

Application of Long-Read Nanopore Sequencing to the Search for Mutations in Hypertrophic Cardiomyopathy

 , and
, and
Abstract
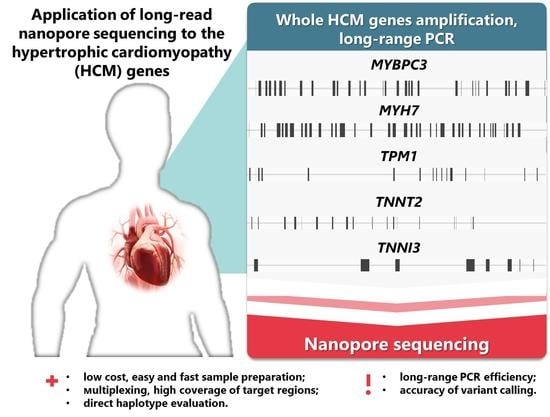
1. Introduction
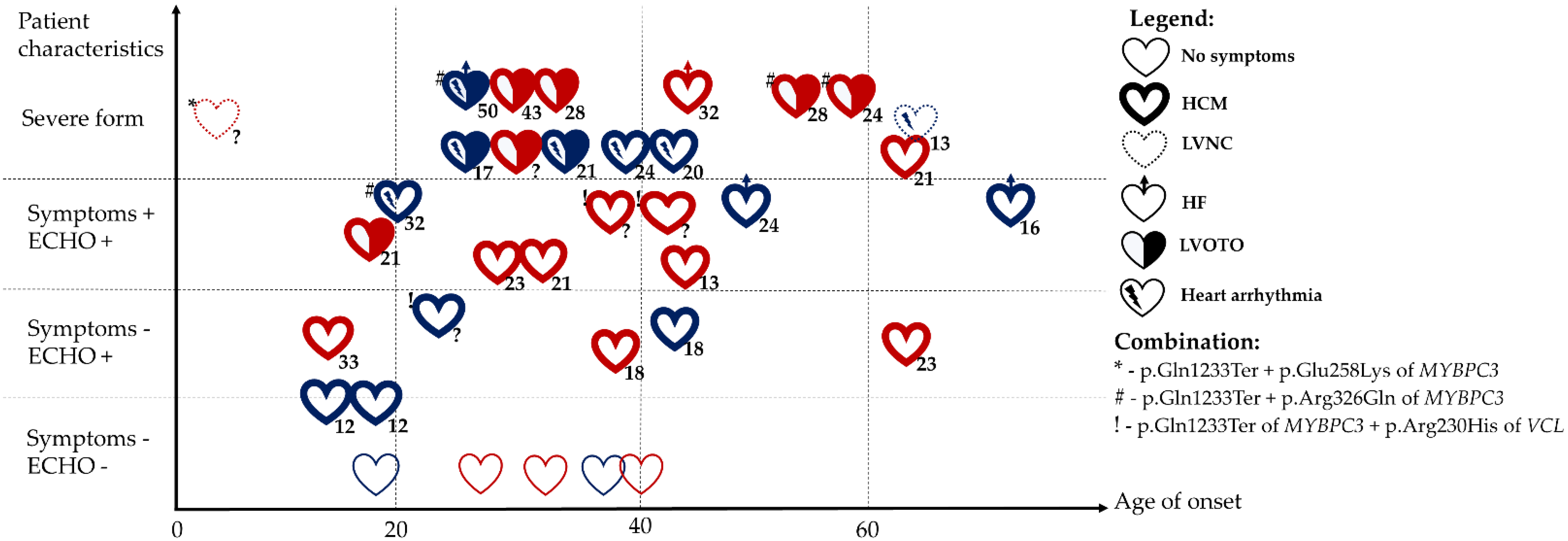
2. Results
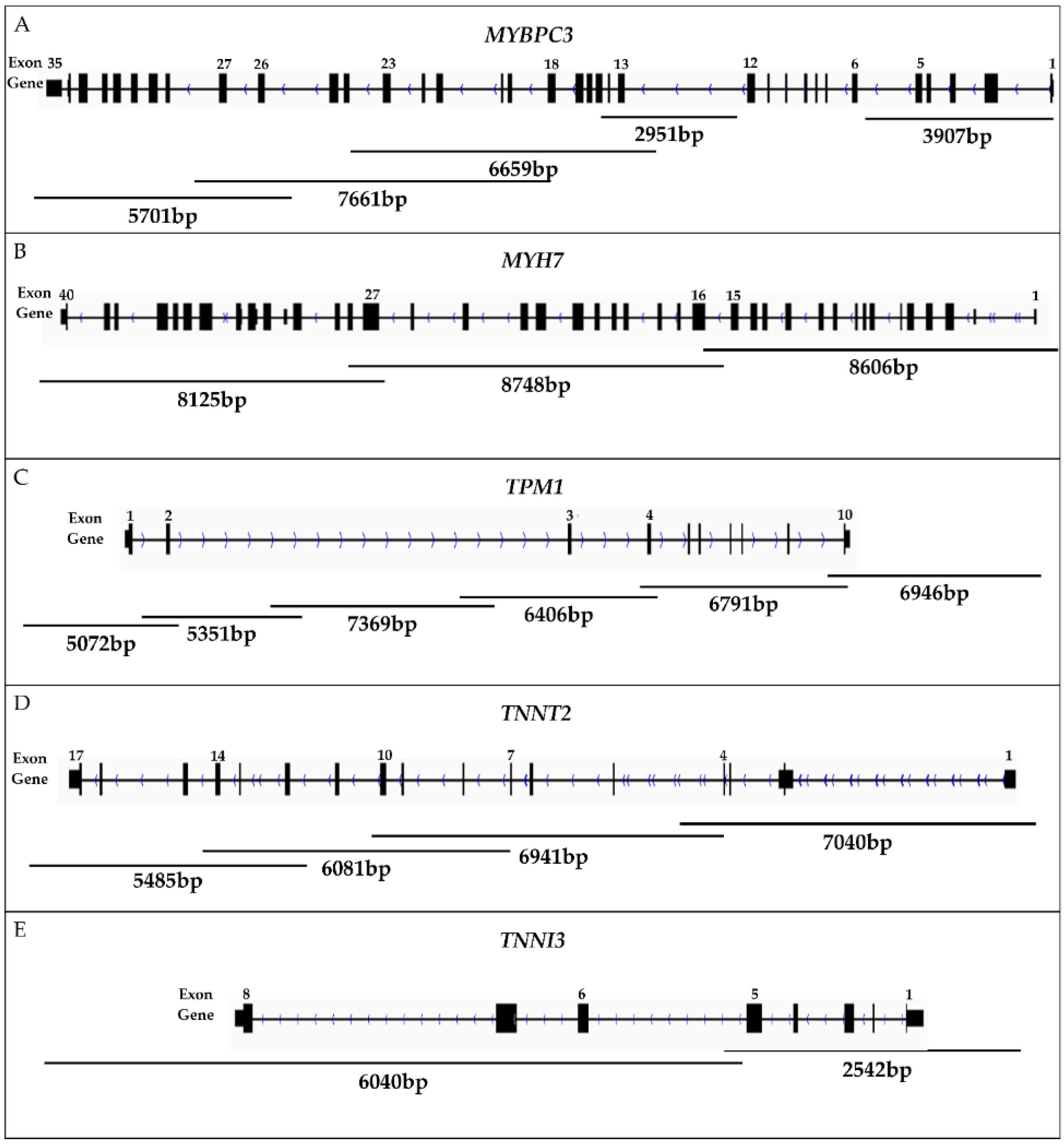
2.1. Long-Range PCR Design
2.2. ONT Sequencing
2.3. Nanopore vs. Illumina Sequencing
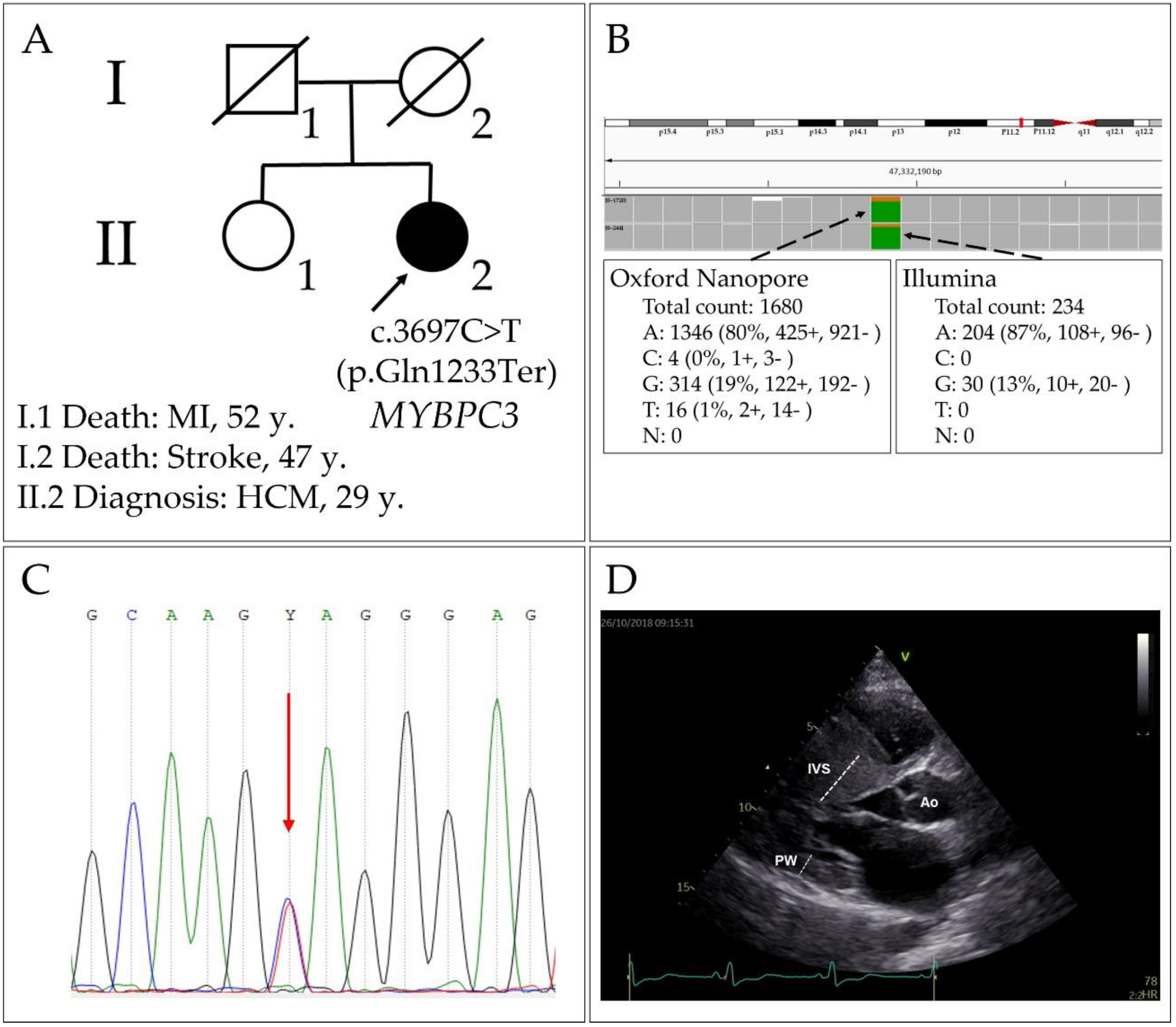
2.4. Pathogenic Variant p.Gln1233Ter in the MYBPC3 Gene
2.5. Haplotype Evaluation
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Isolation and Amplification
4.3. ONT Sequencing
4.4. Processing of ONT Sequencing Data
4.5. Library Preparation, Target Sequencing and Bioinformatic Analysis (Illumina)
4.6. Assessment of Pathogenicity of Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Klaassen, S.; Brodehl, A. The genetic landscape of cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Springer: Cham, Switzerland, 2019; Volume 7, pp. 45–91. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef] [PubMed]
- Coto, E.; Reguero, J.R.; Palacín, M.; Gómez, J.; Alonso, B.; Iglesias, S.; Martín, M.; Tavira, B.; Díaz-Molina, B.; Morales, C.; et al. Resequencing the whole MYH7 gene (including the intronic, promoter, and 3’ UTR sequences) in hypertrophic cardiomyopathy. J. Mol. Diagn. 2012, 14, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Mendes de Almeida, R.; Tavares, J.; Martins, S.; Carvalho, T.; Enguita, F.J.; Brito, D.; Carmo-Fonseca, M.; Lopes, L.R. Whole gene sequencing identifies deep-intronic variants with potential functional impact in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0182946. [Google Scholar] [CrossRef] [PubMed]
- Janin, A.; Chanavat, V.; Rollat-Farnier, P.A.; Bardel, C.; Nguyen, K.; Chevalier, P.; Eicher, J.C.; Faivre, L.; Piard, J.; Albert, E.; et al. Whole MYBPC3 NGS sequencing as a molecular strategy to improve the efficiency of molecular diagnosis of patients with hypertrophic cardiomyopathy. Hum Mutat. 2020, 41, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Barbosa, P.; Torrado, M.; Quinn, E.; Merino, A.; Ochoa, J.P.; Jager, J.; Futema, M.; Carmo-Fonseca, M.; Monserrat, L.; et al. Cryptic splice-altering variants in MYBPC3 are a prevalent cause of hypertrophic cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, e002905. [Google Scholar] [CrossRef] [PubMed]
- Sadayappan, S.; Puckelwartz, M.J.; McNally, E.M. South Asian-Specific MYBPC3Δ25bp Intronic Deletion and Its Role in Cardiomyopathies and Heart Failure. Circ. Genom. Precis. Med. 2020, 13, e002986. [Google Scholar] [CrossRef]
- Torrado, M.; Maneiro, E.; Lamounier Junior, A.; Fernández-Burriel, M.; Sánchez Giralt, S.; Martínez-Carapeto, A.; Cazón, L.; Santiago, E.; Ochoa, J.P.; McKenna, W.J.; et al. Identification of an elusive spliceogenic MYBPC3 variant in an otherwise genotype-negative hypertrophic cardiomyopathy pedigree. Sci. Rep. 2022, 12, 7284. [Google Scholar] [CrossRef]
- Leija-Salazar, M.; Sedlazeck, F.J.; Toffoli, M.; Mullin, S.; Mokretar, K.; Athanasopoulou, M.; Donald, A.; Sharma, R.; Hughes, D.; Schapira, A.H.V.; et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genom. Med. 2019, 7, e564. [Google Scholar] [CrossRef]
- Soufi, M.; Bedenbender, S.; Ruppert, V.; Kurt, B.; Schieffer, B.; Schaefer, J.R. Fast and Easy Nanopore Sequencing Workflow for Rapid Genetic Testing of Familial Hypercholesterolemia. Front. Genet. 2022, 13, 836231. [Google Scholar] [CrossRef]
- Leung, A.W.; Leung, H.C.; Wong, C.L.; Zheng, Z.X.; Lui, W.W.; Luk, H.M.; Lo, I.F.; Luo, R.; Lam, T.W. ECNano: A cost-effective workflow for target enrichment sequencing and accurate variant calling on 4800 clinically significant genes using a single MinION flowcell. BMC Med. Genom. 2022, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Dainis, A.; Tseng, E.; Clark, T.A.; Hon, T.; Wheeler, M.; Ashley, E. Targeted Long-Read RNA Sequencing Demonstrates Transcriptional Diversity Driven by Splice-Site Variation in MYBPC3. Circ. Genom. Precis. Med. 2019, 12, e002464. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Hain, C.; Flottmann, F.; Ratnavadivel, S.; Gaertner, A.; Klauke, B.; Kalinowski, J.; Körperich, H.; Gummert, J.; Paluszkiewicz, L.; et al. The Desmin mutation DES-c.735G>C causes severe restrictive cardiomyopathy by inducing in-frame skipping of exon-3. Biomedicines 2021, 9, 1400. [Google Scholar] [CrossRef]
- Chakova, N.N.; Niyazova, S.S.; Komissarova, S.M.; Sasinovich, M.A.; Goncharenko, M.G. Gln1233* nonsens-mutation and Arg326Gln polymorphism of MYBPC3 gene in patients with hypertrophic cardiomyopathy in Belarus. Med. Genet. 2018, 17, 36–43. (In Russian) [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Helms, A.S.; Davis, F.M.; Coleman, D.; Bartolone, S.N.; Glazier, A.A.; Pagani, F.; Yob, J.M.; Sadayappan, S.; Pedersen, E.; Lyons, R.; et al. Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2014, 7, 434–443. [Google Scholar] [CrossRef]
- Shestak, A.G.; Bukaeva, A.A.; Saber, S.; Zaklyazminskaya, E.V. Allelic dropout is a common phenomenon that reduces the diagnostic yield of PCR-based sequencing of targeted gene panels. Front. Genet. 2021, 12, 620337. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Verweij, N.; Benjamins, J.W.; Morley, M.P.; van de Vegte, Y.J.; Teumer, A.; Trenkwalder, T.; Reinhard, W.; Cappola, T.P.; van der Harst, P. The Genetic Makeup of the Electrocardiogram. Cell Syst. 2020, 11, 229–238.e5. [Google Scholar] [CrossRef]
- Frank, M.; Prenzler, A.; Eils, R.; Graf von der Schulenburg, J.M. Genome sequencing: A systematic review of health economic evidence. Health Econ. Rev. 2013, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, S.; Su, J.; Leung, A.W.; Lam, T.; Luo, R. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. BioRxiv 2021. [Google Scholar] [CrossRef]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef]
- Li, Y.; He, X.Z.; Li, M.H.; Li, B.; Yang, M.J.; Xie, Y.; Zhang, Y.; Ma, X.J. Comparison of third-generation sequencing approaches to identify viral pathogens under public health emergency conditions. Virus Genes 2020, 56, 288–297. [Google Scholar] [CrossRef]
- Erdmann, J.; Raible, J.; Maki-Abadi, J.; Hummel, M.; Hammann, J.; Wollnik, B.; Frantz, E.; Fleck, E.; Hetzer, R.; Regitz-Zagrosek, V. Spectrum of clinical phenotypes and gene variants in cardiac myosin-binding protein C mutation carriers with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2001, 38, 322–330. [Google Scholar] [CrossRef]
- Ingles, J.; Doolan, A.; Chiu, C.; Seidman, J.; Seidman, C.; Semsarian, C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J. Med. Genet. 2005, 42, 59. [Google Scholar] [CrossRef]
- Ehlermann, P.; Weichenhan, D.; Zehelein, J.; Steen, H.; Pribe, R.; Zeller, R.; Lehrke, S.; Zugck, C.; Ivandic, B.T.; Katus, H.A. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med. Genet. 2008, 28, 9–95. [Google Scholar] [CrossRef]
- Fokstuen, S.; Lyle, R.; Munoz, A.; Gehrig, C.; Lerch, R.; Perrot, A.; Osterziel, K.J.; Geier, C.; Beghetti, M.; Mach, F.; et al. A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. Hum Mutat. 2008, 29, 879–885. [Google Scholar] [CrossRef]
- Roncarati, R.; Latronico, M.V.; Musumeci, B.; Aurino, S.; Torella, A.; Bang, M.L.; Jotti, G.S.; Puca, A.A.; Volpe, M.; Nigro, V.; et al. Unexpectedly low mutation rates in beta-myosin heavy chain and cardiac myosin binding protein genes in Italian patients with hypertrophic cardiomyopathy. J. Cell Physiol. 2011, 226, 2894–2900. [Google Scholar] [CrossRef]
- Tóth, T.; Nagy, V.; Faludi, R.; Csanády, M.; Nemes, A.; Simor, T.; Forster, T.; Sepp, R. The Gln1233Ter mutation of the myosin binding protein C gene: Causative mutation or innocent polymorphism in patients with hypertrophic cardiomyopathy? Int. J. Cardiol. 2011, 153, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: A potential link to sudden death in the absence of conventional risk factors. Heart Rhythm. 2012, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Sdvigova, N.A.; Basargina, E.N.; Ryabtsev, D.V.; Savostyanov, K.V.; Pushkov, A.A.; Zhurkova, N.V.; Revunenkov, G.V.; Zharova, O.P. The urgency of genetic verification of non-compaction cardiomyopathy in children: Clinical cases. Curr. Pediatr. 2018, 17, 157–165. [Google Scholar] [CrossRef]
- Dzemeshkevich, S.L.; Motreva, A.P.; Kalmykova, O.V.; Martyanova, Y.B.; Sinitsyn, V.E.; Mershina, E.A.; Nikolaeva, E.V.; Radzhabova, G.M.; Polyak, M.E.; Nikityuk, T.G.; et al. Hypertrophic cardiomyopathy in youth: Phenotype, genotype, and treatment approaches. Clin. Exp. Surg. Petrovsk. J. 2019, 7, 54–62. (In Russian) [Google Scholar] [CrossRef]
- Dementyeva, E.V.; Vyatkin, Y.V.; Kretov, E.I.; Elisaphenko, E.A.; Medvedev, S.P.; Zakian, S.M. Genetic analysis of patients with hypertrophic cardiomyopathy. Genes Cells 2020, 15, 68–73. [Google Scholar] [CrossRef]
- O’Hare, B.J.; Bos, J.M.; Tester, D.J.; Ackerman, M.J. Patients with hypertrophic cardiomyopathy deemed genotype negative based on research grade genetic analysis: Time for repeat diagnostic testing with next-generation sequencing. Circ. Genom. Precis. Med. 2020, 13, e003013. [Google Scholar] [CrossRef]
- Filatova, E.V.; Krylova, N.S.; Kovalevskaya, E.A.; Maslova, M.Y.; Poteshkina, N.G.; Slominsky, P.A.; Shadrina, M.I. The p.Arg230His variant of the VCL protein is not pathogenic and does not affect hypertrophic cardiomyopathy phenotype in russian family carrying the p.Gln1233Ter pathogenic variant in the MYBPC3 gene. JCDR 2021, 12, 1869–1874. [Google Scholar] [CrossRef]
- Blagova, O.; Pavlenko, E.; Sedov, V.; Kogan, E.; Polyak, M.; Zaklyazminskaya, E.; Lutokhina, Y. Different phenotypes of sarcomeric MYBPC3-cardiomyopathy in the same family: Hypertrophic, left ventricular noncompaction and restrictive phenotypes (in association with sarcoidosis). Genes 2022, 13, 1344. [Google Scholar] [CrossRef]
- O’Leary, T.S.; Snyder, J.; Sadayappan, S.; Day, S.M.; Previs, M.J. MYBPC3 truncation mutations enhance actomyosin contractile mechanics in human hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2019, 127, 165–173. [Google Scholar] [CrossRef]
- Salakhov, R.R.; Golubenko, M.V.; Zarubin, A.A.; Pavlyukova, E.N.; Kanev, A.F.; Glotov, O.S.; Alaverdian, D.A.; Tsay, V.V.; Valiakhmetov, N.R.; Nazarenko, M.S. Sequencing of cardiomyopathy genes in patients with hypertrophic cardiomyopathy reveals enrichment for rare variants in the genes for arrhythmogenic right ventricular cardiomyopathy. In Proceedings of the 54th European Society of Human Genetics (ESHG) Conference, Virtual, 28–31 August 2021. [Google Scholar]
- Dementyeva, E.V.; Medvedev, S.P.; Kovalenko, V.R.; Vyatkin, Y.V.; Kretov, E.I.; Slotvitsky, M.M.; Shtokalo, D.N.; Pokushalov, E.A.; Zakian, S.M. Applying Patient-Specific Induced Pluripotent Stem Cells to Create a Model of Hypertrophic Cardiomyopathy. Biochem. Biokhimiia 2019, 84, 291–298. [Google Scholar] [CrossRef]
- Fourey, D.; Care, M.; Siminovitch, K.A.; Weissler-Snir, A.; Hindieh, W.; Chan, R.H.; Gollob, M.H.; Rakowski, H.; Adler, A. Prevalence and Clinical Implication of Double Mutations in Hypertrophic Cardiomyopathy: Revisiting the Gene-Dose Effect. Circ. Cardiovasc. Genet. 2017, 10, e001685. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Qin, S.; Liu, Y.; Tang, L.; Zhao, W.; Pan, C.; Qiu, Z.; Wang, X.; Shu, X. Whole-exome sequencing identifies rare compound heterozygous mutations in the MYBPC3 gene associated with severe familial hypertrophic cardiomyopathy. Eur. J. Med. Genet. 2018, 61, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; the UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- GATK. Available online: https://gatk.broadinstitute.org/hc/en-us (accessed on 21 September 2022).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 September 2022).
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E.; et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: Recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef]
- Walsh, R.; Mazzarotto, F.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Li, N.; Felkin, L.; Ingold, N.; Govind, R.; et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: The case of hypertrophic cardiomyopathy. Genome Med. 2019, 11, 5. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | ONT/Illumina | |||
|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
| Mean mapping quality | 59.77/59.03 | 59.73/58.91 | 59.73/58.89 | 59.81/58.83 |
| Coverage, mean | 3568.7/277.4 | 3205.6/284.9 | 3908.2/347.01 | 4961.2/292.3 |
| General error rate, % | 6.9/1.14 | 6.92/1.23 | 6.91/1.24 | 6.86/1.35 |
| Mismatches | 19,996,414/333,439 | 17,905,385/371,635 | 21,543,356/458,936 | 27,703,185/417,607 |
| Insertions | 3,830,262/3468 | 3,429,705/3464 | 4,150,256/3980 | 5,300,210/3802 |
| Mapped reads with at least one insertion, % | 84.93/0.65 | 83.44/0.54 | 87.6/0.68 | 85.61/0.85 |
| Deletions | 6,608,331/7444 | 5,932,133/6932 | 7,066,882/8140 | 9,173,356/7264 |
| Mapped reads with at least one deletion, % | 85.24/1.39 | 83.7/1.08 | 87.89/1.39 | 85.82/1.64 |
| GC percentage, % | 52.12/52.19 | 51.87/52.1 | 51.42/51.69 | 51.28/51.7 |
| SNVs | Sarcomeric Protein Genes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MYBPC3 | MYH7 | TPM1 | TNNT2 | TNNI3 | ||||||
| ONT | Illumina | ONT | Illumina | ONT | Illumina | ONT | Illumina | ONT | Illumina | |
| Total | 31 | 35 | 28 | 26 | 99 | 100 | 60 | 58 | 13 | 12 |
| Exonic: | 3 | 3 | 3 | 3 | 1 | 1 | 2 | 2 | 0 | 0 |
| Synonymous | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 0 | 0 |
| Nonsynonymous | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| Stopgain | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Splicing sites | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Intronic | 26 | 30 | 23 | 21 | 78 | 80 | 56 | 54 | 11 | 11 |
| 3′UTR & downstream | 2 | 2 | 2 | 2 | 20 | 19 | 2 | 2 | 2 | 1 |
| No. | Position (GRCh38.p13) | Reference/Alternative Allele | Gene | Location | mRNA Variant | Protein Variant | dbSNP rs# | Genotypes | CADD | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | PHRED | ||||||||
| 1 | chr1:201361001 | T/C | TNNT2 | Intron 14/15 | - | - | rs10920181 | 0|1 | 1|0 | 0|1 | 0/0 | 12.43 |
| 2 | chr1: 201361301 | T/C | TNNT2 | Exon 13/15 | c.A788G | p.R263K | rs3730238 | 0|1 | 1|0 | 0|0 | 0/0 | 23.5 |
| 3 | chr1:201362426 | T/G | TNNT2 | Intron 11/15 | - | - | rs1104859 | 1|1 | 1|1 | 1|1 | 1/1 | 19.51 |
| 4 | chr1: 201365254 | G/A | TNNT2 | Exon 9/15 | c.C318T | p.I106I | rs3729547 | 1|1 | 0|1 | 1|1 | 1/1 | 10.80 |
| 5 | chr1:201371368 | G/A | TNNT2 | Intron 3/15 | - | - | rs4915240 | 1|1 | 0|1 | 0|1 | 0/1 | 11.79 |
| 6 | chr1:201376224 | A/G | TNNT2 | Intron 2/16 (1/15) | - | - | rs947485 | 1|1 | 1|1 | 1|1 | 0/1 | 10.25 |
| 7 | chr15:63042489 | G/C | TPM1 | upstream 258 b.p. | - | - | rs35829897 | 0|0 | 0|0 | 0|0 | 0/1 | 18.82 |
| 8 | chr15:63043900 | C/G | TPM1 | Intron 1/8 | - | - | rs62013181 | 0|1 | 0|0 | 0|0 | 0/0 | 18.96 |
| 9 | chr15:63046558 | T/C | TPM1/TPM1-AS | Intron 2/8 | - | - | rs8026502 | 1|0 | 0|0 | 1|0 | 0/1 | 11.46 |
| 10 | chr15:63047328 | A/G | TPM1/TPM1-AS | Intron 2/8 | - | - | - | 0|0 | 0|0 | 0|1 | 0/0 | 15.04 |
| 11 | chr15:63047379 | G/A | TPM1/TPM1-AS | Intron 2/8 | - | - | rs57645645 | 1|0 | 0|0 | 1|0 | 0/1 | 15.48 |
| 12 | chr15:63048408 | T/C | TPM1/TPM1-AS | Intron 2/8 | - | - | rs4075047 | 1|0 | 0|0 | 1|0 | 0/1 | 13.03 |
| 13 | chr15:63048506 | C/T | TPM1/TPM1-AS | Intron 2/8 | - | - | rs111470259 | 1|0 | 0|0 | 1|0 | 0/1 | 14.44 |
| Patient Characteristics | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Age (years old)/sex (f-female; m-male) | 29/f | 54/m | 63/m | 35/m |
| Interventricular septal thickness, mm | 43 | 19 | 12 | 35 |
| Left ventricular posterior wall thickness, mm | 12 | 18 | 13 | 23 |
| Left ventricular outflow tract gradient rest/Valsalva manoeuvre, mmHg | 9.6/5.51 | 66.27/165 | 21.39/51.47 | 11.71/10.51 |
| Left ventricular myocardial mass, g | 213.5 | 184 | 140 | 239 |
| Left ventricular ejection fraction, % | 73 | 61 | 79 | 71 |
| Septal reduction therapy | yes | yes | yes | no |
| Symptoms | Angina, dyspnoea, palpitations | Angina, dyspnoea | Angina, dyspnoea, palpitations | Dyspnoea |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salakhov, R.R.; Golubenko, M.V.; Valiakhmetov, N.R.; Pavlyukova, E.N.; Zarubin, A.A.; Babushkina, N.P.; Kucher, A.N.; Sleptcov, A.A.; Nazarenko, M.S. Application of Long-Read Nanopore Sequencing to the Search for Mutations in Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 15845. https://doi.org/10.3390/ijms232415845
Salakhov RR, Golubenko MV, Valiakhmetov NR, Pavlyukova EN, Zarubin AA, Babushkina NP, Kucher AN, Sleptcov AA, Nazarenko MS. Application of Long-Read Nanopore Sequencing to the Search for Mutations in Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences. 2022; 23(24):15845. https://doi.org/10.3390/ijms232415845
Chicago/Turabian StyleSalakhov, Ramil R., Maria V. Golubenko, Nail R. Valiakhmetov, Elena N. Pavlyukova, Aleksei A. Zarubin, Nadezhda P. Babushkina, Aksana N. Kucher, Aleksei A. Sleptcov, and Maria S. Nazarenko. 2022. "Application of Long-Read Nanopore Sequencing to the Search for Mutations in Hypertrophic Cardiomyopathy" International Journal of Molecular Sciences 23, no. 24: 15845. https://doi.org/10.3390/ijms232415845
APA StyleSalakhov, R. R., Golubenko, M. V., Valiakhmetov, N. R., Pavlyukova, E. N., Zarubin, A. A., Babushkina, N. P., Kucher, A. N., Sleptcov, A. A., & Nazarenko, M. S. (2022). Application of Long-Read Nanopore Sequencing to the Search for Mutations in Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences, 23(24), 15845. https://doi.org/10.3390/ijms232415845