leibowitz, M.D.; Mais, D.A.; Mapes, C.M.; Reifel-Miller, A.; Rungta, D.; Boehm, M.F. Design, synthesis and structure-activity relationship of novel RXR-selective modulators. Bioorganic Med. Chem. Lett. 2004, 14, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.E.; Jurutka, P.W.; Marshall, P.A.; Groy, T.L.; van der Vaart, A.; Ziller, J.W.; Furmick, J.K.; Graeber, M.E.; Matro, E.; Miguel, B.V.; et al. Modeling, Synthesis and Biological Evaluation of Potential Retinoid X Receptor (RXR) Selective Agonists: Novel Analogues of 4-[1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic Acid (Bexarotene). J. Med. Chem. 2009, 52, 5950–5966. [Google Scholar] [CrossRef] [PubMed]
- Furmick, J.K.; Kaneko, I.; Walsh, A.N.; Yang, J.; Bhogal, J.S.; Gray, G.M.; Baso, J.C.; Browder, D.O.; Prentice, J.L.S.; Montano, L.A.; et al. Modeling, Synthesis and Biological Evaluation of Potential Retinoid X Receptor-Selective Agonists: Novel Halogenated Analogues of 4-[1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic Acid (Bexarotene). ChemMedChem 2012, 7, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.F.; Zhang, L.; Zhi, L.; McClurg, M.R.; Berger, E.; Wagoner, M.; Mais, D.E.; Suto, C.M.; Davies, P.J.A.; Heyman, R.A.; et al. Design and Synthesis of Potent Retinoid X Receptor Selective Ligands That Induce Apoptosis in Leukemia Cells. J. Med. Chem. 1995, 38, 3146–3155. [Google Scholar] [CrossRef] [PubMed]
- Jurutka, P.W.; Kaneko, I.; Yang, J.; Bhogal, J.S.; Swierski, J.C.; Tabacaru, C.R.; Montano, L.A.; Huynh, C.C.; Jama, R.A.; Mahelona, R.D.; et al. Modeling, Synthesis, and Biological Evaluation of Potential Retinoid X Receptor (RXR) Selective Agonists: Novel Analogues of 4-[1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic Acid (Bexarotene) and (E)-3-(3-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)-4-hydroxyphenyl)acrylic Acid (CD3254). J. Med. Chem. 2013, 56, 8432–8454. [Google Scholar] [CrossRef]
- Liby, K.; Rendi, M.; Suh, N.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Lamph, W.; Labrie, F.; Krajewski, S.; Xu, X.; et al. The Combination of the Rexinoid, LG100268, and a Selective Estrogen Receptor Modulator, Either Arzoxifene or Acolbifene, Synergizes in the Prevention and Treatment of Mammary Tumors in an Estrogen Receptor–Negative Model of Breast Cancer. Clin. Cancer Res. 2006, 12, 5902–5909. [Google Scholar] [CrossRef]
- Zhang, L.; Badea, B.A.; Enyeart, D.; Berger, E.M.; Mais, D.E.; Boehm, M.F. Syntheses of isotopically labeled 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethenyl]benzoic acid (LGD1069), a potent retinoid x receptor-selective ligand. J. Label. Comp. Radiopharm. 1995, 36, 701–712. [Google Scholar] [CrossRef]
- Faul, M.M.; Ratz, A.M.; Sullivan, K.A.; Trankle, W.G.; Winneroski, L.L. Synthesis of Novel Retinoid X Receptor-Selective Retinoids. J. Org. Chem. 2001, 66, 5772–5782. [Google Scholar] [CrossRef]
- Marshall, P.A.; Jurutka, P.W.; Wagner, C.E.; van der Vaart, A.; Kaneko, I.; Chavez, P.I.; Ma, N.; Bhogal, J.S.; Shahani, P.; Swierski, J.C.; et al. Analysis of differential secondary effects of novel rexinoids: Select rexinoid X receptor ligands demonstrate differentiated side effect profiles. Pharm. Res. Perspect. 2015, 3, e00122. [Google Scholar] [CrossRef]
- Santín, E.P.; Germain, P.; Quillard, F.; Khanwalkar, H.; Rodríguez-Barrios, F.; Gronemeyer, H.; de Lera, Á.R.; Bourguet, W. Modulating Retinoid X Receptor with a Series of (E)-3-[4-Hydroxy-3-(3-alkoxy-5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]acrylic Acids and Their 4-Alkoxy Isomers. J. Med. Chem. 2009, 52, 3150–3158. [Google Scholar] [CrossRef]
- Gianni, M.; Ponzanelli, I.; Mologni, L.; Reichert, U.; Rambaldi, A.; Terao, M.; Garattini, E. Retinoid-dependent growth inhibition, differentiation and apoptosis in acute promyelocytic leukemia cells. Expression and activation of caspases. Cell Death Differ. 2000, 7, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ohsawa, F.; Yamada, S.; Shinozaki, R.; Fukai, R.; Makishima, M.; Enomoto, S.; Tai, A.; Kakuta, H. Modification at the acidic domain of RXR agonists has little effect on permissive RXR-heterodimer activation. Bioorg. Med. Chem. Lett. 2010, 20, 5139–5142. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, F.; Morishita, K.-I.; Yamada, S.; Makishima, M.; Kakuta, H. Modification at the Lipophilic Domain of RXR Agonists Differentially Influences Activation of RXR Heterodimers. ACS Med. Chem. Lett. 2010, 1, 521–525. [Google Scholar] [CrossRef]
- Kakuta, H.; Yakushiji, N.; Shinozaki, R.; Ohsawa, F.; Yamada, S.; Ohta, Y.; Kawata, K.; Nakayama, M.; Hagaya, M.; Fujiwara, C.; et al. RXR Partial Agonist CBt-PMN Exerts Therapeutic Effects on Type 2 Diabetes without the Side Effects of RXR Full Agonists. ACS Med. Chem. Lett. 2012, 3, 427–432. [Google Scholar] [CrossRef]
- Ohsawa, F.; Yamada, S.; Yakushiji, N.; Shinozaki, R.; Nakayama, M.; Kawata, K.; Hagaya, M.; Kobayashi, T.; Kohara, K.; Furusawa, Y.; et al. Mechanism of Retinoid X Receptor Partial Agonistic Action of 1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)-1H-benzotriazole-5-carboxylic Acid and Structural Development To Increase Potency. J. Med. Chem. 2013, 56, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Kagechika, H.; Koichi, S.; Sugioka, T.; Sotome, T.; Nakayama, Y.; Doi, K. Retinoid Activity Regulators. European Patent WO9845242A1, 15 October 1998. [Google Scholar]
- Ohta, K.; Tsuji, M.; Kawachi, E.; Fukasawa, H.; Hashimoto, Y.; Shudo, K.; Kagechika, H. Potent Retinoid Synergists with a Diphenylamine Skeleton. Biol. Pharm. Bull. 1998, 21, 544–546. [Google Scholar] [CrossRef][Green Version]
- Ohta, K.; Kawachi, E.; Fukasawa, H.; Shudo, K.; Kagechika, H. Diphenylamine-based retinoid antagonists: Regulation of RAR and RXR function depending on the N-substituent. Bioorg. Med. Chem. 2011, 19, 2501–2507. [Google Scholar] [CrossRef]
- Heck, M.C.; Wagner, C.E.; Shahani, P.H.; MacNeill, M.; Grozic, A.; Darwaiz, T.; Shimabuku, M.; Deans, D.G.; Robinson, N.M.; Salama, S.H.; et al. Modeling, Synthesis, and Biological Evaluation of Potential Retinoid X Receptor (RXR)-Selective Agonists: Analogues of 4-[1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic Acid (Bexarotene) and 6-(Ethyl(5,5,8,8-tetrahydronaphthalen-2-yl)amino)nicotinic Acid (NEt-TMN). J. Med. Chem. 2016, 59, 8924–8940. [Google Scholar] [CrossRef]
- Ohta, K.; Kawachi, E.; Inoue, N.; Fukasawa, H.; Hashimoto, Y.; Itai, A.; Kagechika, H. Retinoidal pyrimidinecarboxylic acids. Unexpected diaza-substituent effects in retinobenzoic acids. Chem. Pharm. Bull. 2000, 48, 1504–1513. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Stierand, K.; Rarey, M. Drawing the PDB: Protein−Ligand Complexes in Two Dimensions. ACS Med. Chem. Lett. 2010, 1, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Rarey, M. From Modeling to Medicinal Chemistry: Automatic Generation of Two-Dimensional Complex Diagrams. ChemMedChem 2007, 2, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Tachdjian, C.; Guo, J.; Boudjelal, M.; Al-Shamma, H.A.; Giachino, A.F.; Jakubowicz-Jaillardon, K.; Chen, Q.; Zapf, J.W.; Pfahl, M. Preparation of Substituted Isochroman Compounds for the Treatment of Metabolic Disorders, Cancer and Other Diseases. Patent CN1774246A, 17 May 2004. [Google Scholar]
- Jurutka, P.W.; di Martino, O.; Reshi, S.; Mallick, S.; Sabir, Z.L.; Staniszewski, L.J.P.; Warda, A.; Maiorella, E.L.; Minasian, A.; Davidson, J.; et al. Modeling, Synthesis, and Biological Evaluation of Potential Retinoid-X-Receptor (RXR) Selective Agonists: Analogs of 4-[1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahyro-2-naphthyl)ethynyl]benzoic Acid (Bexarotene) and 6-(Ethyl(4-isobutoxy-3-isopropylphenyl)amino)nicotinic Acid (NEt-4IB). Int. J. Mol. Sci. 2021, 22, 1237. [Google Scholar] [CrossRef]
- Egea, P.F.; Mitschler, A.; Rochel, N.; Ruff, M.; Chambon, P.; Moras, D. Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000, 19, 2592–2601. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.; Olson, A.J.; Goodsell, D.S. Automated prediction of ligand-binding sites in proteins. Proteins Struct. Funct. Bioinf. 2008, 70, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, O.; Niu, H.; Hadwiger, G.; Kuusanmaki, H.; Ferris, M.A.; Vu, A.; Beales, J.; Wagner, C.; Menéndez-Gutiérrez, M.P.; Ricote, M.; et al. Endogenous and combination retinoids are active in myelomonocytic leukemias. Haematologica 2021, 106, 1008–1021. [Google Scholar] [CrossRef]
- Di Martino, O.; Ferris, M.A.; Hadwiger, G.; Sarkar, S.; Vu, A.; Menéndez-Gutiérrez, M.P.; Ricote, M.; Welch, J.S. RXRA DT448/9PP generates a dominant active variant capable of inducing maturation in acute myeloid leukemia cells. Haematologica 2021, 107, 417–426. [Google Scholar] [CrossRef]
- Wilson, A.J.C.; Hahn, T.; Wilson, A.J.C.; Shmueli, U.; Crystallography, I.U.O. International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables; Springer: Dordrecht, The Netherland, 1992. [Google Scholar]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
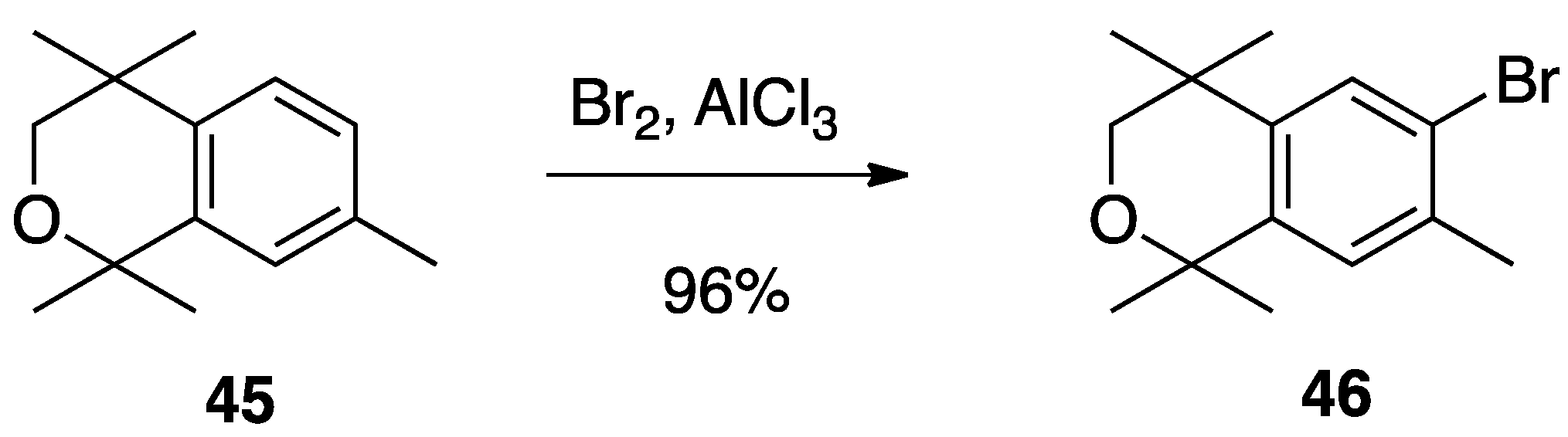{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Auto-Dock Vina Scores (kcal/mol) | EC50 (nM) +/− (SD) | 96 h IC50 (nM) + 100 nM ATRA +/− SD | LXRE Activity (% of Bex) | LHS Score (vs. Bex) | RARE Activity (%ATRA at 10 nM) |
|---|---|---|---|---|---|---|
 1 1 | –12.7 | 17.8 (1.0) | 7.8 (1.1) | 100 | 1 | 37.16 |
 23 23 | –11.5 | 8.6 (0.2) | 3.7 (1.1) | 99.5 | 0.59 | 24.33 |
 24 24 | –10.9 | 6.2 (0.1) | 3.9 (1.1) | 105.98 | 0.95 | 32.04 |
 25 25 | –10.6 | 17.8 (0.2) | 12.9 (1.2) | 80.98 | 0.7 | 14.32 |
 26 26 | –12.4 | 51.0 (0.1) | 60.0 (1.2) | 213.84 | 2.81 | 10.1 |
 27 27 | –9.3 | 65.3 (0.1) | 27.6 (1.2) | 253.64 | 1.76 | 19.45 |
 28 28 | –11.7 | 59.2 (0.1) | 38.4 (1.3) | 208.05 | 1.06 | 15.85 |
 29 29 | –11.5 | 3.9 (0.5) | 3.7 (1.2) | 221.39 | 1.29 | 54.64 |
 30 30 | –11.9 | >1000 | >1000 | 161.38 | 4.03 | 7.05 |
 31 31 | –11.5 | 5.4 (0.1) | 2.1 (1.1) | 114.37 | 1.04 | 48.61 |
 32 32 | –11.4 | 1.8 (1.1) | 2.0 (1.6) | 92.2 | 0.61 | 61.79 |
 33 33 | –11.0 | 5.3 (0.2) | 1.3 (1.1) | 103.99 | 0.72 | 39.94 |
 34 34 | –11.6 | 11.5 (0.2) | 4.1 (1.2) | 77.44 | 0.53 | 18.74 |
 35 35 | –10.6 | 0.54 (0.11) | 4.68 (1.91) | 94.07 | 0.79 | 28.76 |
 36 36 | –11.0 | 75.0 (0.1) | 42.3 (1.2) | 51.57 | 0.69 | 6.25 |
 37 37 | –10.8 | 404.9 (0.1) | >1000 | 62.72 | 1.36 | 4.02 |
 38 38 | –11.0 | 23.7 (0.2) | 11.6 (1.1) | 79.69 | 0.69 | 12.59 |
 39 39 | –10.7 | 151.6 (0.1) | >1000 | 61.52 | 0.92 | 4.96 |
 40 40 | –11.1 | 10.7 (0.2) | 16.6 (1.2) | 73.76 | 0.63 | 11.13 |
 41 41 | –10.7 | 143.4 (0.3) | 105 (1.6) | 75.66 | 0.71 | 5.16 |
 42 42 | –11.4 | 87.7 (0.1) | 136.7 (1.6) | 61.86 | 0.56 | 7.99 |
 43 43 | –11.5 | 23.1 (0.1) | 14.8 (1.2) | 71.09 | 0.55 | 9.6 |
 44 44 | –10.7 | 116.5 (0.1) | ~1000 | 68.04 | 0.68 | 11.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurutka, P.W.; di Martino, O.; Reshi, S.; Mallick, S.; Sausedo, M.A.; Moen, G.A.; Lee, I.J.; Ivan, D.J.; Krall, T.D.; Peoples, S.J.; et al. An Isochroman Analog of CD3254 and Allyl-, Isochroman-Analogs of NEt-TMN Prove to Be More Potent Retinoid-X-Receptor (RXR) Selective Agonists Than Bexarotene. Int. J. Mol. Sci. 2022, 23, 16213. https://doi.org/10.3390/ijms232416213
Jurutka PW, di Martino O, Reshi S, Mallick S, Sausedo MA, Moen GA, Lee IJ, Ivan DJ, Krall TD, Peoples SJ, et al. An Isochroman Analog of CD3254 and Allyl-, Isochroman-Analogs of NEt-TMN Prove to Be More Potent Retinoid-X-Receptor (RXR) Selective Agonists Than Bexarotene. International Journal of Molecular Sciences. 2022; 23(24):16213. https://doi.org/10.3390/ijms232416213
Chicago/Turabian StyleJurutka, Peter W., Orsola di Martino, Sabeeha Reshi, Sanchita Mallick, Michael A. Sausedo, Grant A. Moen, Isaac J. Lee, Dominic J. Ivan, Tyler D. Krall, Samuel J. Peoples, and et al. 2022. "An Isochroman Analog of CD3254 and Allyl-, Isochroman-Analogs of NEt-TMN Prove to Be More Potent Retinoid-X-Receptor (RXR) Selective Agonists Than Bexarotene" International Journal of Molecular Sciences 23, no. 24: 16213. https://doi.org/10.3390/ijms232416213
APA StyleJurutka, P. W., di Martino, O., Reshi, S., Mallick, S., Sausedo, M. A., Moen, G. A., Lee, I. J., Ivan, D. J., Krall, T. D., Peoples, S. J., Perez, A., Tromba, L., Le, A., Khadka, I., Petros, R., Savage, B. M., Salama, E., Salama, J., Ziller, J. W., ... Wagner, C. E. (2022). An Isochroman Analog of CD3254 and Allyl-, Isochroman-Analogs of NEt-TMN Prove to Be More Potent Retinoid-X-Receptor (RXR) Selective Agonists Than Bexarotene. International Journal of Molecular Sciences, 23(24), 16213. https://doi.org/10.3390/ijms232416213