

Semiclassical Theory of Multistage Nonequilibrium Electron Transfer in Macromolecular Compounds in Polar Media with Several Relaxation Timescales


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
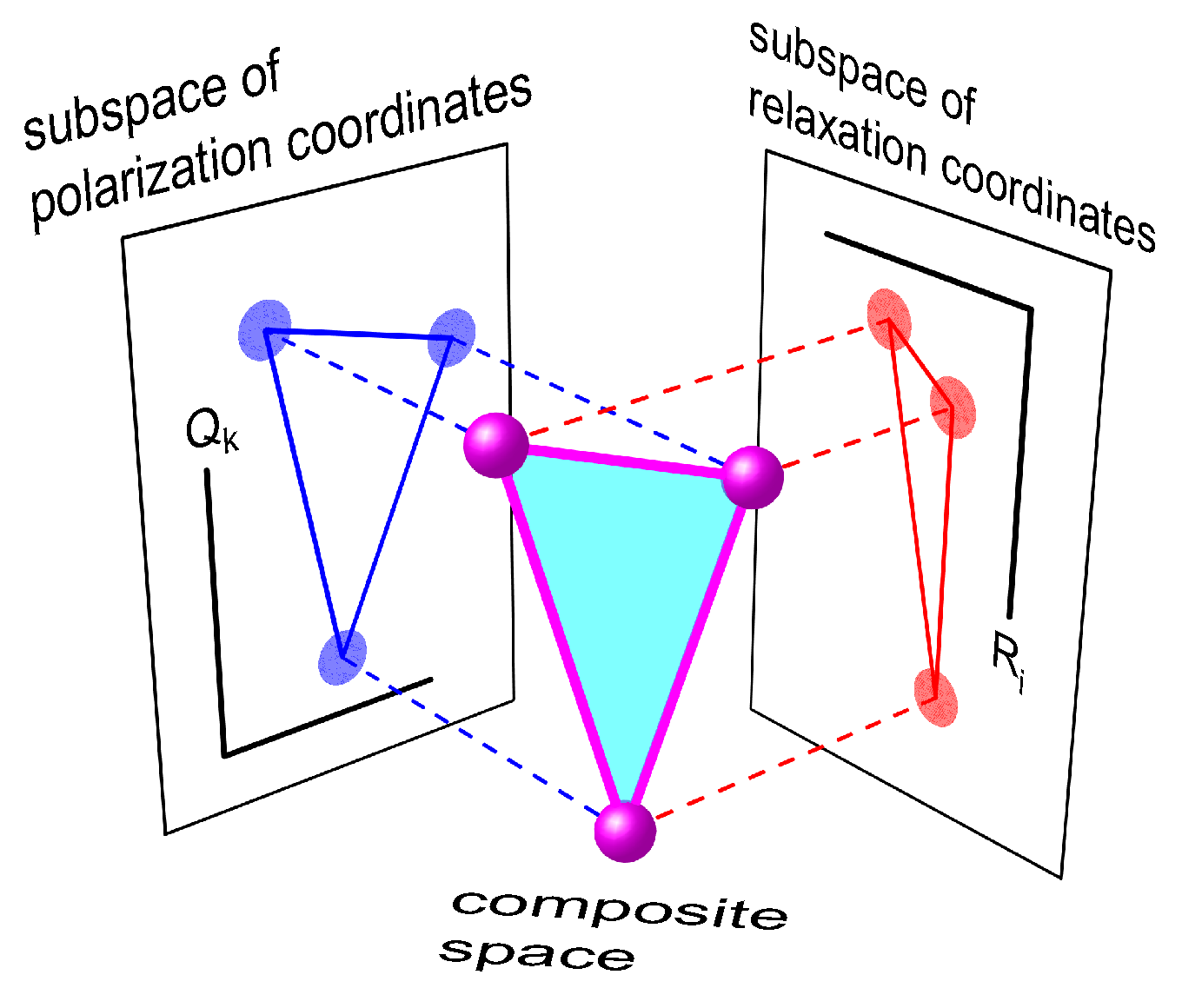
2.1. General Formulation of the Theory
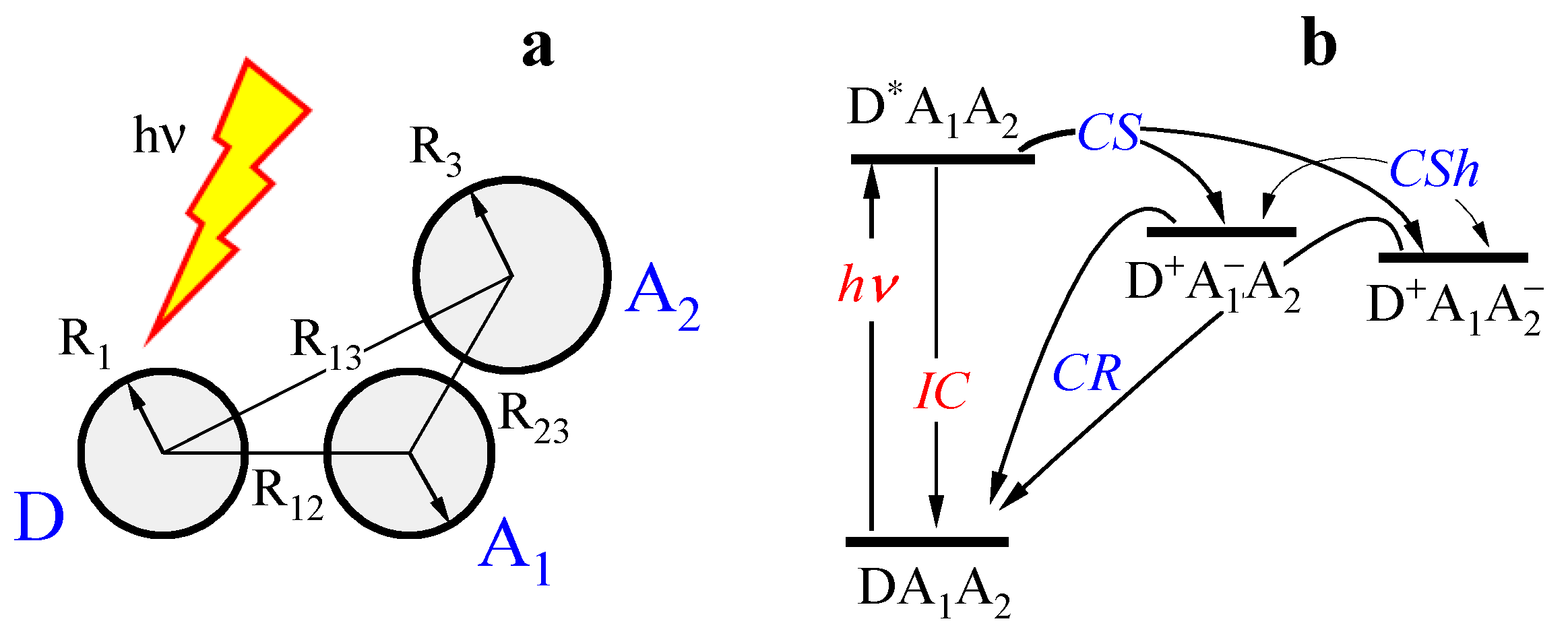
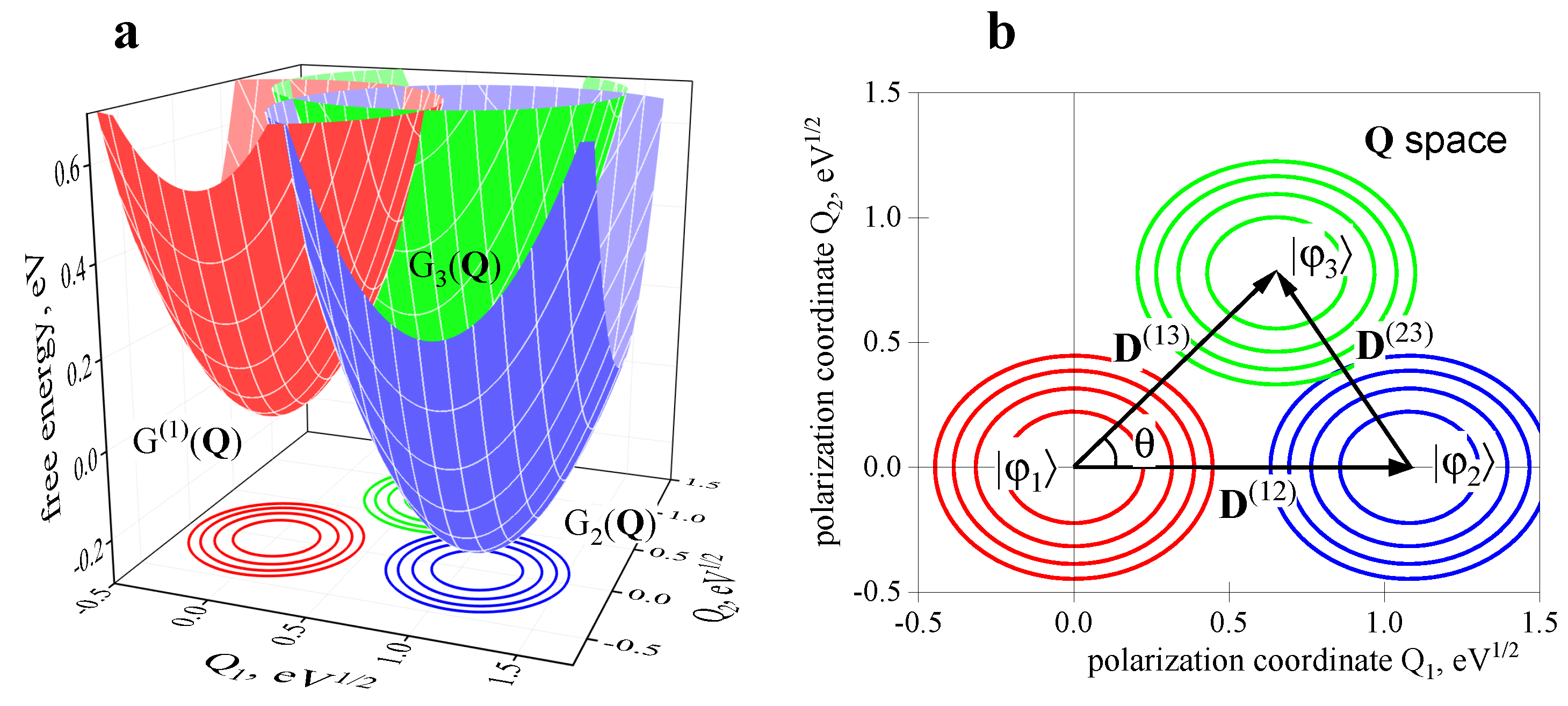
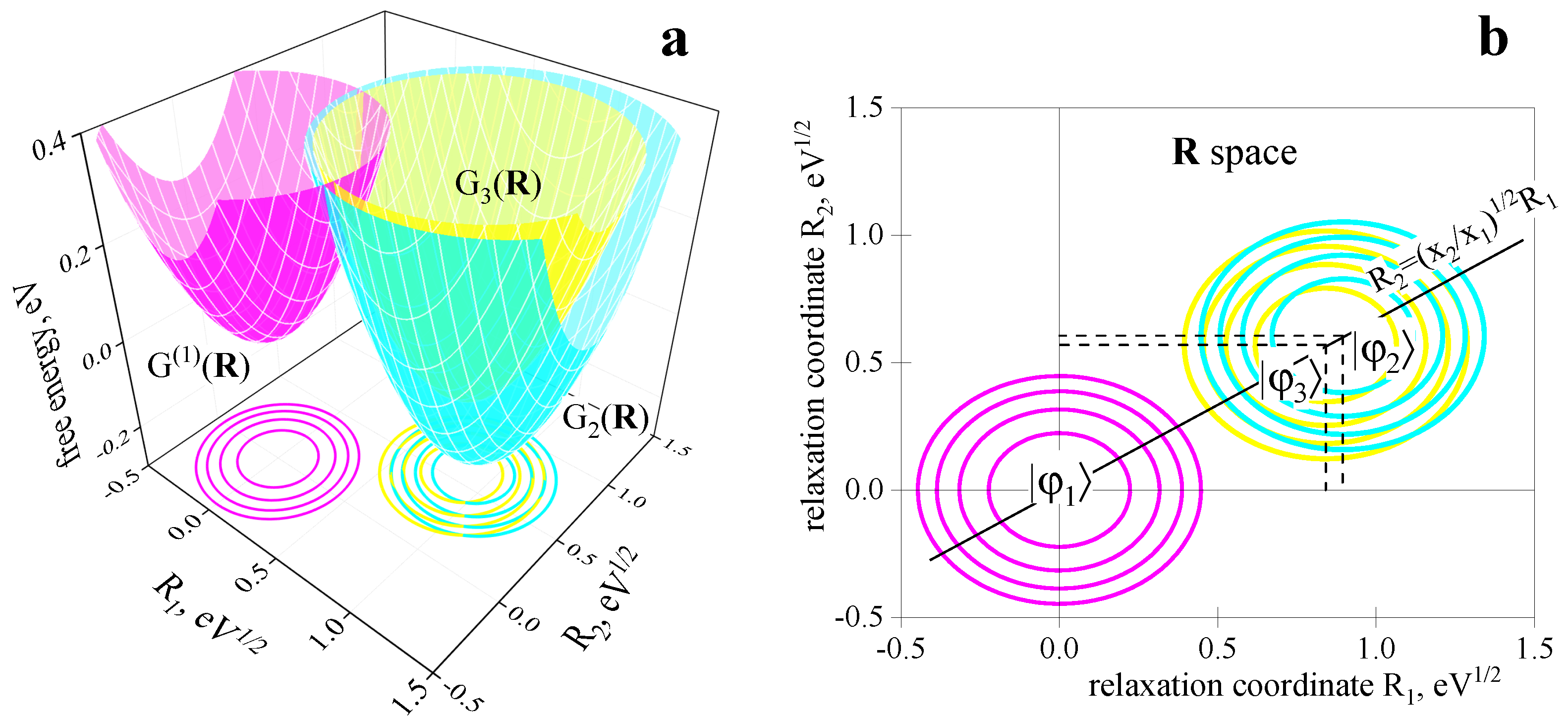
2.2. Three-Center Molecular System in a Two-Component Environment
3. Methods and Materials
Verification of the Theory
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Williams, R.J.P. Overview of Biological Electron Transfer. In Electron Transfer in Biology and the Solid State; American Chemical Society: Washington, DC, USA, 1990; Chapter 1; pp. 3–23. [Google Scholar] [CrossRef]
- Weber, S. Light-driven enzymatic catalysis of DNA repair: A review of recent biophysical studies on photolyase. Biochim. Biophys. Acta 2005, 1707, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Formosinho, S.; Barroso, M. (Eds.) Proton-Coupled Electron Transfer; Catalysis Series; The Royal Society of Chemistry: Cambridge, UK, 2012; pp. 1–157. [Google Scholar] [CrossRef]
- Strumpfer, J.; Sener, M.; Schulten, K. How Quantum Coherence Assists Photosynthetic Light-Harvesting. J. Phys. Chem. Lett. 2012, 3, 536–542. [Google Scholar] [CrossRef]
- Britikov, V.V.; Bocharov, E.V.; Britikova, E.V.; Dergousova, N.I.; Kulikova, O.G.; Solovieva, A.Y.; Shipkov, N.S.; Varfolomeeva, L.A.; Tikhonova, T.V.; Timofeev, V.I.; et al. Unusual Cytochrome c552 from Thioalkalivibrio paradoxus: Solution NMR Structure and Interaction with Thiocyanate Dehydrogenase. Int. J. Mol. Sci. 2022, 23, 9969. [Google Scholar] [CrossRef] [PubMed]
- Zara, V.; De Blasi, G.; Ferramosca, A. Assembly of the Multi-Subunit Cytochrome bc1 Complex in the Yeast Saccharomyces cerevisiae. Int. J. Mol. Sci. 2022, 23, 10537. [Google Scholar] [CrossRef] [PubMed]
- LeBard, D.N.; Martin, D.R.; Lin, S.; Woodbury, N.W.; Matyushov, D.V. Protein dynamics to optimize and control bacterial photosynthesis. Chem. Sci. 2013, 4, 4127–4136. [Google Scholar] [CrossRef]
- Sjulstok, E.; Olsen, J.; Solov’yov, I. Quantifying electron transfer reactions in biological systems: What interactions play the major role? Sci. Rep. 2015, 5, 18446. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Bagchi, B. From structure and dynamics to biomolecular functions: The ubiquitous role of solvent in biology. Curr. Opin. Struct. Biol. 2022, 77, 102462. [Google Scholar] [CrossRef] [PubMed]
- Migliore, A.; Polizzi, N.F.; Therien, M.J.; Beratan, D.N. Biochemistry and Theory of Proton-Coupled Electron Transfer. Chem. Rev. 2014, 114, 3381–3465. [Google Scholar] [CrossRef]
- Ponnu, A.; Sung, J.; Spears, K.G. Ultrafast Electron-Transfer and Solvent Adiabaticity Effects in Viologen Charge-Transfer Complexes. J. Phys. Chem. A 2006, 110, 12372–12384. [Google Scholar] [CrossRef]
- Kundu, M.; He, T.F.; Lu, Y.; Wang, L.; Zhong, D. Short-Range Electron Transfer in Reduced Flavodoxin: Ultrafast Nonequilibrium Dynamics Coupled with Protein Fluctuations. J. Phys. Chem. Lett. 2018, 9, 2782–2790. [Google Scholar] [CrossRef]
- Lu, Y.; Kundu, M.; Zhong, D. Effects of nonequilibrium fluctuations on ultrafast short-range electron transfer dynamics. Nat. Commun. 2020, 11, 2822. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y.; He, T.F.; Lu, Y.; Wang, L.; Ding, B.; Zhong, D. Ultrafast nonequilibrium dynamics of short-range protein electron transfer in flavodoxin. Phys. Chem. Chem. Phys. 2022, 24, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Dhole, K.; Jena, N.K.; Samanta, A.; Ghosh, S.K. Electron transfer reactions in condensed phase: Effect of reversibility. Phys. Rev. E 2012, 85, 026105. [Google Scholar] [CrossRef] [PubMed]
- Rosspeintner, A.; Lang, B.; Vauthey, E. Ultrafast photochemistry in liquids. Annu. Rev. Phys. Chem. 2013, 64, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Zusman, L.D. Outer-sphere electron transfer in polar solvents. Chem. Phys. 1980, 49, 295–304. [Google Scholar] [CrossRef]
- Petersson, J.; Hammarsrtröm, L. Ultrafast Electron Transfer Dynamics in a Series of Porphyrin/Viologen Complexes: Involvement of Electronically Excited Radical Pair Products. J. Phys. Chem. B 2015, 119, 7531–7540. [Google Scholar] [CrossRef]
- Szakacs, Z.; Tasior, M.; Gryko, D.T.; Vauthey, E. Change of Quadrupole Moment upon Excitation and Symmetry Breaking in Multibranched Donor-Acceptor Dyes. ChemPhysChem 2020, 21, 1718–1730. [Google Scholar] [CrossRef]
- Schrauben, J.N.; Dillman, K.L.; Beck, W.F.; McCusker, J.K. Vibrational coherence in the excited state dynamics of Cr(acac)3: Probing the reaction coordinate for ultrafast intersystem crossing. Chem. Sci. 2010, 1, 405–410. [Google Scholar] [CrossRef]
- Yoneda, Y.; Kudisch, B.; Rafiq, S.; Maiuri, M.; Scholes, G.; Miyasaka, H. Vibrational coherence in ultrafast electron transfer reaction observed by broadband transient absorption spectroscopy. EPJ Web Conf. 2019, 205, 09028. [Google Scholar] [CrossRef][Green Version]
- Kim, W.; Musser, A.J. Tracking ultrafast reactions in organic materials through vibrational coherence: Vibronic coupling mechanisms in singlet fission. Adv. Phys. X 2021, 6, 1918022. [Google Scholar] [CrossRef]
- Ennist, N.M.; Zhao, Z.; Stayrook, S.E.; Discher, B.M.; Dutton, P.L.; Moser, C.C. De novo protein design of photochemical reaction centers. Nat. Commun. 2022, 13, 4937. [Google Scholar] [CrossRef] [PubMed]
- Barber, J.; Tran, P.D. From natural to artificial photosynthesis. J. R. Soc. Interface 2013, 10, 20120984. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, R.E. Molecular Mechanisms of Photosynthesis, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2014; p. 312. [Google Scholar]
- Kuznetsov, A.; Ulstrup, J. Electron Transfer in Chemistry and Biology: An Introduction to the Theory; Wiley: Chichester, UK, 1999; p. 350. [Google Scholar]
- Likhtenshtein, G. Electron Transfer Theories. In Solar Energy Conversion; John Wiley & Sons: Hoboken, NJ, USA, 2012; Chapter 1; pp. 1–44. [Google Scholar] [CrossRef]
- Blumberger, J. Recent Advances in the Theory and Molecular Simulation of Biological Electron Transfer Reactions. Chem. Rev. 2015, 115, 11191–11238. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. On the theory of oxidation-reduction reactions involving electron transfer. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron Transfers in Chemistry and Biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Misra, R.; Bhattacharyya, S.P. Intramolecular Charge Transfer: Theory and Applications; Wiley: Weinheim, Germany, 2018; p. 256. [Google Scholar]
- Hatano, S.; Abe, J. A peroxide-bridged imidazole dimer formed from a photochromic naphthalene-bridged imidazole dimer. Phys. Chem. Chem. Phys. 2012, 14, 5855–5860. [Google Scholar] [CrossRef]
- Barzykin, A.V.; Frantsuzov, P.A.; Seki, K.; Tachiya, M. Solvent effects in nonadiabatic electron-transfer reactions: Theoretical aspects. In Advances in Chemical Physics; Wiley: Hoboken, NJ, USA, 2002; Volume 123, Chapter 9; pp. 511–616. [Google Scholar] [CrossRef]
- Feskov, S.V.; Mikhailova, V.A.; Ivanov, A.I. Non-equilibrium effects in ultrafast photoinduced charge transfer kinetics. J. Photochem. Photobiol. C 2016, 29, 48–72. [Google Scholar] [CrossRef]
- Fukuzumi, S. Electron Transfer: Mechanisms and Applications; Wiley: Weinheim, Germany, 2020; p. 230. [Google Scholar]
- Cho, M.; Silbey, R.J. Nonequilibrium photoinduced electron transfer. J. Chem. Phys. 1995, 103, 595–606. [Google Scholar] [CrossRef]
- Najbar, J.; Tachiya, M. Potential energy surfaces for electron transfer in a supramolecular triad system A1-D-A2 in a polar solvent. J. Phys. Chem. 1994, 98, 199–205. [Google Scholar] [CrossRef]
- Khohlova, S.S.; Mikhailova, V.A.; Ivanov, A.I. Three-centered model of ultrafast photoinduced charge transfer: Continuum dielectric approach. J. Chem. Phys. 2006, 124, 114507. [Google Scholar] [CrossRef]
- Newton, M.D. Bridge-Mediated Electron Transfer and Multiple Reaction Coordinates. Isr. J. Chem. 2004, 44, 83–88. [Google Scholar] [CrossRef]
- Motylewski, T.; Najbar, J.; Tachiya, M. Competitive electron transfers in model triad systems: Continuum model approach. Chem. Phys. 1996, 212, 193–206. [Google Scholar] [CrossRef]
- Tang, J.; Norris, J.R. On superexchange electron-transfer reactions involving three paraboloidal potential surfaces in a two-dimensional reaction coordinate. J. Chem. Phys. 1994, 101, 5615–5622. [Google Scholar] [CrossRef]
- Khokhlova, S.S.; Mikhailova, V.A.; Ivanov, A.I. The influence of changes in the dipole moment of reagents on the rate of photoinduced electron transfer. Russ. J. Phys. Chem. A 2008, 82, 1024–1030. [Google Scholar] [CrossRef]
- Feskov, S.V.; Kichigina, A.O.; Ivanov, A.I. Kinetics of nonequilibrium electron transfer in photoexcited ruthenium(II)-cobalt(III) complexes. J. Phys. Chem. A 2011, 115, 1462–1471. [Google Scholar] [CrossRef]
- Feskov, S.V.; Ivanov, A.I. Effect of geometrical parameters of dyad D-A and triad D-A1-A2 on the efficiency of ultrafast intramolecular charge separation from the second excited state. Chem. Phys. 2016, 478, 164–172. [Google Scholar] [CrossRef]
- Feskov, S.V.; Ivanov, A.I. Efficiency of Intramolecular Electron Transfer from the Second Excited State of the Donor in Molecular Triads D-A1-A2. Russ. J. Phys. Chem. 2016, 90, 144–151. [Google Scholar] [CrossRef]
- Feskov, S.V.; Ivanov, A.I. Solvent-assisted multistage nonequilibrium electron transfer in rigid supramolecular systems: Diabatic free energy surfaces and algorithms for numerical simulations. J. Chem. Phys. 2018, 148, 104107. [Google Scholar] [CrossRef]
- Wallin, S.; Monnereau, C.; Blart, E.; Gankou, J.R.; Odobel, F.; Hammarström, L. State-Selective Electron Transfer in an Unsymmetric Acceptor–Zn(II)porphyrin–Acceptor Triad: Toward a Controlled Directionality of Electron Transfer from the Porphyrin S2 and S1 States as a Basis for a Molecular Switch. J. Phys. Chem. A 2010, 114, 1709–1721. [Google Scholar] [CrossRef]
- Robotham, B.; Lastman, K.A.; Langford, S.J.; Ghiggino, K.P. Ultrafast Electron Transfer in a Porphyrin-Amino Naphthalene Diimide Dyad. J. Photochem. Photobiol. A 2013, 251, 167–174. [Google Scholar] [CrossRef]
- Zusman, L.D. The theory of electron transfer reactions in solvents with two characteristic relaxation times. Chem. Phys. 1988, 119, 51–61. [Google Scholar] [CrossRef]
- Jimenez, R.; Fleming, G.R.; Kumar, P.V.; Maroncelli, M. Femtosecond solvation dynamics of water. Nature 1994, 369, 471–473. [Google Scholar] [CrossRef]
- Maroncelli, M.; Kumar, V.P.; Papazyan, A. A simple interpretation of polar solvation dynamics. J. Phys. Chem. 1993, 97, 13–17. [Google Scholar] [CrossRef]
- Nazarov, A.E.; Ivanov, A.I.; Rosspeintner, A.; Angulo, G. Full relaxation dynamics recovery from ultrafast fluorescence experiments by means of the stochastic model: Does the solvent response dynamics depend on the fluorophore nature? J. Mol. Liq. 2022, 360, 119387. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Maigurov, A. Reaction flux deviation from the saddle point in fast electron transfer processes. Russ. J. Phys. Chem. 2003, 77, 243–248. [Google Scholar]
- Nazarov, A.E.; Ivanov, A.I. Excitation Frequency Dependence of Ultrafast Photoinduced Charge Transfer Dynamics. Int. J. Chem. Kinet. 2017, 49, 810–820. [Google Scholar] [CrossRef]
- Feskov, S.V.; Malykhin, R.E.; Ivanov, A.I. The Efficiency of Photoinduced Intramolecular Charge Separation from the Second Excited State: What Factors Can Control It? J. Phys. Chem. B 2020, 124, 10442–10455. [Google Scholar] [CrossRef]
- Siplivy, N.B.; Feskov, S.V.; Ivanov, A.I. Quantum yield and energy efficiency of photoinduced intramolecular charge separation. J. Chem. Phys. 2020, 153, 044301. [Google Scholar] [CrossRef]
- Fedunov, R.G.; Yermolenko, I.P.; Nazarov, A.E.; Ivanov, A.I.; Rosspeintner, A.; Angulo, G. Theory of fluorescence spectrum dynamics and its application to determining the relaxation characteristics of the solvent and intramolecular vibrations. J. Mol. Liq. 2020, 298, 112016. [Google Scholar] [CrossRef]
- Nazarov, A.E.; Ivanov, A.I. Principles of modeling the fluorescence spectral dynamics of dye molecules in solutions. Comput. Phys. Commun. 2022, 270, 108178. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feskov, S.V. Semiclassical Theory of Multistage Nonequilibrium Electron Transfer in Macromolecular Compounds in Polar Media with Several Relaxation Timescales. Int. J. Mol. Sci. 2022, 23, 15793. https://doi.org/10.3390/ijms232415793
Feskov SV. Semiclassical Theory of Multistage Nonequilibrium Electron Transfer in Macromolecular Compounds in Polar Media with Several Relaxation Timescales. International Journal of Molecular Sciences. 2022; 23(24):15793. https://doi.org/10.3390/ijms232415793
Chicago/Turabian StyleFeskov, Serguei V. 2022. "Semiclassical Theory of Multistage Nonequilibrium Electron Transfer in Macromolecular Compounds in Polar Media with Several Relaxation Timescales" International Journal of Molecular Sciences 23, no. 24: 15793. https://doi.org/10.3390/ijms232415793
APA StyleFeskov, S. V. (2022). Semiclassical Theory of Multistage Nonequilibrium Electron Transfer in Macromolecular Compounds in Polar Media with Several Relaxation Timescales. International Journal of Molecular Sciences, 23(24), 15793. https://doi.org/10.3390/ijms232415793