


Protein Interactome Profiling of Stable Molecular Complexes in Biomaterial Lysate

 , , , ,
, , , ,
Abstract
1. Introduction
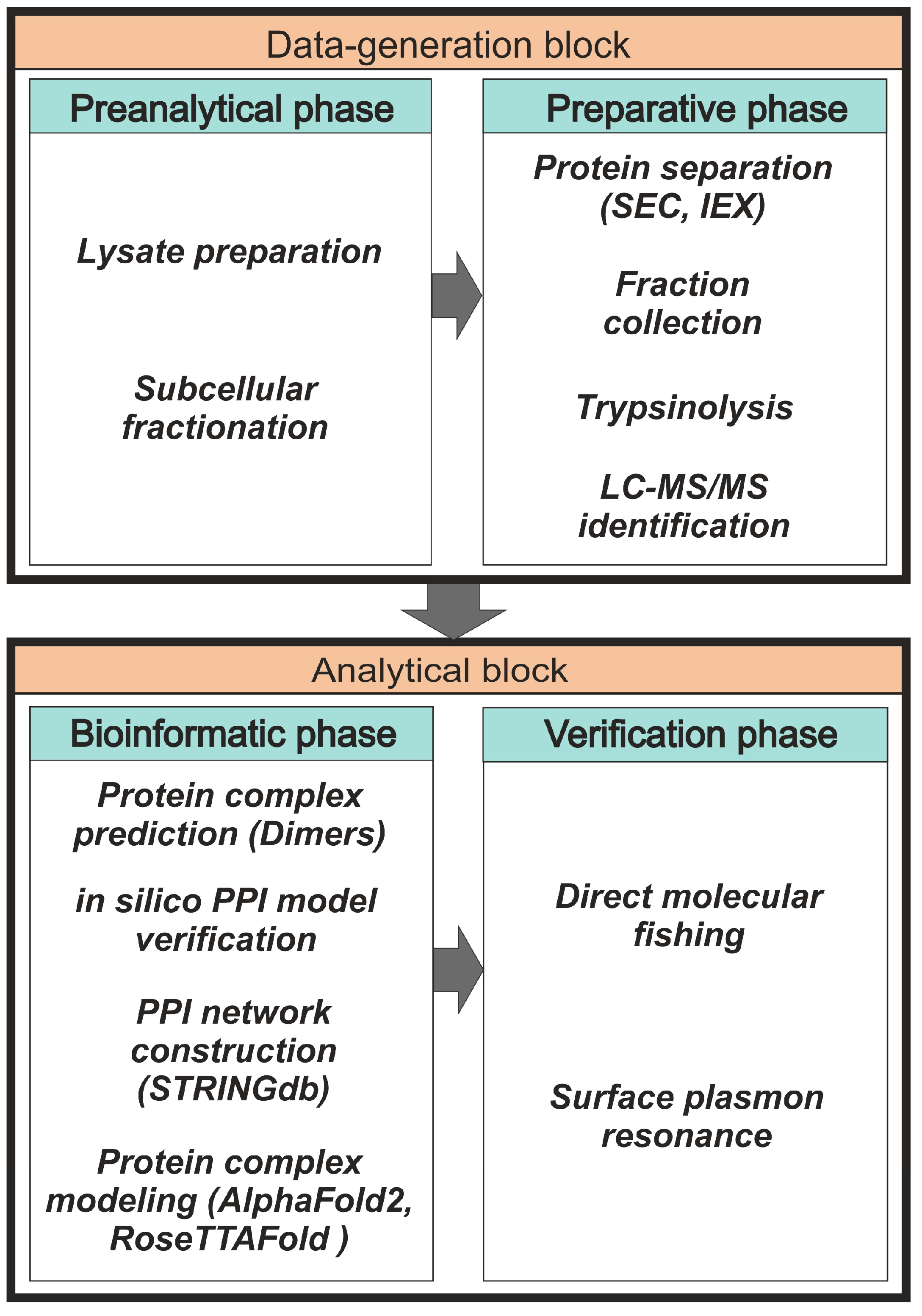
2. Interactome Profiling of Stable Protein Complexes
2.1. Preanalytical and Preparative Phases
2.2. Bioinformatic Phase
2.3. Experimental Verification of Possible Protein Complexes
3. Biomedical Aspects of Protein Interactome Profiling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stadmiller, S.S.; Pielak, G.J. Protein-Complex Stability in Cells and in Vitro under Crowded Conditions. Curr. Opin. Struct. Biol. 2021, 66, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Nooren, I.M.A.; Thornton, J.M. Diversity of Protein-Protein Interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef] [PubMed]
- Hemmerich, P.; Schmiedeberg, L.; Diekmann, S. Dynamic as Well as Stable Protein Interactions Contribute to Genome Function and Maintenance. Chromosome Res. 2011, 19, 131–151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garlick, J.M.; Mapp, A.K. Selective Modulation of Dynamic Protein Complexes. Cell Chem. Biol. 2020, 27, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Drew, K.; Wallingford, J.B.; Marcotte, E.M.H. MAP 2.0: Integration of over 15,000 Proteomic Experiments Builds a Global Compendium of Human Multiprotein Assemblies. Mol. Syst. Biol. 2021, 17, e10016. [Google Scholar] [CrossRef] [PubMed]
- Giurgiu, M.; Reinhard, J.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The Comprehensive Resource of Mammalian Protein Complexes-2019. Nucleic. Acids Res. 2019, 47, D559–D563. [Google Scholar] [CrossRef] [PubMed]
- Meldal, B.H.M.; Bye-A-Jee, H.; Gajdoš, L.; Hammerová, Z.; Horácková, A.; Melicher, F.; Perfetto, L.; Pokorný, D.; Lopez, M.R.; Türková, A.; et al. Complex Portal 2018: Extended Content and Enhanced Visualization Tools for Macromolecular Complexes. Nucleic Acids Res. 2019, 47, D550–D558. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Scott, N.E.; Prudova, A.; Kerr, C.H.; Stoynov, N.; Stacey, R.G.; Chan, Q.W.T.; Rattray, D.; Gsponer, J.; Foster, L.J. An Atlas of Protein-Protein Interactions across Mouse Tissues. Cell 2021, 184, 4073–4089.e17. [Google Scholar] [CrossRef]
- Kito, K.; Kawaguchi, N.; Okada, S.; Ito, T. Discrimination between Stable and Dynamic Components of Protein Complexes by Means of Quantitative Proteomics. Proteomics 2008, 8, 2366–2370. [Google Scholar] [CrossRef]
- Paiano, A.; Margiotta, A.; De Luca, M.; Bucci, C. Yeast Two-Hybrid Assay to Identify Interacting Proteins. Curr. Protoc. Protein. Sci. 2019, 95, e70. [Google Scholar] [CrossRef]
- Chavez, J.D.; Bruce, J.E. Chemical Cross-Linking with Mass Spectrometry: A Tool for Systems Structural Biology. Curr. Opin. Chem. Biol. 2019, 48, 8–18. [Google Scholar] [CrossRef]
- Yu, C.; Huang, L. Cross-Linking Mass Spectrometry: An Emerging Technology for Interactomics and Structural Biology. Anal. Chem. 2018, 90, 144–165. [Google Scholar] [CrossRef] [PubMed]
- Jasiecki, J.; Szczoczarz, A.; Cysewski, D.; Lewandowski, K.; Skowron, P.; Waleron, K.; Wasąg, B. Butyrylcholinesterase-Protein Interactions in Human Serum. Int. J. Mol. Sci. 2021, 22, 10662. [Google Scholar] [CrossRef]
- Kosuge, H.; Nakakido, M.; Nagatoishi, S.; Fukuda, T.; Bando, Y.; Ohnuma, S.-I.; Tsumoto, K. Proteomic Identification and Validation of Novel Interactions of the Putative Tumor Suppressor PRELP with Membrane Proteins Including IGFI-R and P75NTR. J. Biol. Chem. 2021, 296, 100278. [Google Scholar] [CrossRef] [PubMed]
- Göös, H.; Kinnunen, M.; Salokas, K.; Tan, Z.; Liu, X.; Yadav, L.; Zhang, Q.; Wei, G.-H.; Varjosalo, M. Human Transcription Factor Protein Interaction Networks. Nat. Commun. 2022, 13, 766. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Madero-Pérez, J.; Swaney, D.L.; Chang, T.S.; Moritz, M.; Konrad, C.; Ward, M.E.; Stevenson, E.; Hüttenhain, R.; Kauwe, G.; et al. Tau Interactome Maps Synaptic and Mitochondrial Processes Associated with Neurodegeneration. Cell 2022, 185, 712–728.e14. [Google Scholar] [CrossRef]
- Vincis Pereira Sanglard, L.; Colas des Francs-Small, C. High-Throughput BN-PAGE for Mitochondrial Respiratory Complexes. Methods Mol. Biol. 2022, 2363, 111–119. [Google Scholar] [CrossRef]
- Woytash, J.; Inigo, J.R.; Chandra, D. Assessing Oligomerization Status of Mitochondrial OXPHOS Complexes Via Blue Native Page. Methods Mol. Biol. 2022, 2413, 55–62. [Google Scholar] [CrossRef]
- Salas, D.; Stacey, R.G.; Akinlaja, M.; Foster, L.J. Next-Generation Interactomics: Considerations for the Use of Co-Elution to Measure Protein Interaction Networks. Mol. Cell Proteom. 2020, 19, 1–10. [Google Scholar] [CrossRef]
- Tiambeng, T.N.; Wu, Z.; Melby, J.A.; Ge, Y. Size Exclusion Chromatography Strategies and MASH Explorer for Large Proteoform Characterization. Methods Mol. Biol. 2022, 2500, 15–30. [Google Scholar] [CrossRef]
- Ventouri, I.K.; Malheiro, D.B.A.; Voeten, R.L.C.; Kok, S.; Honing, M.; Somsen, G.W.; Haselberg, R. Probing Protein Denaturation during Size-Exclusion Chromatography Using Native Mass Spectrometry. Anal. Chem. 2020, 92, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- McBride, Z.; Chen, D.; Reick, C.; Xie, J.; Szymanski, D.B. Global Analysis of Membrane-Associated Protein Oligomerization Using Protein Correlation Profiling. Mol. Cell Proteom. 2017, 16, 1972–1989. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Chang, H.-Y.; Rappsilber, J.; Ishihama, Y. Isolation of Acetylated and Unmodified Protein N-Terminal Peptides by Strong Cation Exchange Chromatographic Separation of TrypN-Digested Peptides. Mol. Cell Proteom. 2021, 20, 100003. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, M.P.; Cutillas, P.R. In-Depth Analysis of Protein Phosphorylation by Multidimensional Ion Exchange Chromatography and Mass Spectrometry. Methods Mol. Biol. 2010, 658, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Valeja, S.G.; Alpert, A.J.; Jin, S.; Ge, Y. Effective Protein Separation by Coupling Hydrophobic Interaction and Reverse Phase Chromatography for Top-down Proteomics. Anal. Chem. 2014, 86, 7899–7906. [Google Scholar] [CrossRef]
- Caudron-Herger, M.; Wassmer, E.; Nasa, I.; Schultz, A.-S.; Seiler, J.; Kettenbach, A.N.; Diederichs, S. Identification, Quantification and Bioinformatic Analysis of RNA-Dependent Proteins by RNase Treatment and Density Gradient Ultracentrifugation Using R-DeeP. Nat. Protoc. 2020, 15, 1338–1370. [Google Scholar] [CrossRef]
- Deng, L.; Handler, D.C.L.; Multari, D.H.; Haynes, P.A. Comparison of Protein and Peptide Fractionation Approaches in Protein Identification and Quantification from Saccharomyces Cerevisiae. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1162, 122453. [Google Scholar] [CrossRef]
- Some, D.; Amartely, H.; Tsadok, A.; Lebendiker, M. Characterization of Proteins by Size-Exclusion Chromatography Coupled to Multi-Angle Light Scattering (SEC-MALS). J. Vis. Exp. 2019, e59615. [Google Scholar] [CrossRef]
- Velours, C.; Zhou, J.; Zecchin, P.; He, N.; Salameh, M.; Golinelli-Cohen, M.-P.; Golinelli-Pimpaneau, B. Determination of the Absolute Molar Mass of [Fe-S]-Containing Proteins Using Size Exclusion Chromatography-Multi-Angle Light Scattering (SEC-MALS). Biomolecules 2022, 12, 270. [Google Scholar] [CrossRef]
- Fossati, A.; Frommelt, F.; Uliana, F.; Martelli, C.; Vizovisek, M.; Gillet, L.; Collins, B.; Gstaiger, M.; Aebersold, R. System-Wide Profiling of Protein Complexes Via Size Exclusion Chromatography-Mass Spectrometry (SEC-MS). Methods Mol. Biol. 2021, 2259, 269–294. [Google Scholar] [CrossRef]
- Tucholski, T.; Knott, S.J.; Chen, B.; Pistono, P.; Lin, Z.; Ge, Y. A Top-Down Proteomics Platform Coupling Serial Size Exclusion Chromatography and Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2019, 91, 3835–3844. [Google Scholar] [CrossRef] [PubMed]
- Páleníková, P.; Harbour, M.E.; Ding, S.; Fearnley, I.M.; Van Haute, L.; Rorbach, J.; Scavetta, R.; Minczuk, M.; Rebelo-Guiomar, P. Quantitative Density Gradient Analysis. by Mass Spectrometry (QDGMS) and Complexome Profiling Analysis (ComPrAn) R Package for the Study of Macromolecular Complexes. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148399. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, A.R.; Foster, L.J. Protein Correlation Profiling-SILAC to Study Protein-Protein Interactions. Methods Mol. Biol. 2014, 1188, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, S.; Ji, Y.; Guo, X.; Yang, F. Quantitative Proteomics Using SILAC: Principles, Applications, and Developments. Proteomics 2015, 15, 3175–3192. [Google Scholar] [CrossRef] [PubMed]
- Snider, J.; Wang, D.; Bogenhagen, D.F.; Haley, J.D. Pulse SILAC Approaches to the Measurement of Cellular Dynamics. Adv. Exp. Med. Biol. 2019, 1140, 575–583. [Google Scholar] [CrossRef]
- Florinskaya, A.V.; Ershov, P.V.; Mezentsev, Y.V.; Kaluzhskiy, L.A.; Yablokov, E.O.; Buneeva, O.A.; Zgoda, V.G.; Medvedev, A.E.; Ivanov, A.S. The Analysis of Participation of Individual Proteins in the Protein Interactome Formation. Biochem. Moscow. Suppl. Ser. B 2018, 12, 241–246. [Google Scholar] [CrossRef]
- Wittig, I.; Malacarne, P.F. Complexome Profiling: Assembly and Remodeling of Protein Complexes. Int. J. Mol. Sci. 2021, 22, 7809. [Google Scholar] [CrossRef]
- Zahiri, J.; Emamjomeh, A.; Bagheri, S.; Ivazeh, A.; Mahdevar, G.; Sepasi Tehrani, H.; Mirzaie, M.; Fakheri, B.A.; Mohammad-Noori, M. Protein Complex Prediction: A Survey. Genomics 2020, 112, 174–183. [Google Scholar] [CrossRef]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially Modified Protein Abundance Index (EmPAI) for Estimation of Absolute Protein Amount in Proteomics by the Number of Sequenced Peptides per Protein. Mol. Cell Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef]
- Yu, Y.; Kong, D. Protein Complexes Detection Based on Node Local Properties and Gene Expression in PPI Weighted Networks. BMC Bioinform. 2022, 23, 24. [Google Scholar] [CrossRef]
- Dilmaghani, S.; Brust, M.R.; Ribeiro, C.H.C.; Kieffer, E.; Danoy, G.; Bouvry, P. From Communities to Protein Complexes: A Local Community Detection Algorithm on PPI Networks. PLoS ONE 2022, 17, e0260484. [Google Scholar] [CrossRef] [PubMed]
- Kanitkar, T.R.; Sen, N.; Nair, S.; Soni, N.; Amritkar, K.; Ramtirtha, Y.; Madhusudhan, M.S. Methods for Molecular Modelling of Protein Complexes. Methods Mol. Biol. 2021, 2305, 53–80. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating Molecular Docking and Molecular Dynamics Simulations. Methods Mol. Biol. 2019, 2053, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for Protein Complex Modeling Reveals Accuracy Determinants. Protein. Sci. 2022, 31, e4379. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. Artificial Intelligence Powers Protein-Folding Predictions. Nature 2021, 599, 706–708. [Google Scholar] [CrossRef]
- Miteva, Y.V.; Budayeva, H.G.; Cristea, I.M. Proteomics-Based Methods for Discovery, Quantification, and Validation of Protein-Protein Interactions. Anal. Chem. 2013, 85, 749–768. [Google Scholar] [CrossRef]
- Dunham, W.H.; Mullin, M.; Gingras, A.-C. Affinity-Purification Coupled to Mass Spectrometry: Basic Principles and Strategies. Proteomics 2012, 12, 1576–1590. [Google Scholar] [CrossRef]
- Florinskaya, A.; Ershov, P.; Mezentsev, Y.; Kaluzhskiy, L.; Yablokov, E.; Medvedev, A.; Ivanov, A. SPR Biosensors in Direct Molecular Fishing: Implications for Protein Interactomics. Sensors 2018, 18, 1616. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Medvedev, A.; Ershov, P.; Molnar, A.; Mezentsev, Y.; Yablokov, E.; Kaluzhsky, L.; Gnedenko, O.; Buneeva, O.; Haidukevich, I.; et al. Protein Interactomics Based on Direct Molecular Fishing on Paramagnetic Particles: Practical Realization and Further SPR Validation. Proteomics 2014, 14, 2261–2274. [Google Scholar] [CrossRef]
- Ershov, P.V.; Mezentsev, Y.V.; Yablokov, E.O.; Kaluzhskiy, L.A.; Florinskaya, A.V.; Gnedenko, O.V.; Zgoda, V.G.; Vakhrushev, I.V.; Raeva, O.S.; Yarygin, K.N.; et al. Direct Molecular Fishing of Protein Partners for Proteins Encoded by Genes of Human Chromosome 18 in HepG2 Cell Lysate. Russ. J. Bioorg. Chem. 2018, 44, 759–768. [Google Scholar] [CrossRef]
- Rodriguez-Zavala, J.S.; Weiner, H. Structural Aspects of Aldehyde Dehydrogenase That Influence Dimer-Tetramer Formation. Biochemistry 2002, 41, 8229–8237. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the Human Interactome Defines Protein Communities and Disease Networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Revennaugh, B.; Rusnak, L.; Gonzalez-Pecchi, V.; Mo, X.; Johns, M.A.; Du, Y.; Cooper, L.A.D.; Moreno, C.S.; Khuri, F.R.; et al. The OncoPPi Portal: An Integrative Resource to Explore and Prioritize Protein-Protein Interactions for Cancer Target Discovery. Bioinformatics 2018, 34, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Eckhardt, M.; Krogan, N.J. Mass Spectrometry-Based Protein-Protein Interaction Networks for the Study of Human Diseases. Mol. Syst Biol. 2021, 17, e8792. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.E.; Hedrick, V.; Paschoal Sobreira, T.J.; Dykhuizen, E.C.; Aryal, U.K. Analysis of Human Nuclear Protein Complexes by Quantitative Mass Spectrometry Profiling. Proteomics 2018, 18, e1700427. [Google Scholar] [CrossRef] [PubMed]
- Park, C.G.; Choi, S.-H.; Lee, S.Y.; Eun, K.; Park, M.G.; Jang, J.; Jeong, H.J.; Kim, S.J.; Jeong, S.; Lee, K.; et al. Cytoplasmic LMO2-LDB1 Complex Activates STAT3 Signaling through Interaction with Gp130-JAK in Glioma Stem Cells. Cells 2022, 11, 2031. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Duncan, J.L.; Lee, C.; Kuchel, P.W.; Matthews, J.M. Assembly of the Oncogenic DNA-Binding Complex LMO2-Ldb1-TAL1-E12. Proteins 2008, 70, 1461–1474. [Google Scholar] [CrossRef]
- Nakata, K.; Ohuchida, K.; Nagai, E.; Hayashi, A.; Miyasaka, Y.; Kayashima, T.; Yu, J.; Aishima, S.; Oda, Y.; Mizumoto, K.; et al. LMO2 Is a Novel Predictive Marker for a Better Prognosis in Pancreatic Cancer. Neoplasia 2009, 11, 712–719. [Google Scholar] [CrossRef]
- García, S.A.; Swiersy, A.; Radhakrishnan, P.; Branchi, V.; Kanth Nanduri, L.; Győrffy, B.; Betzler, A.M.; Bork, U.; Kahlert, C.; Reißfelder, C.; et al. LDB1 Overexpression Is a Negative Prognostic Factor in Colorectal Cancer. Oncotarget 2016, 7, 84258–84270. [Google Scholar] [CrossRef]
- Tanaka, T.; Sewell, H.; Waters, S.; Phillips, S.E.V.; Rabbitts, T.H. Single Domain Intracellular Antibodies from Diverse Libraries: Emphasizing Dual Functions of LMO2 Protein Interactions Using a Single VH Domain. J. Biol. Chem. 2011, 286, 3707–3716. [Google Scholar] [CrossRef]
- Grossman, S.R. P300/CBP/P53 Interaction and Regulation of the P53 Response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive Characterization of Protein-Protein Interactions Perturbed by Disease Mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Kaur, P.; Lang, J.E. EP300 Knockdown Reduces Cancer Stem Cell Phenotype, Tumor Growth and Metastasis in Triple Negative Breast Cancer. BMC Cancer 2020, 20, 1076. [Google Scholar] [CrossRef]
- Paur, J.; Valler, M.; Sienel, R.; Taxauer, K.; Holzmann, K.; Marian, B.; Unterberger, A.; Mohr, T.; Berger, W.; Gvozdenovich, A.; et al. Interaction of FGF9 with FGFR3-IIIb/IIIc, a Putative Driver of Growth and Aggressive Behaviour of Hepatocellular Carcinoma. Liver Int. 2020, 40, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Knowles, M.A. Altered Splicing of FGFR1 Is Associated with High Tumor Grade and Stage and Leads to Increased Sensitivity to FGF1 in Bladder Cancer. Am. J. Pathol. 2010, 177, 2379–2386. [Google Scholar] [CrossRef]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a Promising Druggable Target in Cancer: Molecular Biology and New Drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef]
- Melino, G. P63 Is a Suppressor of Tumorigenesis and Metastasis Interacting with Mutant P53. Cell Death Differ. 2011, 18, 1487–1499. [Google Scholar] [CrossRef]
- Bankhead, A.; McMaster, T.; Wang, Y.; Boonstra, P.S.; Palmbos, P.L. TP63 Isoform Expression Is Linked with Distinct Clinical Outcomes in Cancer. EBioMedicine 2020, 51, 102561. [Google Scholar] [CrossRef]
- Wang, Z.; Strasser, A.; Kelly, G.L. Should Mutant TP53 Be Targeted for Cancer Therapy? Cell Death Differ. 2022, 29, 911–920. [Google Scholar] [CrossRef]
- Ryan, D.P.; Matthews, J.M. Protein-Protein Interactions in Human Disease. Curr. Opin. Struct. Biol. 2005, 15, 441–446. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; Tortelli, R.; Johnson, E.B.; Wijeratne, P.A.; Arridge, M.; De Vita, E.; Ghazaleh, N.; Houghton, R.; Furby, H.; et al. Mutant Huntingtin and Neurofilament Light Have Distinct Longitudinal Dynamics in Huntington’s Disease. Sci Transl Med. 2020, 12, eabc2888. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Fujimaki, M.; Rogers, P.; Rubinsztein, D.C. Huntingtin-Lowering Strategies for Huntington’s Disease. Expert. Opin. Investig. Drugs 2020, 29, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Samant, R.S.; Batista, S.; Larance, M.; Ozer, B.; Milton, C.I.; Bludau, I.; Biggins, L.; Andrews, S.; Hervieu, A.; Johnston, H.E.; et al. Native Size Exclusion Chromatography-Based Mass Spectrometry (SEC-MS) Identifies Novel Components of the Heat Shock Protein 90-Dependent Proteome. BioRxiv 2022, 1–28. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, H.; Bi, X.; Hou, G.; Liu, A.; Zhao, Y.; Wang, G.; Cao, X. Src Acts as the Target of Matrine to Inhibit the Proliferation of Cancer Cells by Regulating Phosphorylation Signaling Pathways. Cell Death Dis. 2021, 12, 931. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qu, M.; Pan, M.; Zheng, X.; Sheng, Y.; Ji, Y.; You, C.; Dai, X. Chemical Proteomic Profiling of UTP-Binding Proteins in Human Cells. Anal. Chim. Acta 2021, 1168, 338607. [Google Scholar] [CrossRef]
- Lenz, T.; Stühler, K. Small Molecule Arranged Thermal Proximity Coaggregation (SmarTPCA)-A Novel Approach to Characterize Protein-Protein Interactions in Living Cells by Similar Isothermal Dose-Responses. Int. J. Mol. Sci. 2022, 23, 5605. [Google Scholar] [CrossRef]
- Wang, S.; Tian, Y.; Wang, M.; Wang, M.; Sun, G.-B.; Sun, X.-B. Advanced Activity-Based Protein Profiling Application Strategies for Drug Development. Front. Pharmacol. 2018, 9, 353. [Google Scholar] [CrossRef]
- Ershov, P.V.; Mezentsev, Y.V.; Yablokov, E.O.; Kaluzhskiy, L.A.; Florinskaya, A.V.; Svirid, A.V.; Gilep, A.A.; Usanov, S.A.; Medvedev, A.E.; Ivanov, A.S. Specificity of Isatin Interaction with Cytochromes P450. Biochem. Moscow. Suppl. Ser. B 2018, 12, 130–135. [Google Scholar] [CrossRef]
- Bludau, I.; Aebersold, R. Proteomic and Interactomic Insights into the Molecular Basis of Cell Functional Diversity. Nat. Rev. Mol. Cell Biol. 2020, 21, 327–340. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MW (kDa) of Identified Proteins | Mean MW OF Fraction (kDa) | |||||
|---|---|---|---|---|---|---|
| 400 | 290 | 130 | 90 | 60 | 48 | |
| <11 | 5 | 12 | 1 | 4 | 4 | 2 |
| 11–21 | 33 | 42 | 37 | 18 | 13 | 25 |
| 21–31 | 86 | 75 | 53 | 45 | 34 | 80 |
| 31–41 | 117 | 110 | 55 | 56 | 68 | 84 |
| 41–51 | 93 | 106 | 56 | 54 | 70 | 78 |
| 51–61 | 77 | 88 | 43 | 53 | 59 | 65 |
| 61–71 | 29 | 32 | 25 | 28 | 28 | 29 |
| 71–81 | 22 | 24 | 14 | 14 | 22 | 20 |
| 81–91 | 12 | 10 | 4 | 3 | 7 | 3 |
| 91–101 | 10 | 9 | 10 | 7 | 4 | 5 |
| 101–111 | 11 | 11 | 6 | 9 | 5 | 5 |
| 111–121 | 10 | 3 | 4 | 3 | 5 | 0 |
| 121–131 | 7 | 4 | 5 | 7 | 3 | 4 |
| 131–141 | 4 | 4 | 0 | 0 | 0 | 0 |
| 141–151 | 6 | 5 | 1 | 1 | 1 | 0 |
| 151–176 | 4 | 2 | 1 | 1 | 1 | 1 |
| 176–201 | 4 | 5 | 3 | 1 | 0 | 0 |
| 201–226 | 0 | 0 | 1 | 0 | 1 | 1 |
| 226–251 | 0 | 0 | 0 | 2 | 2 | 1 |
| 251–301 | 8 | 4 | 2 | 1 | 1 | 1 |
| 301–351 | 0 | 0 | 0 | 0 | 0 | 0 |
| 351–401 | 0 | 0 | 0 | 0 | 1 | 0 |
| 401–451 | 0 | 0 | 0 | 0 | 0 | 0 |
| 451–501 | 0 | 0 | 0 | 0 | 0 | 0 |
| 501–601 | 1 | 1 | 0 | 0 | 1 | 1 |
| 501–701 | 1 | 1 | 1 | 1 | 0 | 1 |
| Total | 540 | 548 | 322 | 307 | 330 | 406 |
| Type | Description | Examples * | Protein Name |
|---|---|---|---|
| I | Monomeric form only ** |  | Apoptosis-inducing factor 1 (mitochondrial), O95831, 66.9 kDa, monomer |
| II | Homodimers and heterodimers |  | 3-hydroxyisobutyrate dehydrogenase (mitochondrial), P31937, 35.3 kDa, homodimer |
| III | Homooligomers and heterooligomers *** |  | Very long chain specific acyl-CoA dehydrogenase (mitochondrial), P49748, 70.4 kDa, homodimer |
| IV | I and II |  | Aspartate aminotransferase (mitochondrial), P00505, 47.5 kDa, homodimer |
| V | I and III |  | Apolipoprotein A–I, P02647, 30.8 kDa, homodimer |
| VI | I, II, and III |  | Peroxisomal multifunctional enzyme type 2, P51659, 79.7 kDa, homodimer |
| Protein Complex | Disease | Prognostic Value | Drug Target |
|---|---|---|---|
| LMO2/LDB1 *, LMO2/LDB1/TAL1/E12 | Cancer [56,57] | [58,59] | [60] |
| TP53/EP300 | Cancer [61] | [62] | [63] |
| FGFx/FGFRx | Cancer [64] | [65] | [66] |
| TP63/mutTP53 | Cancer [67] | [68] | [69] |
| HTT/HAP-1 | Neurological disorders [70] | [71] | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezentsev, Y.; Ershov, P.; Yablokov, E.; Kaluzhskiy, L.; Kupriyanov, K.; Gnedenko, O.; Ivanov, A. Protein Interactome Profiling of Stable Molecular Complexes in Biomaterial Lysate. Int. J. Mol. Sci. 2022, 23, 15697. https://doi.org/10.3390/ijms232415697
Mezentsev Y, Ershov P, Yablokov E, Kaluzhskiy L, Kupriyanov K, Gnedenko O, Ivanov A. Protein Interactome Profiling of Stable Molecular Complexes in Biomaterial Lysate. International Journal of Molecular Sciences. 2022; 23(24):15697. https://doi.org/10.3390/ijms232415697
Chicago/Turabian StyleMezentsev, Yuri, Pavel Ershov, Evgeniy Yablokov, Leonid Kaluzhskiy, Konstantin Kupriyanov, Oksana Gnedenko, and Alexis Ivanov. 2022. "Protein Interactome Profiling of Stable Molecular Complexes in Biomaterial Lysate" International Journal of Molecular Sciences 23, no. 24: 15697. https://doi.org/10.3390/ijms232415697
APA StyleMezentsev, Y., Ershov, P., Yablokov, E., Kaluzhskiy, L., Kupriyanov, K., Gnedenko, O., & Ivanov, A. (2022). Protein Interactome Profiling of Stable Molecular Complexes in Biomaterial Lysate. International Journal of Molecular Sciences, 23(24), 15697. https://doi.org/10.3390/ijms232415697