
Molecular Basis for Non-Covalent, Non-Competitive FAAH Inhibition

 ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Results
2.1. Computational Studies
2.1.1. Identification of Putative Binding Sites of TPA14
2.1.2. Docking of TPA14 in rFAAH and mFAAH
2.1.3. Molecular Dynamics Simulations (MDs) of TPA14 in rFAAH
2.1.4. Non-Competitive Inhibition Mechanism of TPA14
2.1.5. Thermodynamic Integration (TI) Calculations
2.1.6. Molecular Dynamics Simulations (MDs) of TPA14 in mFAAH
3. Discussion and Conclusions
4. Materials and Methods
4.1. Structural Models
4.2. Molecular Docking
4.3. Molecular Dynamics Simulations (MDs)
4.4. Free-Energy Calculations
4.5. QM/MM Calculations
4.6. Thermodynamic Integration (TI)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Maione, S.; Costa, B.; Di Marzo, V. Endocannabinoids: A Unique Opportunity to Develop Multitarget Analgesics. Pain 2013, 154, S87–S93. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Hohmann, A. The Endocannabinoid System and Pain. CNS Neurol. Disord. Drug Targets 2012, 8, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Wendler, D.S. Problems with the Consensus Definition of the Therapeutic Misconception. J. Clin. Ethics 2013, 24, 387–394. [Google Scholar] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the Expanded Endocannabinoid System in Neurological Disorders. Nat. Rev. Neurol. 2019, 16, 9–29. [Google Scholar] [CrossRef]
- Pertwee, R.G. Endocannabinoids and Their Pharmacological Actions. In Endocannabinoids; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2015; Volume 231, pp. 1–37. [Google Scholar]
- Oláh, A.; Szekanecz, Z.; Bíró, T. Targeting Cannabinoid Signaling in the Immune System: “High”-Ly Exciting Questions, Possibilities, and Challenges. Front. Immunol. 2017, 8, 1487. [Google Scholar] [CrossRef]
- Polini, B.; Cervetto, C.; Carpi, S.; Pelassa, S.; Gado, F.; Ferrisi, R.; Bertini, S.; Nieri, P.; Marcoli, M.; Manera, C. Positive Allosteric Modulation of CB1 and CB2 Cannabinoid Receptors Enhances the Neuroprotective Activity of a Dual CB1R/CB2R Orthosteric Agonist. Life 2020, 10, 333. [Google Scholar] [CrossRef]
- Maccarrone, M. Metabolism of the Endocannabinoid Anandamide: Open Questions after 25 Years. Front. Mol. Neurosci. 2017, 10, 166. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. COX-2-Derived Endocannabinoid Metabolites as Novel Inflammatory Mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef]
- Palermo, G.; Rothlisberger, U.; Cavalli, A.; De Vivo, M. Computational insights into function and inhibition of fatty acid amide hydrolase. Eur. J. Med. Chem. 2014, 91, 15–26. [Google Scholar] [CrossRef]
- Tripathi, R.K.P. A Perspective Review on Fatty Acid Amide Hydrolase (FAAH) Inhibitors as Potential Therapeutic Agents. Eur. J. Med. Chem. 2020, 188, 111953. [Google Scholar] [CrossRef]
- Van Egmond, N.; Straub, V.M.; Van Der Stelt, M. Targeting Endocannabinoid Signaling: FAAH and MAG Lipase Inhibitors. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 441–463. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Pasquini, S.; Contri, C.; Gemma, S.; Campiani, G.; Butini, S.; Varani, K.; Vincenzi, F. Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition. Cells 2022, 11, 471. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Hill, M.N.; Cheer, J.F.; Wotjak, C.T.; Holmes, A. Neuroscience and Biobehavioral Reviews The Endocannabinoid System as a Target for Novel Anxiolytic Drugs. Neurosci. Biobehav. Rev. 2017, 76, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J. The Potential of Inhibitors of Endocannabinoid Metabolism as Anxiolytic and Antidepressive Drugs—A Practical View. Eur. Neuropsychopharmacol. 2015, 25, 749–762. [Google Scholar] [CrossRef]
- Ogawa, S.; Kunugi, H. Inhibitors of Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase: New Targets for Future Antidepressants. Curr. Neuropharmacol. 2015, 13, 760–775. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X. FAAH Inhibition Produces Antidepressant-like Efforts of Mice to Acute Stress via Synaptic Long-Term Depression. Behav. Brain Res. 2017, 324, 138–145. [Google Scholar] [CrossRef]
- Winkler, K.; Ramer, R.; Dithmer, S.; Ivanov, I.; Merkord, J.; Hinz, B. Fatty Acid Amide Hydrolase Inhibitors Confer Anti-Invasive and Antimetastatic Effects on Lung Cancer Cells. Oncotarget 2016, 7, 15047–15064. [Google Scholar] [CrossRef]
- Ravi, J.; Sneh, A.; Shilo, K.; Nasser, M.W.; Ganju, R.K. FAAH Inhibition Enhances Anandamide Mediated Anti-Tumorigenic Effects in Non-Small Cell Lung Cancer by Downregulating the EGF/EGFR Pathway. Oncotarget 2014, 5, 2475–2486. [Google Scholar] [CrossRef]
- Ogasawara, D.; Shpak, G.; Van Der Kroeg, M.; Kantae, V.; Baggelaar, M.P.; De Vrij, F.M.S.; Deng, H.; Allarà, M.; Fezza, F.; Lin, Z.; et al. Activity-Based Protein Profiling Reveals off-Target Proteins of the FAAH Inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. [Google Scholar]
- Janssen Research & Development, L. A Safety and Efficacy Study of JNJ-42165279 in Participants with Social Anxiety Disorder. Available online: https://clinicaltrials.gov/ct2/show/NCT02432703?term=JNJ-42165279&rank=1 (accessed on 2 September 2021).
- Pawsey, S.; Wood, M.; Browne, H.; Donaldson, K.; Christie, M.; Warrington, S. Safety, Tolerability and Pharmacokinetics of FAAH Inhibitor V158866: A Double-Blind, Randomised, Placebo-Controlled Phase I Study in Healthy Volunteers. Drugs R D 2016, 16, 181–191. [Google Scholar] [CrossRef]
- Tuo, W.; Leleu-chavain, N.; Spencer, J.; Sansook, S.; Chavatte, P. Therapeutic Potential of Fatty Acid Amide Hydrolase, Monoacylglycerol Lipase, and N—Acylethanolamine Acid Amidase Inhibitors. J. Med. Chem. 2017, 60, 4–46. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.W.; Tian, G.; Yu, X.H.; Paschetto, K.A.; Wilkins, D.E.; Meury, L.; Cao, C.Q.; Varnes, J.; Edwards, P.D. Biochemical characterization and in vitro activity of AZ513, a noncovalent, reversible, and noncompetitive inhibitor of fatty acid amide hydrolase. Eur. J. Pharmacol. 2011, 667, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Deplano, A.; Morgillo, C.M.; Demurtas, M.; Björklund, E.; Cipriano, M.; Svensson, M.; Hashemian, S.; Smaldone, G.; Pedone, E.; Luque, F.J.; et al. Novel Propanamides as Fatty Acid Amide Hydrolase Inhibitors. Eur. J. Med. Chem. 2017, 136, 523–542. [Google Scholar] [CrossRef]
- Gustin, D.J.; Ma, Z.; Min, X.; Li, Y.; Hedberg, C.; Guimaraes, C.; Porter, A.C.; Lindstrom, M.; Lester-Zeiner, D.; Xu, G.; et al. Identification of Potent, Noncovalent Fatty Acid Amide Hydrolase (FAAH) Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2492–2496. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Bertolacci, L.; Romeo, E.; Veronesi, M.; Magotti, P.; Albani, C.; Dionisi, M.; Lambruschini, C.; Scarpelli, R.; Cavalli, A.; De Vivo, M.; et al. A Binding Site for Nonsteroidal Anti-Inflammatory Drugs in Fatty Acid Amide Hydrolase. J. Am. Chem. Soc. 2013, 135, 22–25. [Google Scholar] [CrossRef]
- Otrubova, K.; Brown, M.; McCormick, M.S.; Han, G.W.; O’Neal, S.T.; Cravatt, B.F.; Stevens, R.C.; Lichtman, A.H.; Boger, D.L. Rational Design of Fatty Acid Amide Hydrolase Inhibitors That Act by Covalently Bonding to Two Active Site Residues. J. Am. Chem. Soc. 2013, 135, 6289–6299. [Google Scholar] [CrossRef][Green Version]
- Karlsson, J.; Morgillo, C.M.; Deplano, A.; Smaldone, G.; Pedone, E.; Luque, F.J.; Svensson, M.; Novellino, E.; Congiu, C.; Onnis, V.; et al. Interaction of the N-(3-Methylpyridin-2-Yl) Amide Derivatives of Flurbiprofen and Ibuprofen with Faah: Enantiomeric Selectivity and Binding Mode. PLoS ONE 2015, 10, e0142711. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Bracey, M.H.; Hanson, M.A.; Masuda, K.R.; Stevens, R.C.; Cravatt, B.F. Structural Adaptations in a Membrane Enzyme That Terminates Endocannabinoid Signaling. Science 2002, 298, 1793–1796. [Google Scholar] [CrossRef]
- Deplano, A.; Cipriano, M.; Moraca, F.; Novellino, E.; Catalanotti, B.; Fowler, C.J.; Onnis, V. Benzylamides and Piperazinoarylamides of Ibuprofen as Fatty Acid Amide Hydrolase Inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Deplano, A.; Karlsson, J.; Moraca, F.; Svensson, M.; Cristiano, C.; Morgillo, C.M.; Fowler, C.J.; Catalanotti, B.; Onnis, V.; Deplano, A.; et al. Design, Synthesis and in Vitro and in Vivo Biological Evaluation of Flurbiprofen Amides as New Fatty Acid Amide Hydrolase/Cyclooxygenase-2 Dual Inhibitory Potential Analgesic Agents. J. Enzym. Inhib. Med. Chem. 2021, 36, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Deplano, A.; Karlsson, J.; Svensson, M.; Moraca, F.; Catalanotti, B.; Fowler, C.J.; Onnis, V. Exploring the Fatty Acid Amide Hydrolase and Cyclooxygenase Inhibitory Properties of Novel Amide Derivatives of Ibuprofen. J. Enzym. Inhib. Med. Chem. 2020, 35, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Mileni, M.; Johnson, D.S.; Wang, Z.; Everdeen, D.S.; Liimatta, M.; Pabst, B.; Bhattacharya, K.; Nugent, R.A.; Kamtekar, S.; Cravatt, B.F.; et al. Structure-Guided Inhibitor Design for Human FAAH by Interspecies Active Site Conversion. Proc. Natl. Acad. Sci. USA 2008, 105, 12820–12824. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, T.; Joung, I.; Case, D.A. Soft-Core Potentials in Thermodynamic Integration: Comparing One- and Two-Step Transformations. J. Comput. Chem. 2011, 32, 3253–3263. [Google Scholar] [CrossRef] [PubMed]
- Mileni, M.; Garfunkle, J.; Ezzili, C.; Kimball, F.S.; Cravatt, B.F.; Stevens, R.C.; Boger, D.L. X-Ray Crystallographic Analysis of α-Ketoheterocycle Inhibitors Bound to a Humanized Variant of Fatty Acid Amide Hydrolase. J. Med. Chem. 2010, 53, 230–240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling Protein Tertiary and Quaternary Structure Using Evolutionary Information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Min, X.; Thibault, S.T.; Porter, A.C.; Gustin, D.J.; Carlson, T.J.; Xu, H.; Lindstrom, M.; Xu, G.; Uyeda, C.; Ma, Z.; et al. Discovery and molecular basis of potent noncovalent inhibitors of fatty acid amide hydrolase (FAAH). Proc. Natl. Acad. Sci. USA 2011, 108, 7379–7384. [Google Scholar] [CrossRef]
- Palermo, G.; Campomanes, P.; Neri, M.; Piomelli, D.; Cavalli, A.; Rothlisberger, U.; De Vivo, M. Wagging the Tail: Essential Role of Substrate Flexibility in FAAH Catalysis. J. Chem. Theory Comput. 2013, 9, 1202–1213. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall III, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tufféry, P. Fpocket: Online Tools for Protein Ensemble Pocket Detection and Tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. Amber 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, A.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.1; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An Analysis of the Accuracy of Langevin and Molecular Dynamics Algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Brown, P.M.; Steers, J.; Hui, S.W.; Yeagle, P.L.; Silvius, J.R. Role of Head Group Structure in the Phase Behavior of Amino Phospholipids. 2. Lamellar and Nonlamellar Phases of Unsaturated Phosphatidylethanolamine Analogues. Biochemistry 1986, 25, 4259–4267. [Google Scholar] [CrossRef]
- Yeagle, P.L.; Bennett, M.; Lemaître, V.; Watts, A. Transmembrane Helices of Membrane Proteins May Flex to Satisfy Hydrophobic Mismatch. Biochim. Biophys. Acta 2007, 1768, 530–537. [Google Scholar] [CrossRef]
- Rosso, L.; Gould, I.R. Structure and Dynamics of Phospholipid Bilayers Using Recently Developed General All-Atom Force Fields. J. Comput. Chem. 2008, 29, 24–37. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
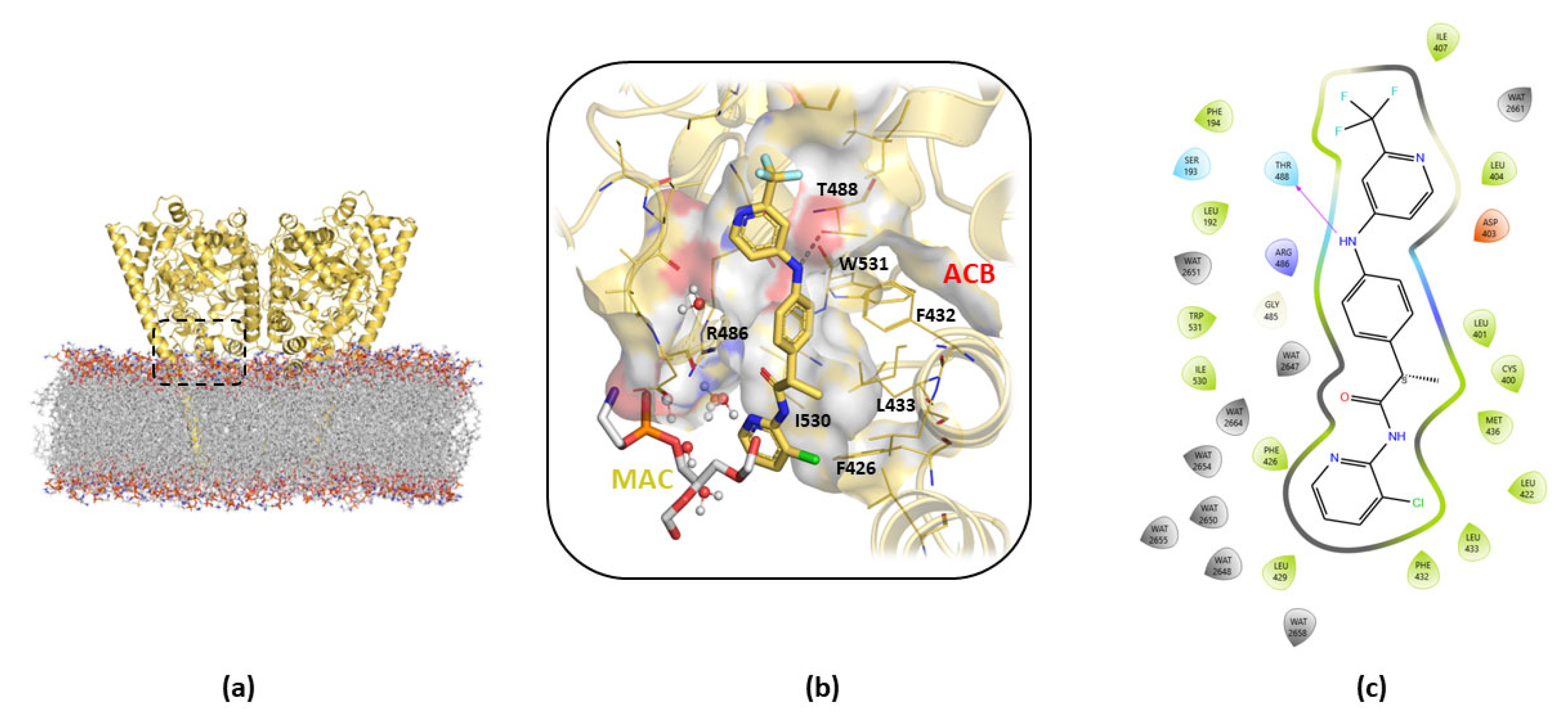{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand/Species | Box | IC50 (µM) | Pose | Cluster Size (%) | ADscore (kcal/mol) |
|---|---|---|---|---|---|
| TPA14/rFAAH | 1 | 0.058 | A2db1 B2db1 Ddb1 B3db1 A3db1 | 61 25 9 4 1 | −8.9 −8.7 −8.7 −8.2 −8.1 |
| TPA14/rFAAH | 2 | 0.058 | A3db2 B2db2 B1db2 A1db2 A2db2 B3db2 | 55 20 3 4 2 5 | −8.7 −8.5 −8.2 −8.0 −7.9 −7.8 |
| TPA14/mFAAH | 1 | 0.48 | B2db1 A2db1 A1db1 B1db1 B3db1 | 43 47 4 4 2 | −9.3 −8.9 −8.8 −8.5 −8.0 |
| Pose | Cluster | Cluster Size (%) | Time (ns) | MM/GBSA | QM-MM/GBSA |
|---|---|---|---|---|---|
| A3db1 | a0 a1 a2 b0 b1 | 44.3 31.1 9.6 64.0 25.5 | 88–93 95–100 73–78 58–63 95–100 | −38.8(±0.4) −44.0(±0.4) n.d −41.9(±0.3) −38.2(±0.3) | n.d −52.5 n.d n.d n.d |
| B3db1 | a0 b0 | 99.7 100 | 80.5–85.5 95–100 | −43.7(±0.4) −49.3(±0.4) | n.d −66.0 |
| TPA1 Pose | QM-MM/GBSA |
|---|---|
| A1 | −71.62 |
| B3 | −61.28 |
| TPA14 Pose | Cluster | Cluster Size (%) | Time (ns) | MM/GBSA | QM-MM/GBSA |
|---|---|---|---|---|---|
| A2db1 | a0 b0 b1 | 92.4 58.3 40.3 | 95–100 95–100 38–43 | −42.54 (±0.31) −45.77 (±0.34) −41.08 (±0.35) | n.d. −59.58 n.d |
| B2db1 | a0 b1 b0 b1 b2 | 64.7 21.8 47.5 41.6 10.6 | 95–100 27–32 95–100 48–53 5–10 | −46.34 (±0.35) −43.98 (±0.39) −42.34 (±0.36) −43.79 (±0.35) −42.3 (±0.31) | −51.97 n.d n.d n.d n.d |
| A3db1 | a0 a1 b0 b1 b2 | 81.3 11 39.5 27.3 21.3 | 95–100 29–34 92–95 21–26 47–52 | −47.82 (±0.37) −44.26 (0.4) −41.73 (0.32) −39.54 (0.39) −37.83 (0.32) | −45.84 n.d n.d n.d n.d |
| B3db1 | a0 b0 b1 | 95.5 62 18.5 | 95–100 95–100 18–23 | −48.27 (0.35) −39.01 (0.37) −37.32 (0.28) | −46.98 n.d n.d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgillo, C.M.; Lupia, A.; Deplano, A.; Pirone, L.; Fiorillo, B.; Pedone, E.; Luque, F.J.; Onnis, V.; Moraca, F.; Catalanotti, B. Molecular Basis for Non-Covalent, Non-Competitive FAAH Inhibition. Int. J. Mol. Sci. 2022, 23, 15502. https://doi.org/10.3390/ijms232415502
Morgillo CM, Lupia A, Deplano A, Pirone L, Fiorillo B, Pedone E, Luque FJ, Onnis V, Moraca F, Catalanotti B. Molecular Basis for Non-Covalent, Non-Competitive FAAH Inhibition. International Journal of Molecular Sciences. 2022; 23(24):15502. https://doi.org/10.3390/ijms232415502
Chicago/Turabian StyleMorgillo, Carmine Marco, Antonio Lupia, Alessandro Deplano, Luciano Pirone, Bianca Fiorillo, Emilia Pedone, F. Javier Luque, Valentina Onnis, Federica Moraca, and Bruno Catalanotti. 2022. "Molecular Basis for Non-Covalent, Non-Competitive FAAH Inhibition" International Journal of Molecular Sciences 23, no. 24: 15502. https://doi.org/10.3390/ijms232415502
APA StyleMorgillo, C. M., Lupia, A., Deplano, A., Pirone, L., Fiorillo, B., Pedone, E., Luque, F. J., Onnis, V., Moraca, F., & Catalanotti, B. (2022). Molecular Basis for Non-Covalent, Non-Competitive FAAH Inhibition. International Journal of Molecular Sciences, 23(24), 15502. https://doi.org/10.3390/ijms232415502