
Pharmacophore-Oriented Identification of Potential Leads as CCR5 Inhibitors to Block HIV Cellular Entry


 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Pharmacophore Model Generation
2.2. Validation of Pharmacophores
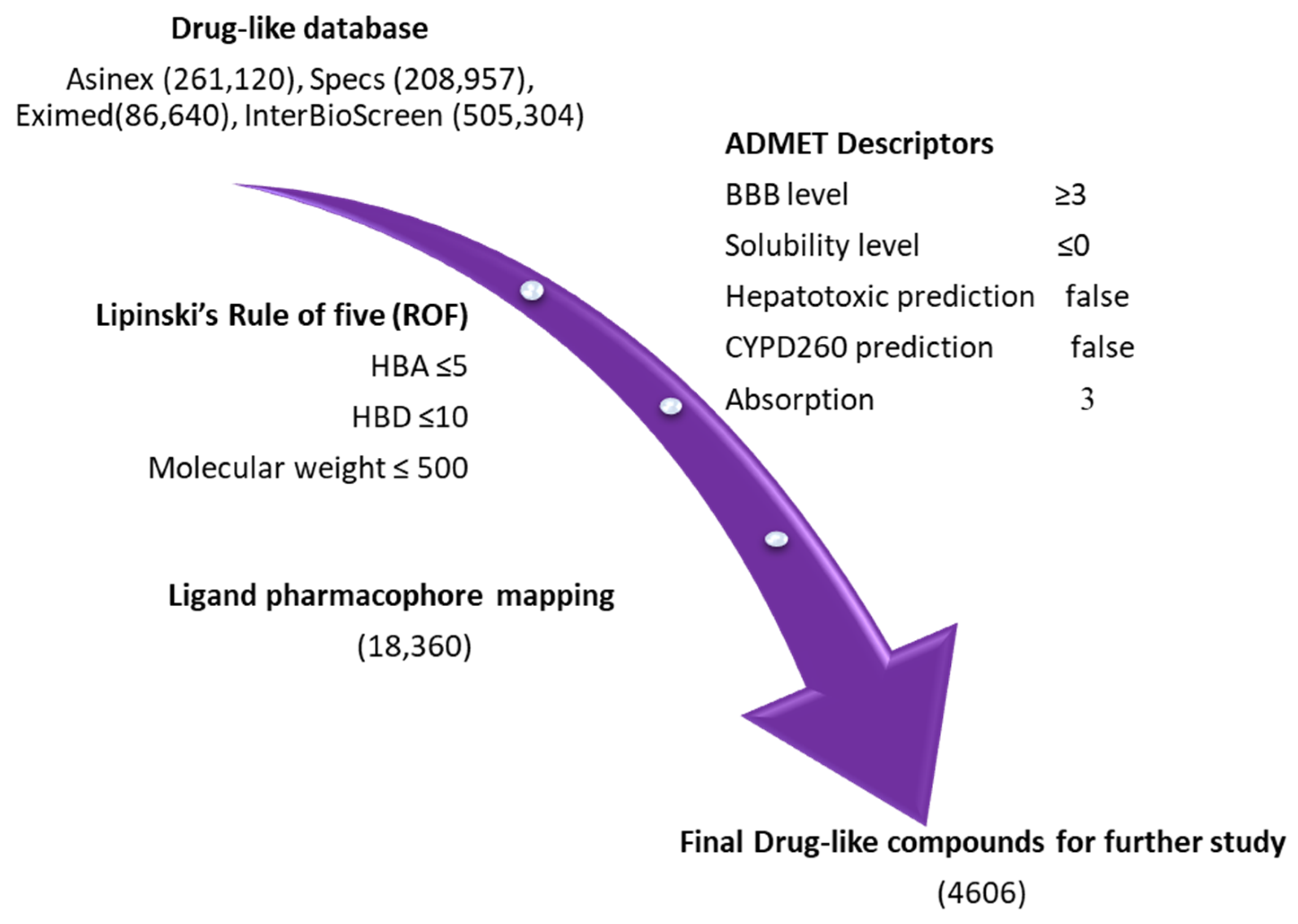
2.3. Drug-like Database Generation and Virtual Screening
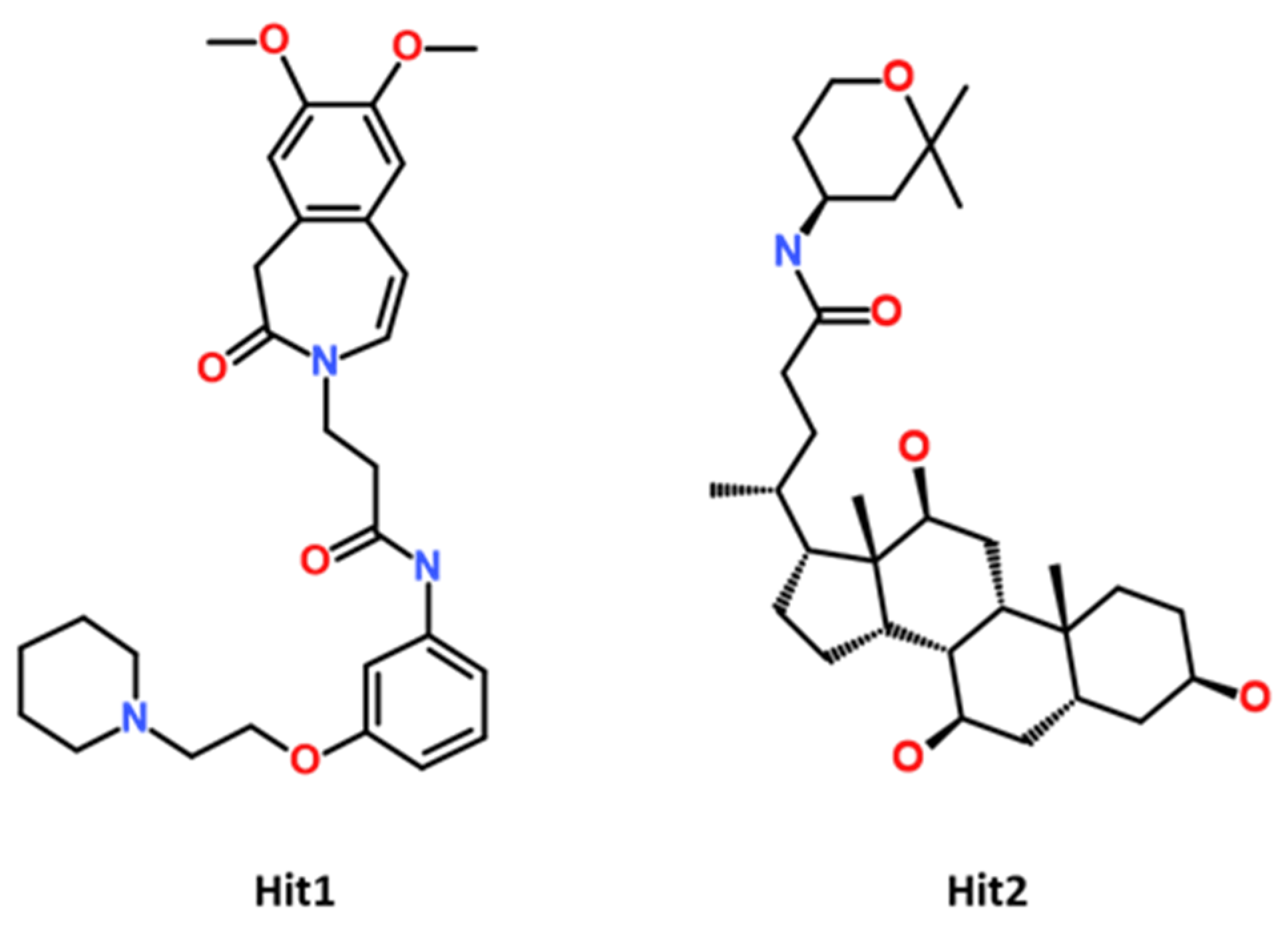
2.4. Molecular Docking of Potential Compounds with CCR5
2.5. Molecular Dynamics Simulations
2.5.1. Stability of MD Simulation Systems
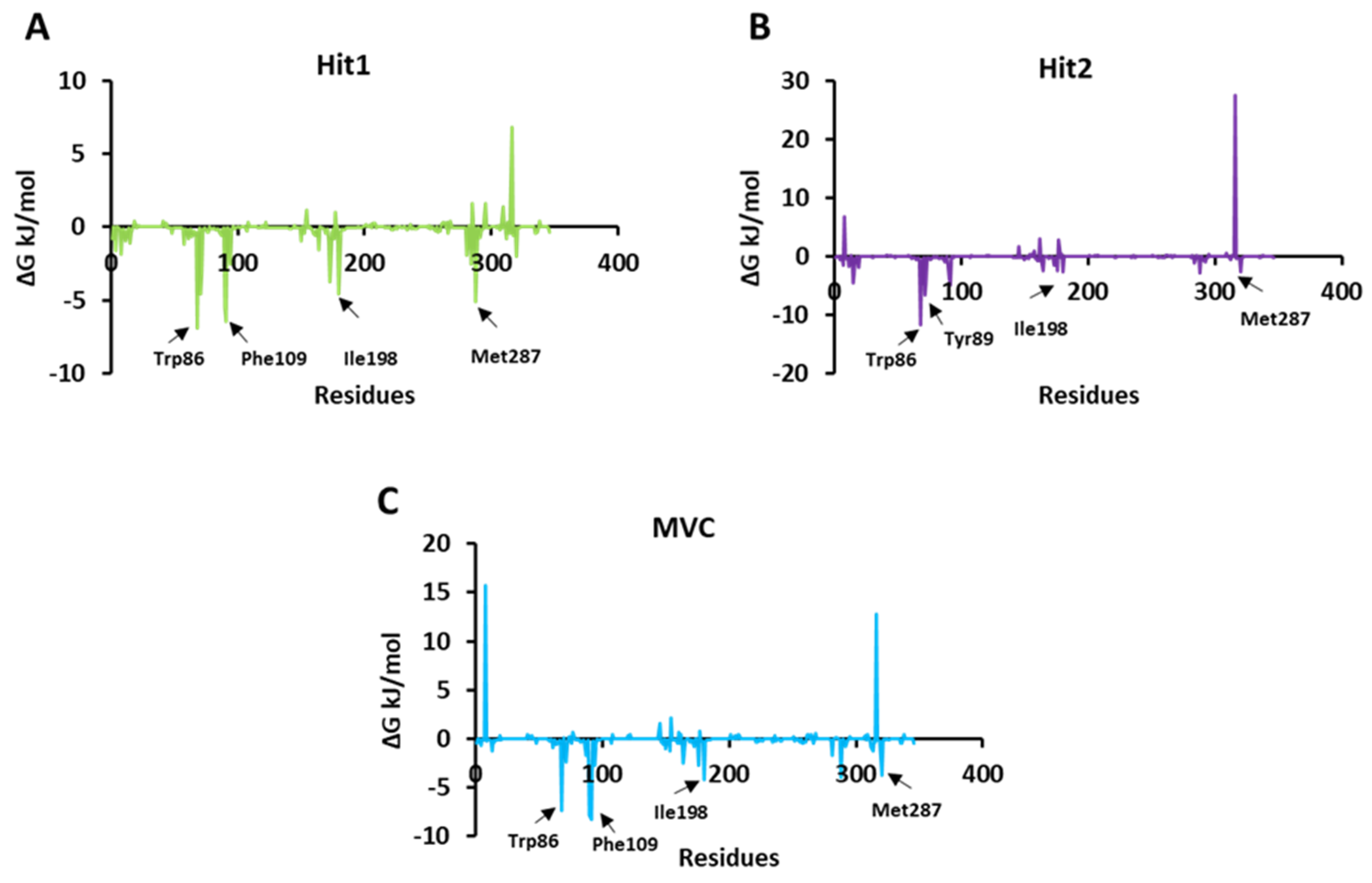
2.5.2. Calculation of Binding Free Energy by MM-PBSA Method
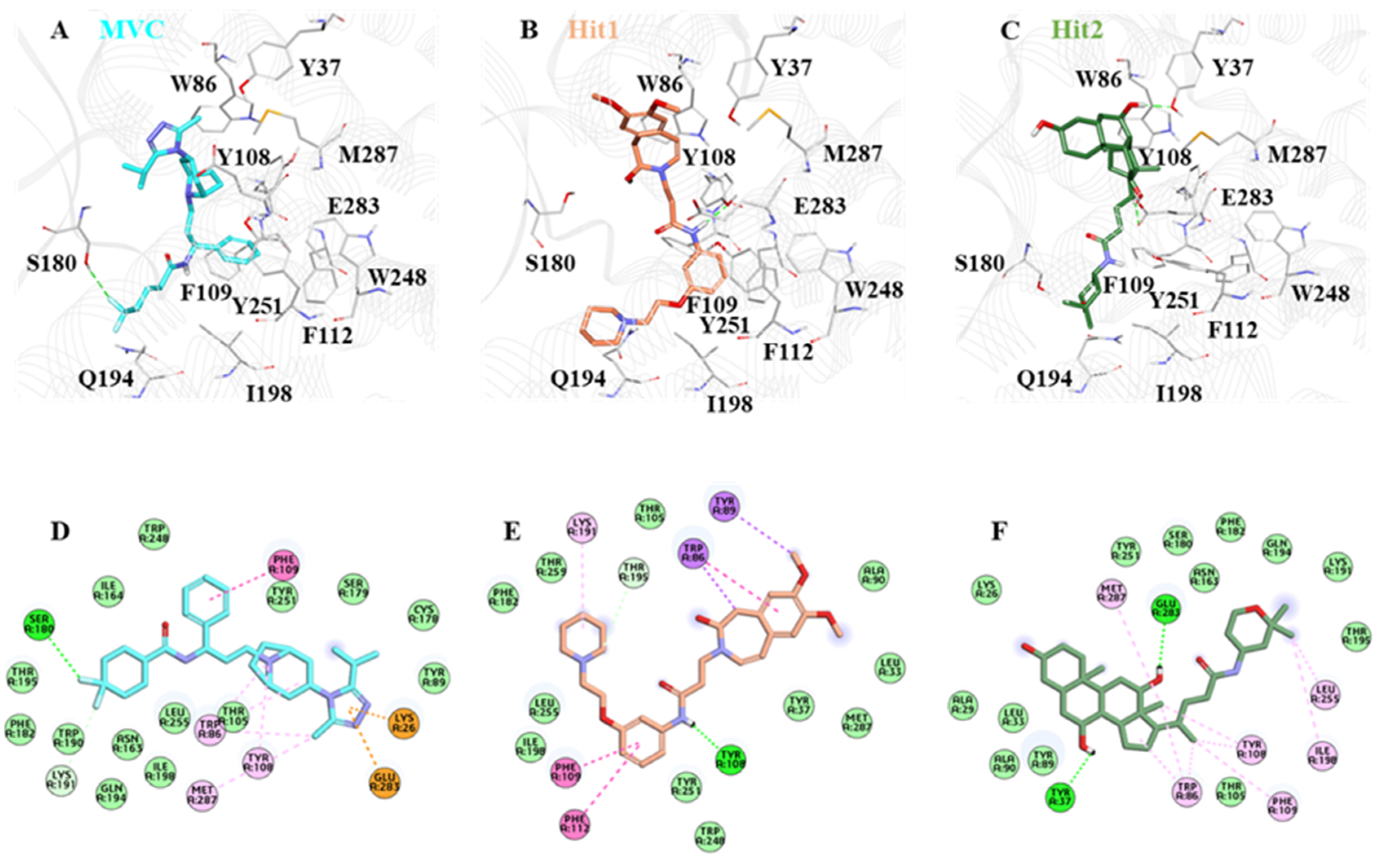
2.6. Binding Mode and Intermolecular Interaction Analysis
2.7. Pharmacokinetic Properties Prediction of via pkCSM
3. Materials and Methods
3.1. Generation of Common Feature Pharmacophore
3.2. Validation of Pharmacophore Model
3.3. Generation of Drug-like Database and Virtual Screening
3.4. Molecular Docking Studies
3.5. Molecular Dynamics Simulations
Root Mean Square Deviation (RMSD) and H-Bond Analysis
3.6. Binding Free Energy Calculation
3.7. Prediction of Pharmacokinetic Properties via pkCSM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deeks, S.G.; Archin, N.; Cannon, P.; Collins, S.; Jones, R.B.; de Jong, M.A.W.P.; Lambotte, O.; Lamplough, R.; Ndung’u, T.; Sugarman, J.; et al. Research Priorities for an HIV Cure: International AIDS Society Global Scientific Strategy 2021. Nat. Med. 2021, 27, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A. Variability of HIV Infections. J. Theor. Biol. 1992, 155, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Perno, C.F. HIV-1 Genetic Variability and Clinical Implications. ISRN Microbiol. 2013, 2013, 481314. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Fang, Q.; Liu, S.; Hou, W.; Li, J.; Huang, Y.; Shi, J. Advances of CCR5 Antagonists: From Small Molecules to Macromolecules. Eur. J. Med. Chem. 2020, 208, 112819. [Google Scholar] [CrossRef] [PubMed]
- United Nations Programme on HIV/aids. In UNAIDS Data 2021; UNAIDS: Geneva, Switzerland, 2021; pp. 4–38.
- Lopalco, L. CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses 2010, 2, 574–600. [Google Scholar] [CrossRef]
- Moore, J.P.; Trkola, A.; Dragic, T. Co-Receptors for HIV-1 Entry. Curr. Opin. Immunol. 1997, 9, 551–562. [Google Scholar] [CrossRef]
- Rao, P.K.S. CCR5 Inhibitors: Emerging Promising HIV Therapeutic Strategy. Indian J. Sex. Transm. Dis. AIDS 2009, 30, 1–9. [Google Scholar] [CrossRef]
- Clotet, B. CCR5 Inhibitors: Promising yet Challenging. J. Infect. Dis. 2007, 196, 178–180. [Google Scholar] [CrossRef][Green Version]
- Latinovic, O.S.; Reitz, M.; Heredia, A. CCR5 Inhibitors and HIV-1 Infection. J. AIDS HIV Treat. 2019, 1, 1–5. [Google Scholar] [CrossRef]
- Zheng, Y.; Han, G.W.; Abagyan, R.; Wu, B.; Stevens, R.C.; Cherezov, V.; Kufareva, I.; Handel, T.M. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 2017, 46, 1005–1017.e5. [Google Scholar] [CrossRef]
- Zeng, Z.; Lan, T.; Wei, Y.; Wei, X. CCL5/CCR5 Axis in Human Diseases and Related Treatments. Genes Dis. 2022, 9, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Tong, P.; Wang, J.; Zhao, L.; Li, J.; Yu, Y.; Chen, Y.-H.; Wang, J. The Membrane-Proximal Region of C–C Chemokine Receptor Type 5 Participates in the Infection of HIV-1. Front. Immunol. 2017, 8, 478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, K.; Tan, Q.; Shao, Q.; Han, S.; Zhang, C.; Yi, C.; Chu, X.; Zhu, Y.; Xu, Y.; et al. Structural Basis for Chemokine Recognition and Receptor Activation of Chemokine Receptor CCR5. Nat. Commun. 2021, 12, 4151. [Google Scholar] [CrossRef] [PubMed]
- Salmas, R.E.; Yurtsever, M.; Durdagi, S. Investigation of Inhibition Mechanism of Chemokine Receptor CCR5 by Micro-Second Molecular Dynamics Simulations. Sci. Rep. 2015, 5, 13180. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Rabin, R.L.; Arthos, J.; Rubbert, A.; Dybul, M.; Swofford, R.; Venkatesan, S.; Farber, J.M.; Fauci, A.S. Macrophage-Tropic HIV and SIV Envelope Proteins Induce a Signal through the CCR5 Chemokine Receptor. Nature 1997, 389, 981–985. [Google Scholar] [CrossRef]
- Venuti, A.; Pastori, C.; Lopalco, L. The Role of Natural Antibodies to CC Chemokine Receptor 5 in HIV Infection. Front. Immunol. 2017, 8, 1358. [Google Scholar] [CrossRef]
- Alkhatib, G. The Biology of CCR5 and CXCR4. Curr. Opin. HIV AIDS 2009, 4, 96–103. [Google Scholar] [CrossRef]
- Flanagan, C.A. Receptor Conformation and Constitutive Activity in CCR5 Chemokine Receptor Function and HIV Infection. Adv. Pharmacol. 2014, 70, 215–263. [Google Scholar] [CrossRef]
- Mueller, A.; Kelly, E.; Strange, P.G. Pathways for Internalization and Recycling of the Chemokine Receptor CCR5. Blood 2002, 99, 785–791. [Google Scholar] [CrossRef]
- Ganju, R.K.; Brubaker, S.A.; Chernock, R.D.; Avraham, S.; Groopman, J.E. β-Chemokine Receptor CCR5 Signals through SHP1, SHP2, and Syk *. J. Biol. Chem. 2000, 275, 17263–17268. [Google Scholar] [CrossRef]
- Carter, G.C.; Bernstone, L.; Sangani, D.; Bee, J.W.; Harder, T.; James, W. HIV Entry in Macrophages Is Dependent on Intact Lipid Rafts. Virology 2009, 386, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M. Chemokine Receptor CCR5: Insights into Structure, Function, and Regulation. Cell. Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Shideman, C.R.; Hu, S.; Peterson, P.K.; Thayer, S.A. CCL5 Evokes Calcium Signals in Microglia through a Kinase-, Phosphoinositide-, and Nucleotide-Dependent Mechanism. J. Neurosci. Res. 2006, 83, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.; Thompson, M.; Kumar, P.; Piliero, P.; Davey, R.; Patterson, K.; Shachoy-Clark, A.; Adkison, K.; Demarest, J.; Lou, Y.; et al. Antiviral Activity and Safety of 873140, a Novel CCR5 Antagonist, during Short-Term Monotherapy in HIV-Infected Adults. AIDS 2005, 19, 1443–1448. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A Randomized, Placebo-Controlled Trial of Cenicriviroc for Treatment of Nonalcoholic Steatohepatitis with Fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef]
- Qian, K.; Morris-Natschke, S.L.; Lee, K.-H. HIV Entry Inhibitors and Their Potential in HIV Therapy. Med. Res. Rev. 2009, 29, 369–393. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Ho, Y.; Liu, H.-L. Structure-Based Pharmacophore Modeling to Discover Novel CCR5 Inhibitors for HIV-1/Cancers Therapy. J. Biomed. Sci. Eng. 2019, 12, 10–30. [Google Scholar] [CrossRef]
- Wang, Y.; Suzek, T.; Zhang, J.; Wang, J.; He, S.; Cheng, T.; Shoemaker, B.A.; Gindulyte, A.; Bryant, S.H. PubChem BioAssay: 2014 Update. Nucleic Acids Res. 2014, 42, D1075–D1082. [Google Scholar] [CrossRef]
- Studio, D. Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Accelrys Software Inc.: San Diego, CA, USA, 2015. [Google Scholar] [CrossRef]
- Niu, M.; Wang, F.; Li, F.; Dong, Y.; Gu, Y. Establishment of a Screening Protocol for Identification of Aminopeptidase N Inhibitors. J. Taiwan Inst. Chem. Eng. 2015, 49, 19–26. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, R.; Parate, S.; Yoon, S.; Lee, G.; Kim, D.; Lee, K.W. Identification of ACK1 Inhibitors as Anticancer Agents by Using Computer-Aided Drug Designing. J. Mol. Struct. 2021, 1235, 130200. [Google Scholar] [CrossRef]
- Guner, O.; Waldman, M.; Hoffman, R.; Kim, J.-H. Strategies for Database Mining and Pharmacophore Development. In Pharmacophore Perception, Development, and Use in Drug Design; International University Line: Portland, OR, USA, 2000; pp. 193–212. ISBN 0-9936817-6-1. [Google Scholar]
- Pal, S.; Kumar, V.; Kundu, B.; Bhattacharya, D.; Preethy, N.; Reddy, M.P.; Talukdar, A. Ligand-Based Pharmacophore Modeling, Virtual Screening and Molecular Docking Studies for Discovery of Potential Topoisomerase I Inhibitors. Comput. Struct. Biotechnol. J. 2019, 17, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Computational Investigation Identified Potential Chemical Scaffolds for Heparanase as Anticancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 5311. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-Y. Pharmacophore Modeling and Applications in Drug Discovery: Challenges and Recent Advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Arooj, M.; Sakkiah, S.; Kim, S.; Arulalapperumal, V.; Lee, K.W. A Combination of Receptor-Based Pharmacophore Modeling & QM Techniques for Identification of Human Chymase Inhibitors. PLoS ONE 2013, 8, e63030. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular Dynamics Simulations and Novel Drug Discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Sakano, T.; Mahamood, M.I.; Yamashita, T.; Fujitani, H. Molecular Dynamics Analysis to Evaluate Docking Pose Prediction. Biophys. Physicobiol. 2016, 13, 181–194. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, V.; Lee, K.W. A Computational Drug Repurposing Approach in Identifying the Cephalosporin Antibiotic and Anti-Hepatitis C Drug Derivatives for COVID-19 Treatment. Comput. Biol. Med. 2021, 130, 104186. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef] [PubMed]
- Rifai, E.A.; van Dijk, M.; Vermeulen, N.P.E.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1–Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Kumbhar, N.; Nimal, S.; Barale, S.; Kamble, S.; Bavi, R.; Sonawane, K.; Gacche, R. Identification of Novel Leads as Potent Inhibitors of HDAC3 Using Ligand-Based Pharmacophore Modeling and MD Simulation. Sci. Rep. 2022, 12, 1712. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Chan Hong, J.; Lee, K.W. Investigating Natural Compounds against Oncogenic RET Tyrosine Kinase Using Pharmacoinformatic Approaches for Cancer Therapeutics. RSC Adv. 2022, 12, 1194–1207. [Google Scholar] [CrossRef]
- Viola, A.; Luster, A.D. Chemokines and Their Receptors: Drug Targets in Immunity and Inflammation. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 171–197. [Google Scholar] [CrossRef]
- Kothandan, G.; Gadhe, C.G.; Cho, S.J. Structural Insights from Binding Poses of CCR2 and CCR5 with Clinically Important Antagonists: A Combined In Silico Study. PLoS ONE 2012, 7, e32864. [Google Scholar] [CrossRef]
- Gu, W.-G.; Zhang, X.; Yuan, J.-F. Anti-HIV Drug Development through Computational Methods. AAPS J. 2014, 16, 674–680. [Google Scholar] [CrossRef]
- Prokofjeva, M.M.; Kochetkov, S.N.; Prassolov, V.S. Therapy of HIV Infection: Current Approaches and Prospects. Acta Nat. 2016, 8, 23–32. [Google Scholar] [CrossRef]
- Alqahtani, S. In Silico ADME-Tox Modeling: Progress and Prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.; Pillans, P. P-Glycoprotein and Its Role in Drug-Drug Interactions. Aust. Prescr. 2014, 37, 137–139. [Google Scholar] [CrossRef]
- Kumar, V.; Parate, S.; Thakur, G.; Lee, G.; Ro, H.-S.; Kim, Y.; Kim, H.J.; Kim, M.O.; Lee, K.W. Identification of CDK7 Inhibitors from Natural Sources Using Pharmacoinformatics and Molecular Dynamics Simulations. Biomedicines 2021, 9, 1197. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.L. P-Glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-Glycoprotein in Pharmacokinetics: Clinical Implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Murad, N.; Pasikanti, K.K.; Madej, B.D.; Minnich, A.; McComas, J.M.; Crouch, S.; Polli, J.W.; Weber, A.D. Predicting Volume of Distribution in Humans: Performance of In Silico Methods for a Large Set of Structurally Diverse Clinical Compounds. Drug Metab. Dispos. 2021, 49, 169–178. [Google Scholar] [CrossRef]
- Fagerholm, U.; Hellberg, S.; Alvarsson, J.; Arvidsson McShane, S.; Spjuth, O. In Silico Prediction of Volume of Distribution of Drugs in Man Using Conformal Prediction Performs on Par with Animal Data-Based Models. Xenobiotica. 2021, 51, 1366–1371. [Google Scholar] [CrossRef]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Gifford, E. ADMET in Silico Modelling: Towards Prediction Paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Adane, L.; Bharatam, P.V.; Sharma, V. A Common Feature-Based 3D-Pharmacophore Model Generation and Virtual Screening: Identification of Potential PfDHFR Inhibitors. J. Enzyme Inhib. Med. Chem. 2010, 25, 635–645. [Google Scholar] [CrossRef]
- Chandrasekaran, B.; Agrawal, N.; Kaushik, S. Pharmacophore Development. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 677–687. ISBN 978-0-12-811432-2. [Google Scholar]
- Guner, F.O. History and Evolution of the Pharmacophore Concept in Computer-Aided Drug Design. Curr. Top. Med. Chem. 2002, 2, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz Kenneth, M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Sapay, N.; Tieleman, D.P. Combination of the CHARMM27 Force Field with United-Atom Lipid Force Fields. J. Comput. Chem. 2011, 32, 1400–1410. [Google Scholar] [CrossRef]
- Salsbury, F.R. Molecular Dynamics Simulations of Protein Dynamics and Their Relevance to Drug Discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Singh, R.; Singh, P.; Purohit, R.; Kumar, S. Elimination of Bitter-off Taste of Stevioside through Structure Modification and Computational Interventions. J. Theor. Biol. 2020, 486, 110094. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kulkarni, A.M.; Kumar, V.; Parate, S.; Lee, G.; Yoon, S.; Lee, K.W. Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods. Int. J. Mol. Sci. 2022, 23, 1309. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure Based Pharmacophore Modeling, Virtual Screening, Molecular Docking and ADMET Approaches for Identification of Natural Anti-Cancer Agents Targeting XIAP Protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Paissoni, C.; Spiliotopoulos, D.; Musco, G.; Spitaleri, A. GMXPBSA 2.1: A GROMACS Tool to Perform MM/PBSA and Computational Alanine Scanning. Comput. Phys. Commun. 2015, 186, 105–107. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Madan, A.K.; Dureja, H. Prediction of Pharmacokinetic Parameters. Methods Mol. Biol. 2012, 929, 337–357. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Features a | Rank b | Direct Hit c | Partial Hit d | Max Fit |
|---|---|---|---|---|---|
| 01 | ZZZHHD | 136.074 | 111111111 | 000000000 | 6 |
| 02 | ZZZHHD | 132.219 | 111111111 | 000000000 | 6 |
| 03 | ZZHHD | 132.123 | 111111111 | 000000000 | 5 |
| 04 | ZZZHHD | 131.485 | 111111111 | 000000000 | 6 |
| 05 | ZZZHD | 130.851 | 111111111 | 000000000 | 5 |
| 06 | ZZZHHD | 130.473 | 111111111 | 000000000 | 6 |
| 07 | RZHHD | 130.470 | 111111111 | 000000000 | 5 |
| 08 | ZZHHD | 130.421 | 111111111 | 000000000 | 5 |
| 09 | ZZHAD | 130.323 | 111111111 | 000000000 | 5 |
| 10 | ZZHHD | 130.309 | 111111111 | 000000000 | 5 |
| Sr. No. | Parameters | Calculated Values |
|---|---|---|
| 1 | Total number of molecules in the database (D) | 265 |
| 2 | Total number of active molecules in the database (A) | 20 |
| 3 | Total number of active molecules in the retrieved hits (Ht) | 25 |
| 4 | Number of retrieved hits by pharmacophore (Ha) | 19 |
| 5 | % Yield of actives [(Ha/Ht) × 100] | 76% |
| 6 | % Ratio of actives [(Ha/A × 100)] | 95% |
| 7 | False negative [A-Ha] | 1 |
| 8 | False positive [Ht-Ha] | 6 |
| 9 | Goodness of fit | 0.77 |
| 10 | Enrichment factor (EF) | 10.07 |
| Name | Hydrogen Bond Interactions | van der Waals Interactions | π -π /π-Alkyl Interactions | |||
|---|---|---|---|---|---|---|
| Amino Acid | Amino Acid Atom | Ligand Atom | Distance (<3.5 Å) | |||
| Hit1 | Tyr108 | OH | H45 | 2.23 | Leu33, Tyr37, Ala90, Met287, Thr259, Phe182, Leu255, Lle198, Tyr251 | Phe109, Phe112, Trp86 |
| Hit2 | Glu283 | OE1 | H64 | 2.22 | Lys26, Ala29, Leu33, Ala90, Tyr89, Thr105, Asn163, Ser180, Phe182, Lys191, Gln194, Thr195, Tyr251 | Trp86, Tyr108, Phe109, Ile198, Leu255, Met287 |
| Tyr37 | OH | H65 | 2.05 | |||
| MVC | Ser180 | HG1 | F2 | 2.95 | Tyr89, Cys178, Ser179, Thr105, Leu255, Asn163, Gln194, Ile198, Trp190, Phe182, Thr195, Ile164 | Met287, Trp86, Tyr108, Phe109 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.; Kumar, V.; Lee, G.; Jung, T.S.; Ha, M.W.; Hong, J.C.; Lee, K.W. Pharmacophore-Oriented Identification of Potential Leads as CCR5 Inhibitors to Block HIV Cellular Entry. Int. J. Mol. Sci. 2022, 23, 16122. https://doi.org/10.3390/ijms232416122
Singh P, Kumar V, Lee G, Jung TS, Ha MW, Hong JC, Lee KW. Pharmacophore-Oriented Identification of Potential Leads as CCR5 Inhibitors to Block HIV Cellular Entry. International Journal of Molecular Sciences. 2022; 23(24):16122. https://doi.org/10.3390/ijms232416122
Chicago/Turabian StyleSingh, Pooja, Vikas Kumar, Gihwan Lee, Tae Sung Jung, Min Woo Ha, Jong Chan Hong, and Keun Woo Lee. 2022. "Pharmacophore-Oriented Identification of Potential Leads as CCR5 Inhibitors to Block HIV Cellular Entry" International Journal of Molecular Sciences 23, no. 24: 16122. https://doi.org/10.3390/ijms232416122
APA StyleSingh, P., Kumar, V., Lee, G., Jung, T. S., Ha, M. W., Hong, J. C., & Lee, K. W. (2022). Pharmacophore-Oriented Identification of Potential Leads as CCR5 Inhibitors to Block HIV Cellular Entry. International Journal of Molecular Sciences, 23(24), 16122. https://doi.org/10.3390/ijms232416122