
A Novel Tool for the Analysis and Detection of Copy Number Variants Associated with Haemoglobinopathies

 , , ,
, , , 
 and
and 
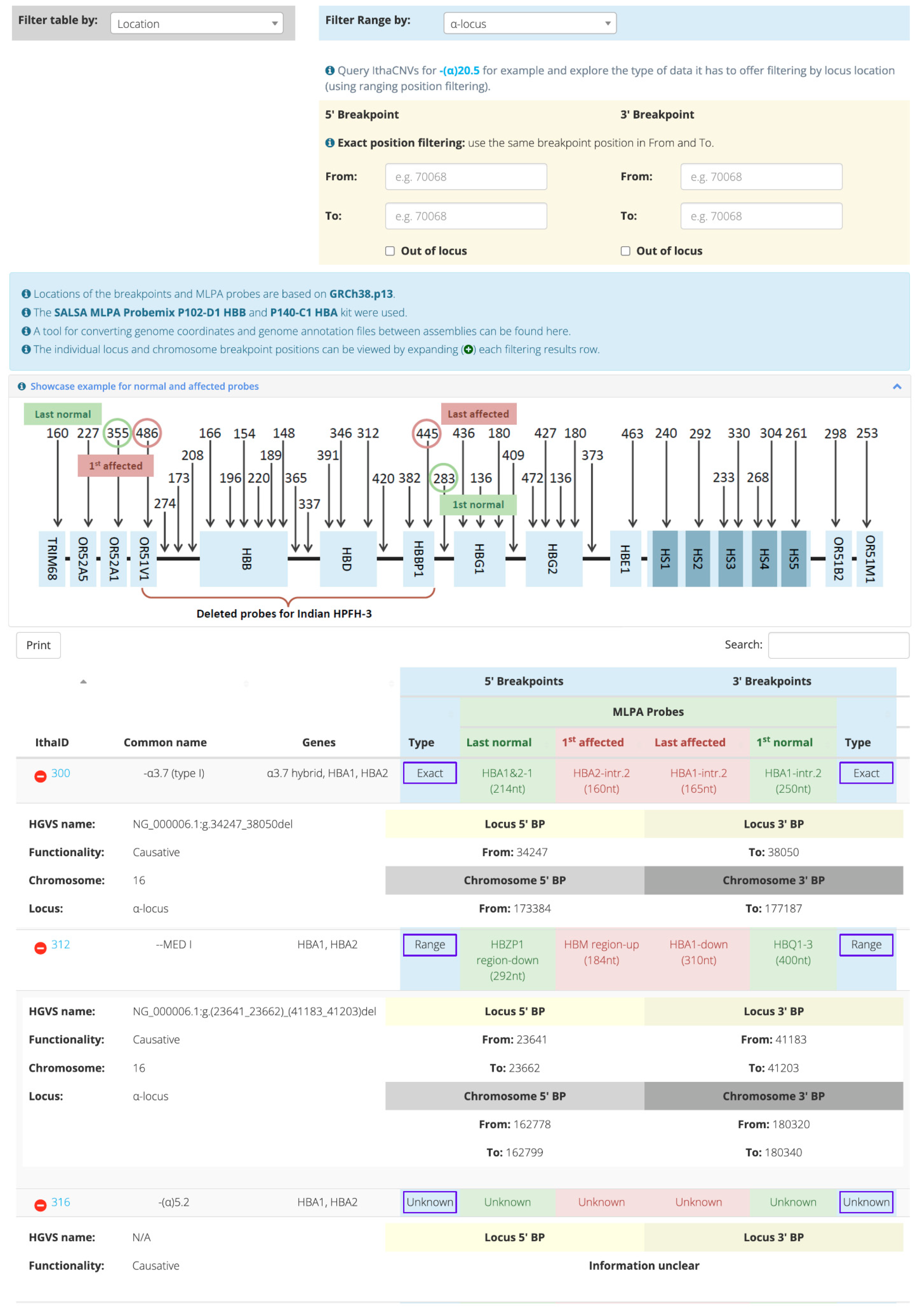{kind=link}
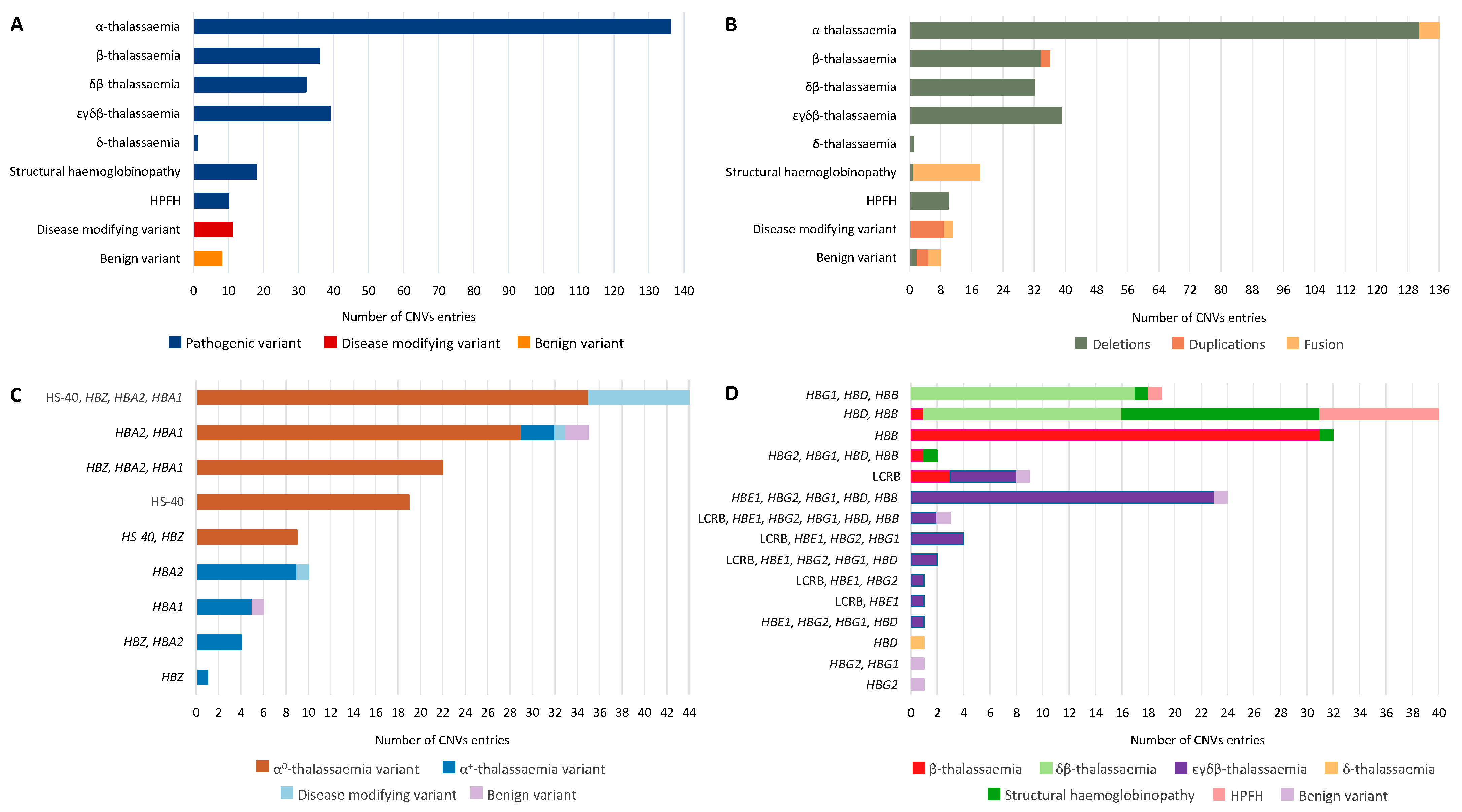{kind=link}
Abstract
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Modell, B.; Darlison, M. Global Epidemiology of Haemoglobin Disorders and Derived Service Indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J. The Thalassaemias. BMJ 1997, 314, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Old, J. Haemoglobinopathies. Prenat. Diagn. 1996, 16, 1181–1186. [Google Scholar] [CrossRef]
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An Interactive Database for Haemoglobin Variations and Epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef]
- Kountouris, P.; Michailidou, K.; Christou, S.; Hadjigavriel, M.; Sitarou, M.; Kolnagou, A.; Kleanthous, M.; Telfer, P. Effect of HBB Genotype on Survival in a Cohort of Transfusion-Dependent Thalassemia Patients in Cyprus. Haematologica 2021, 106, 2458–2468. [Google Scholar] [CrossRef]
- Harteveld, C.L.; Voskamp, A.; Phylipsen, M.; Akkermans, N.; den Dunnen, J.T.; White, S.J.; Giordano, P.C. Nine Unknown Rearrangements in 16p13.3 and 11p15.4 Causing Alpha- and Beta-Thalassaemia Characterised by High Resolution Multiplex Ligation-Dependent Probe Amplification. J. Med. Genet. 2005, 42, 922–931. [Google Scholar] [CrossRef]
- Thein, S.L. The Molecular Basis of β-Thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef]
- Colosimo, A.; Gatta, V.; Guida, V.; Leodori, E.; Foglietta, E.; Rinaldi, S.; Cappabianca, M.P.; Amato, A.; Stuppia, L.; Dallapiccola, B. Application of MLPA Assay to Characterize Unsolved α-Globin Gene Rearrangements. Blood Cells Mol. Dis. 2011, 46, 139–144. [Google Scholar] [CrossRef]
- Rooks, H.; Clark, B.; Best, S.; Rushton, P.; Oakley, M.; Thein, O.S.; Cuthbert, A.C.; Britland, A.; Ruf, A.; Thein, S.L. A Novel 506kb Deletion Causing Εγδβ Thalassemia. Blood Cells Mol. Dis. 2012, 49, 121–127. [Google Scholar] [CrossRef]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A Copy Number Variation Map of the Human Genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Efremov, G.D.; Simjanovska, L.; Plaseska-Karanfilska, D.; Stanojevic, E.; Petkov, G.H. Hb Jambol: A New Hyperunstable Hemoglobin Causing Severe Hemolytic Anemia. Acta Haematol. 2007, 117, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, S.H.; Cho, S.I.; Song, S.H.; Hattori, Y.; Park, S.-K.; Song, J.; Choi, Y.; Yang, M.; Park, H.; et al. Molecular Identification of the Novel Gγ-β Hybrid Hemoglobin: Hb Gγ-β Ulsan (Gγ through 13; β from 19). Blood Cells Mol. Dis. 2010, 45, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Shang, X.; Yi, S.; Cai, R.; Li, Z.; Liu, C.; Liang, Y.; Cai, D.; Zhang, F.; Xu, X. Two Novel Copy Number Variations Involving the α-Globin Gene Cluster on Chromosome 16 Cause Thalassemia in Two Chinese Families. Mol. Genet. Genom. MGG 2016, 291, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Amid, A.; Cheong, M.; Eng, B.; Hanna, M.; Hohenadel, B.-A.; Nakamura, L.M.; Walker, L.; Odame, I.; Kirby-Allen, M.; Waye, J.S. Hb S/Β+-Thalassemia Due to Hb Sickle and a Novel Deletion of DNase I Hypersensitive Sites HS3 and HS4 of the β Locus Control Region. Haematologica 2015, 100, e166–e168. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wei, X.; Cai, D.; Pang, D.; Zhong, J.; Liang, M.; Zuo, Y.; Xu, X.; Shang, X. A Novel 223 Kb Deletion in the Beta-Globin Gene Cluster Was Identified in a Chinese Thalassemia Major Patient. Int. J. Lab. Hematol. 2019, 41, 456–460. [Google Scholar] [CrossRef]
- Kulozik, A.E.; Bail, S.; Bellan-Koch, A.; Bartram, C.R.; Kohne, E.; Kleihauer, E. The Proximal Element of the Beta Globin Locus Control Region Is Not Functionally Required in Vivo. J. Clin. Investig. 1991, 87, 2142–2146. [Google Scholar] [CrossRef][Green Version]
- Calzolari, R.; McMorrow, T.; Yannoutsos, N.; Langeveld, A.; Grosveld, F. Deletion of a Region That Is a Candidate for the Difference between the Deletion Forms of Hereditary Persistence of Fetal Hemoglobin and Deltabeta-Thalassemia Affects Beta- but Not Gamma-Globin Gene Expression. EMBO J. 1999, 18, 949–958. [Google Scholar] [CrossRef][Green Version]
- Mayuranathan, T.; Rayabaram, J.; Das, R.; Arora, N.; Edison, E.S.; Chandy, M.; Srivastava, A.; Velayudhan, S.R. Identification of Rare and Novel Deletions That Cause (Δβ)0-Thalassaemia and Hereditary Persistence of Foetal Haemoglobin in Indian Population. Eur. J. Haematol. 2014, 92, 514–520. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Xu, J.; Byron, R.; Greisman, H.A.; Fisher, C.; Weatherall, D.J.; Sabath, D.E.; Groudine, M.; Orkin, S.H.; Premawardhena, A.; et al. A Functional Element Necessary for Fetal Hemoglobin Silencing. N. Engl. J. Med. 2011, 365, 807–814. [Google Scholar] [CrossRef]
- Bollekens, J.A.; Forget, B.G. Delta Beta Thalassemia and Hereditary Persistence of Fetal Hemoglobin. Hematol. Oncol. Clin. N. Am. 1991, 5, 399–422. [Google Scholar] [CrossRef]
- Ghedira, E.S.; Lecerf, L.; Faubert, E.; Costes, B.; Moradkhani, K.; Bachir, D.; Galactéros, F.; Pissard, S. Estimation of the Difference in HbF Expression Due to Loss of the 5’ δ-Globin BCL11A Binding Region. Haematologica 2013, 98, 305–308. [Google Scholar] [CrossRef] [PubMed][Green Version]
- So, C.C.; So, A.C.Y.; Chan, A.Y.Y.; Tsang, S.T.Y.; Ma, E.S.K.; Chan, L.C. Detection and Characterisation of Beta-Globin Gene Cluster Deletions in Chinese Using Multiplex Ligation-Dependent Probe Amplification. J. Clin. Pathol. 2009, 62, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Pissard, S.; Raclin, V.; Lacan, P.; Garcia, C.; Aguilar-Martinez, P.; Francina, A.; Joly, P. Characterization of Three New Deletions in the β-Globin Gene Cluster during a Screening Survey in Two French Urban Areas. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 415, 35–40. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wei, Y.; Lin, L.; Chen, Q.; Yi, S.; Zuo, Y.; Wei, H.; Zheng, C.; Chen, B.; Qiu, X. The Prevalence and Molecular Characterization of (Δβ)0-Thalassemia and Hereditary Persistence of Fetal Hemoglobin in the Chinese Zhuang Population. J. Clin. Lab. Anal. 2018, 32, e22304. [Google Scholar] [CrossRef] [PubMed]
- Cardiero, G.; Prezioso, R.; Dembech, S.; Del Vecchio Blanco, F.; Scarano, C.; Lacerra, G. Identification and Molecular Characterization of a Novel 163 Kb Deletion: The Italian (Εγδβ)(0)-Thalassemia. Hematol. Amst. Neth. 2016, 21, 317–324. [Google Scholar] [CrossRef]
- Shalev, H.; Landau, D.; Pissard, S.; Krasnov, T.; Kapelushnik, J.; Gilad, O.; Broides, A.; Dgany, O.; Tamary, H. A Novel Epsilon Gamma Delta Beta Thalassemia Presenting with Pregnancy Complications and Severe Neonatal Anemia. Eur. J. Haematol. 2013, 90, 127–133. [Google Scholar] [CrossRef]
- Repnikova, E.; Roberts, J.; Mc Dermott, S.; Farooqi, M.S.; Iqbal, N.T.; Silvey, M.; Nolen, J.; Taboada, E.; Li, W. Clinical and Molecular Characterization of Novel Deletions Causing Epsilon Gamma Delta Beta Thalassemia: Report of Two Cases. Pathol. Res. Pract. 2019, 215, 152578. [Google Scholar] [CrossRef]
- Kaminsky, E.B.; Kaul, V.; Paschall, J.; Church, D.M.; Bunke, B.; Kunig, D.; Moreno-De-Luca, D.; Moreno-De-Luca, A.; Mulle, J.G.; Warren, S.T.; et al. An Evidence-Based Approach to Establish the Functional and Clinical Significance of Copy Number Variants in Intellectual and Developmental Disabilities. Genet. Med. Off. J. Am. Coll. Med. Genet. 2011, 13, 777–784. [Google Scholar] [CrossRef]
- Munkongdee, T.; Chen, P.; Winichagoon, P.; Fucharoen, S.; Paiboonsukwong, K. Update in Laboratory Diagnosis of Thalassemia. Front. Mol. Biosci. 2020, 7, 74. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, Y.; Liu, C.; Lu, J.; Wang, J.; Qin, D.; Liu, L.; Wu, J.; Zhao, X.; Fang, L.; et al. Characterisation of Two Unusual Cases of Haemoglobin Bart’s Hydrops Foetalis Caused by -SEA and Large Novel α-Globin Gene Cluster Deletions. J. Int. Med. Res. 2021, 49, 300060521993642. [Google Scholar] [CrossRef]
- Clark, B.; Shooter, C.; Smith, F.; Brawand, D.; Steedman, L.; Oakley, M.; Rushton, P.; Rooks, H.; Wang, X.; Drousiotou, A.; et al. Beta Thalassaemia Intermedia Due to Co-Inheritance of Three Unique Alpha Globin Cluster Duplications Characterised by next Generation Sequencing Analysis. Br. J. Haematol. 2018, 180, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.-M.; Yang, X.; Huang, L.-M.; Ouyang, G.-J.; Yang, X.-X.; Li, M. Simultaneous Detection of Target CNVs and SNVs of Thalassemia by Multiplex PCR and Next-generation Sequencing. Mol. Med. Rep. 2019, 19, 2837–2848. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Xu, L.-L.; Tang, X.-W.; Li, D.-Z. A Novel Deletion of the Major Regulatory Element Flanking the α-Globin Gene Cluster as a Cause of A0-Thalassemia. Int. J. Lab. Hematol. 2021, 43, O190–O192. [Google Scholar] [CrossRef] [PubMed]
- Grimholt, R.M.; Fjeld, B.; Klingenberg, O. Hemoglobinopathy Gone Astray-Three Novel Forms of α-Thalassemia in Norwegian Patients Characterized by Quantitative Real-Time PCR and DNA Sequencing. Scand. J. Clin. Lab. Investig. 2021, 81, 670–678. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Traeger-Synodinos, J.; Bento, C.; Harteveld, C.L.; Fylaktou, E.; Koopmann, T.T.; Halim-Fikri, H.; Michailidou, K.; et al. Adapting the ACMG/AMP Variant Classification Framework: A Perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Hum. Mutat. 2022, 43, 1089–1096. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minaidou, A.; Tamana, S.; Stephanou, C.; Xenophontos, M.; Harteveld, C.L.; Bento, C.; Kleanthous, M.; Kountouris, P. A Novel Tool for the Analysis and Detection of Copy Number Variants Associated with Haemoglobinopathies. Int. J. Mol. Sci. 2022, 23, 15920. https://doi.org/10.3390/ijms232415920
Minaidou A, Tamana S, Stephanou C, Xenophontos M, Harteveld CL, Bento C, Kleanthous M, Kountouris P. A Novel Tool for the Analysis and Detection of Copy Number Variants Associated with Haemoglobinopathies. International Journal of Molecular Sciences. 2022; 23(24):15920. https://doi.org/10.3390/ijms232415920
Chicago/Turabian StyleMinaidou, Anna, Stella Tamana, Coralea Stephanou, Maria Xenophontos, Cornelis L. Harteveld, Celeste Bento, Marina Kleanthous, and Petros Kountouris. 2022. "A Novel Tool for the Analysis and Detection of Copy Number Variants Associated with Haemoglobinopathies" International Journal of Molecular Sciences 23, no. 24: 15920. https://doi.org/10.3390/ijms232415920
APA StyleMinaidou, A., Tamana, S., Stephanou, C., Xenophontos, M., Harteveld, C. L., Bento, C., Kleanthous, M., & Kountouris, P. (2022). A Novel Tool for the Analysis and Detection of Copy Number Variants Associated with Haemoglobinopathies. International Journal of Molecular Sciences, 23(24), 15920. https://doi.org/10.3390/ijms232415920