

3-Bromopyruvate Suppresses the Malignant Phenotype of Vemurafenib-Resistant Melanoma Cells

 , , , , and
, , , , and
Abstract
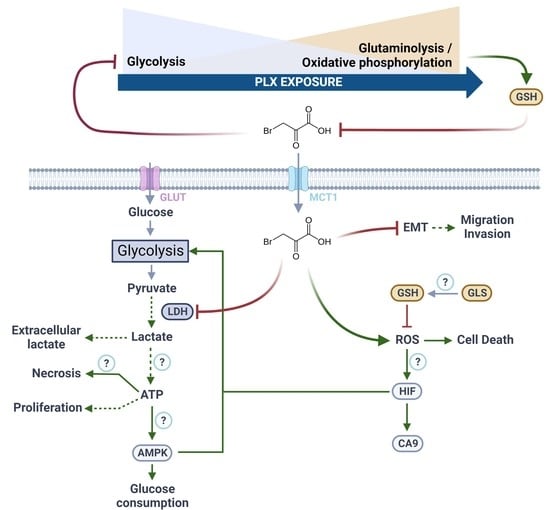
{kind=link}
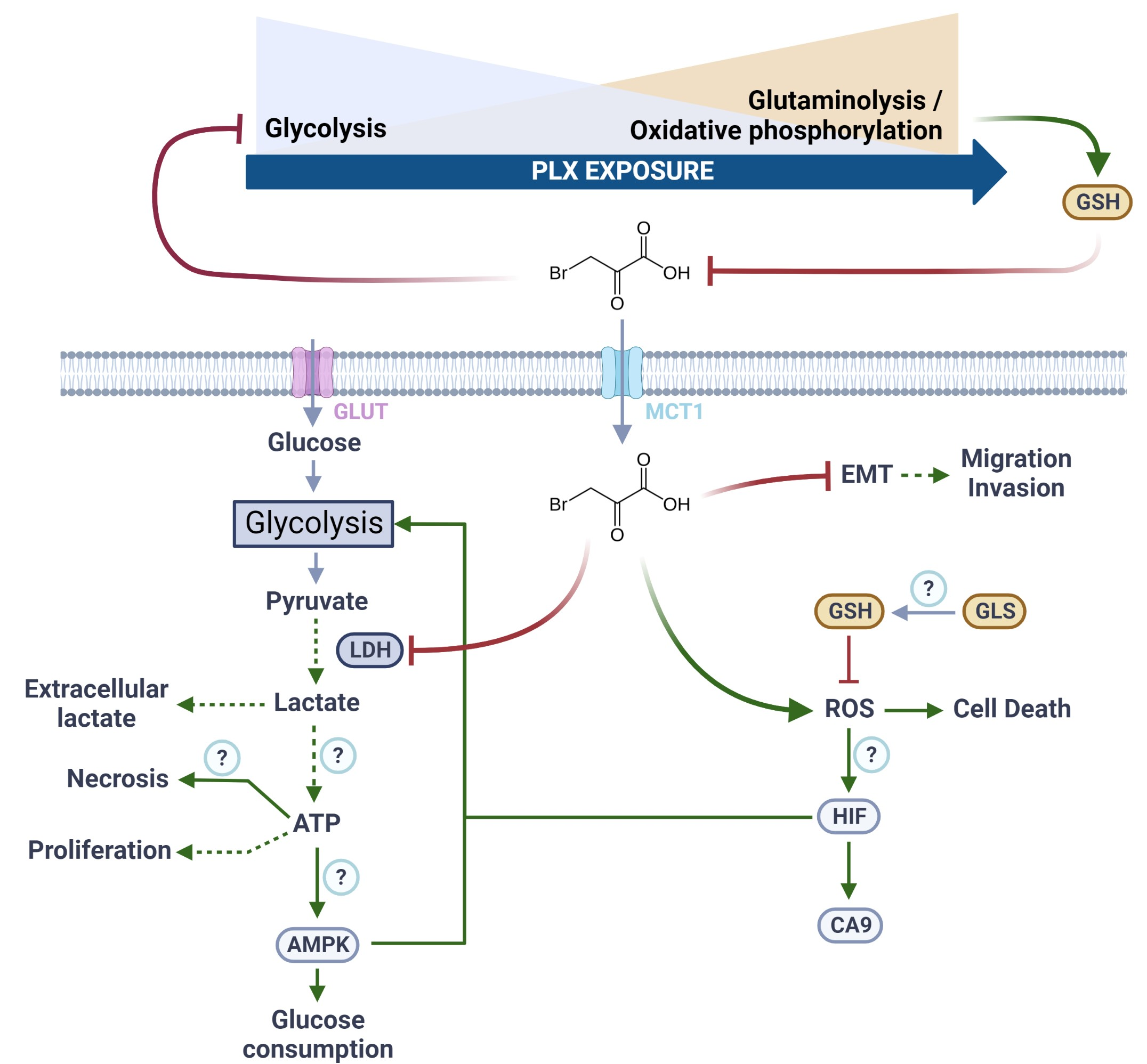
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
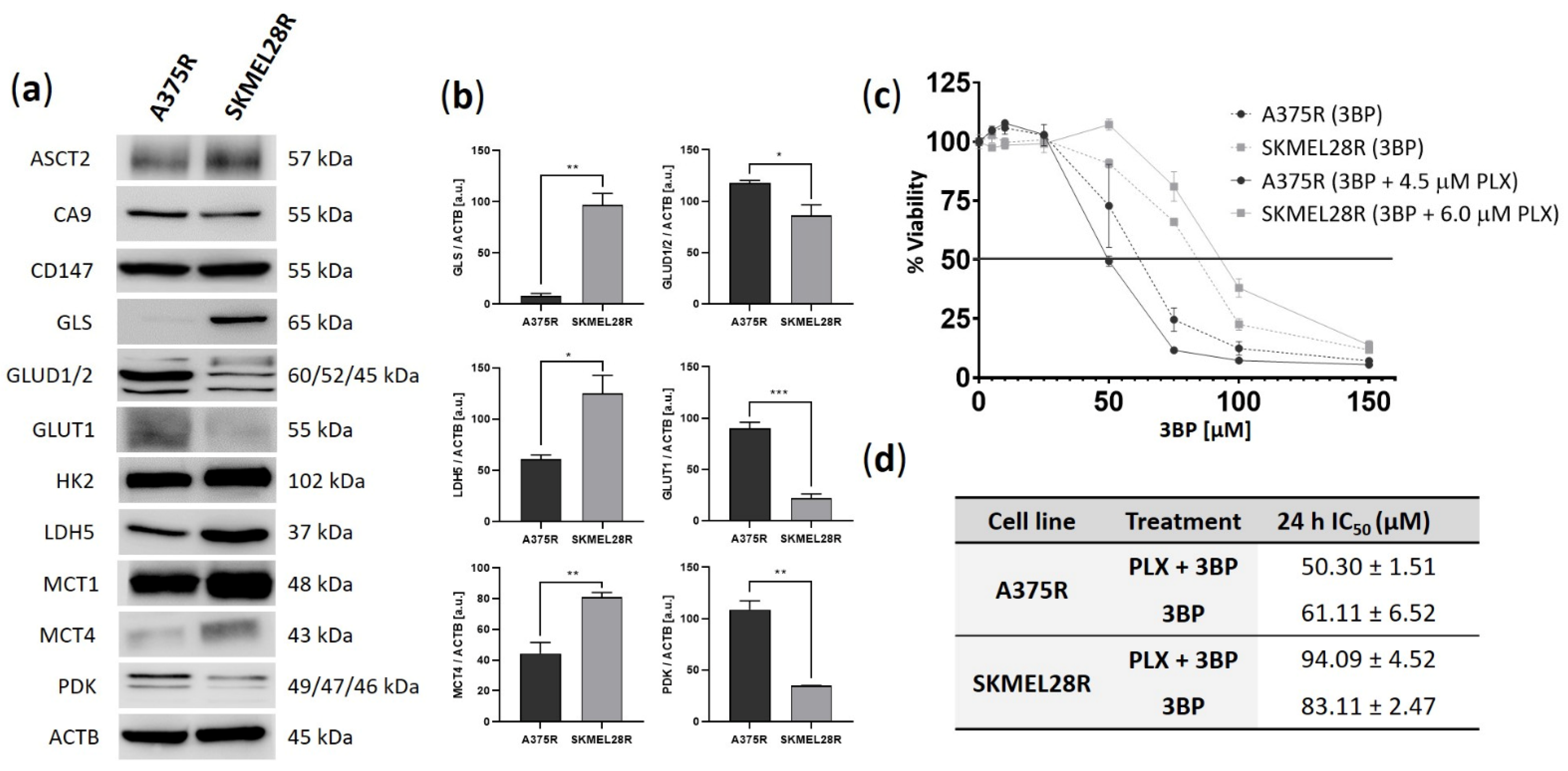
2.1. A375R and SKMEL28R Cells Present Different Metabolic Profiles with Different Sensitivities to 3-Bromopyruvate
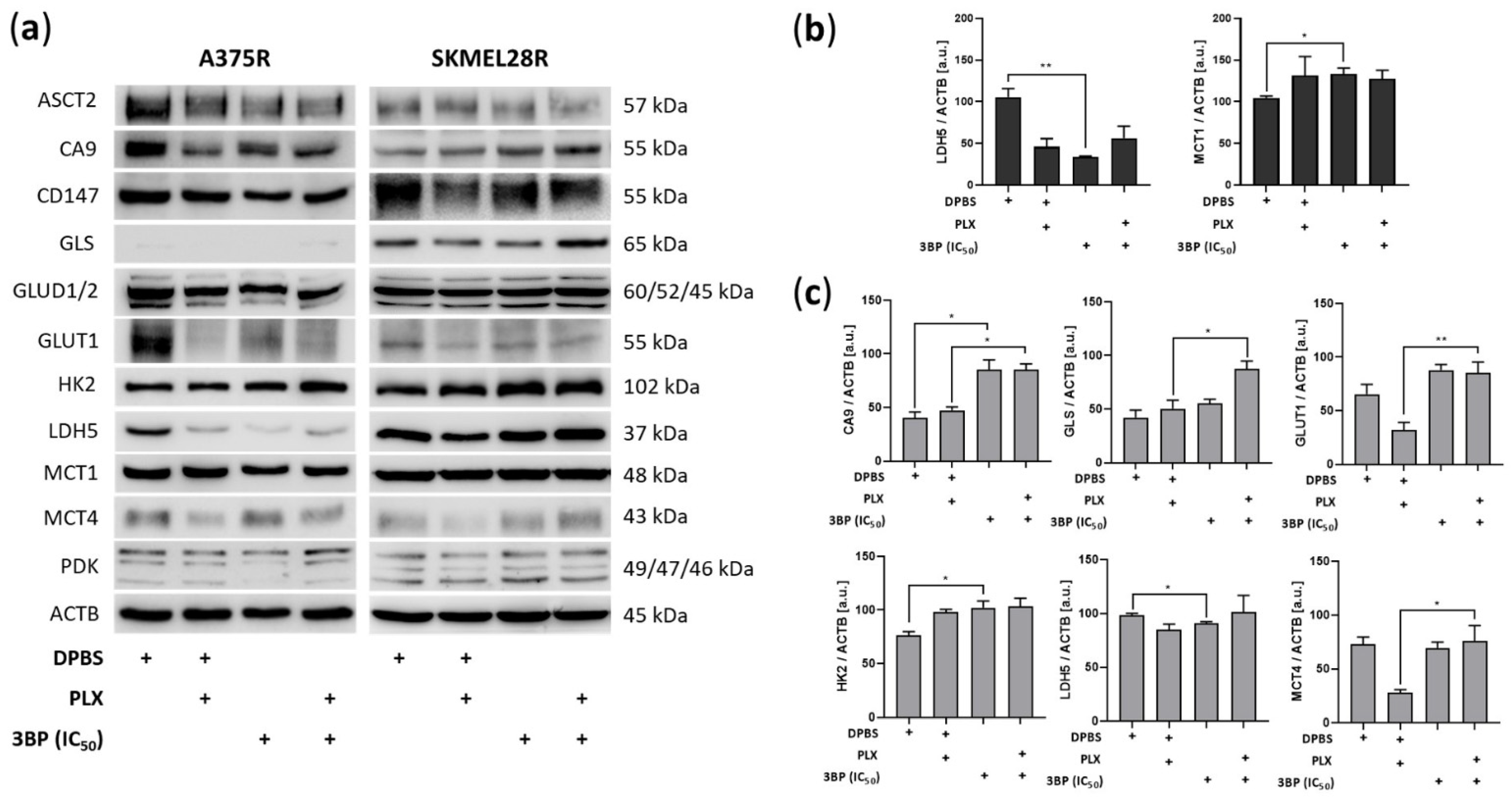
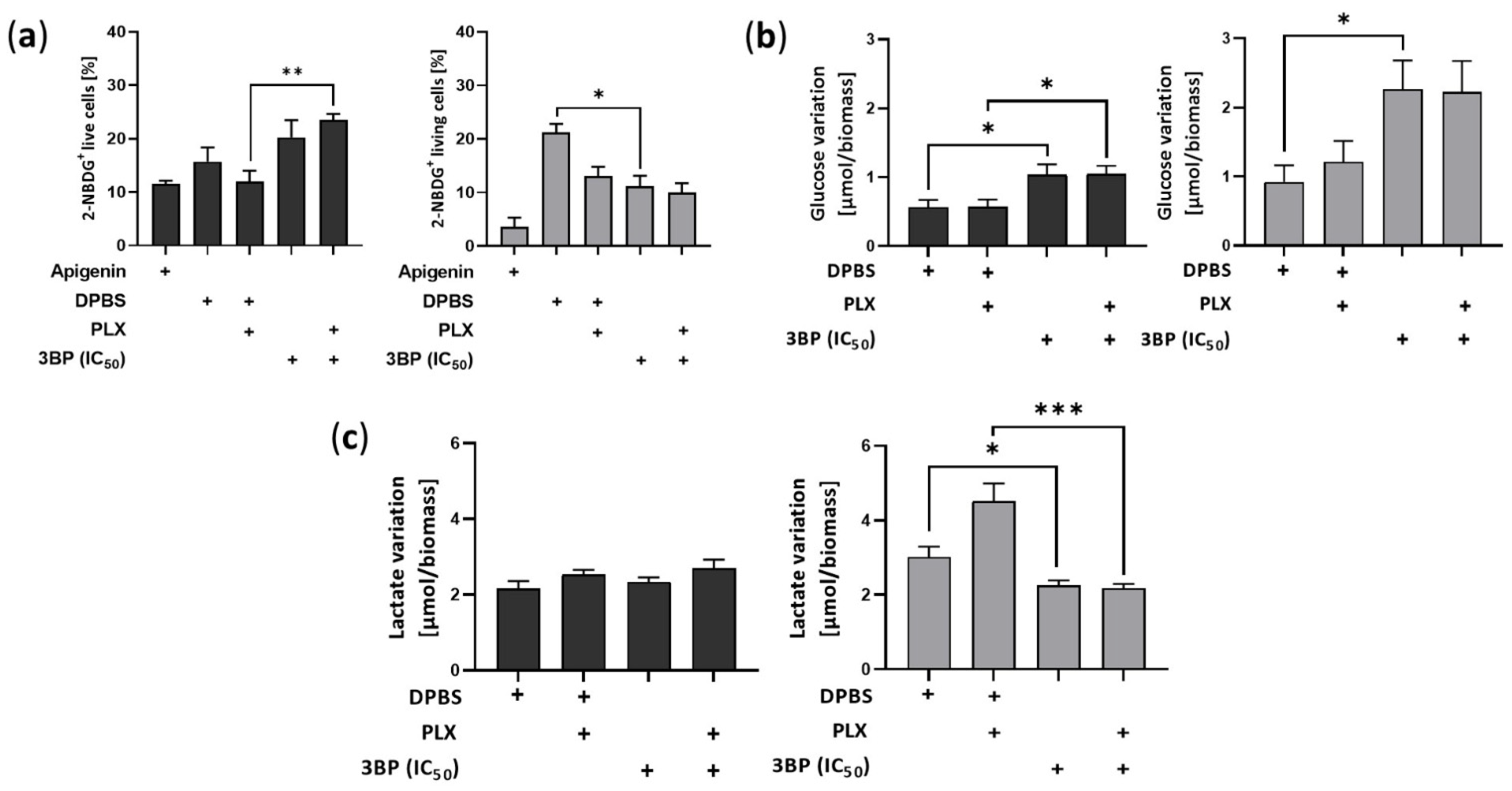
2.2. 3-Bromopyruvate Modulates the Metabolic Profile of Vemurafenib-Resistant Melanoma Cells
2.3. 3-Bromopyruvate Decreases Cell Division and Induces Cell Death in Vemurafenib-Resistant Melanoma Cells
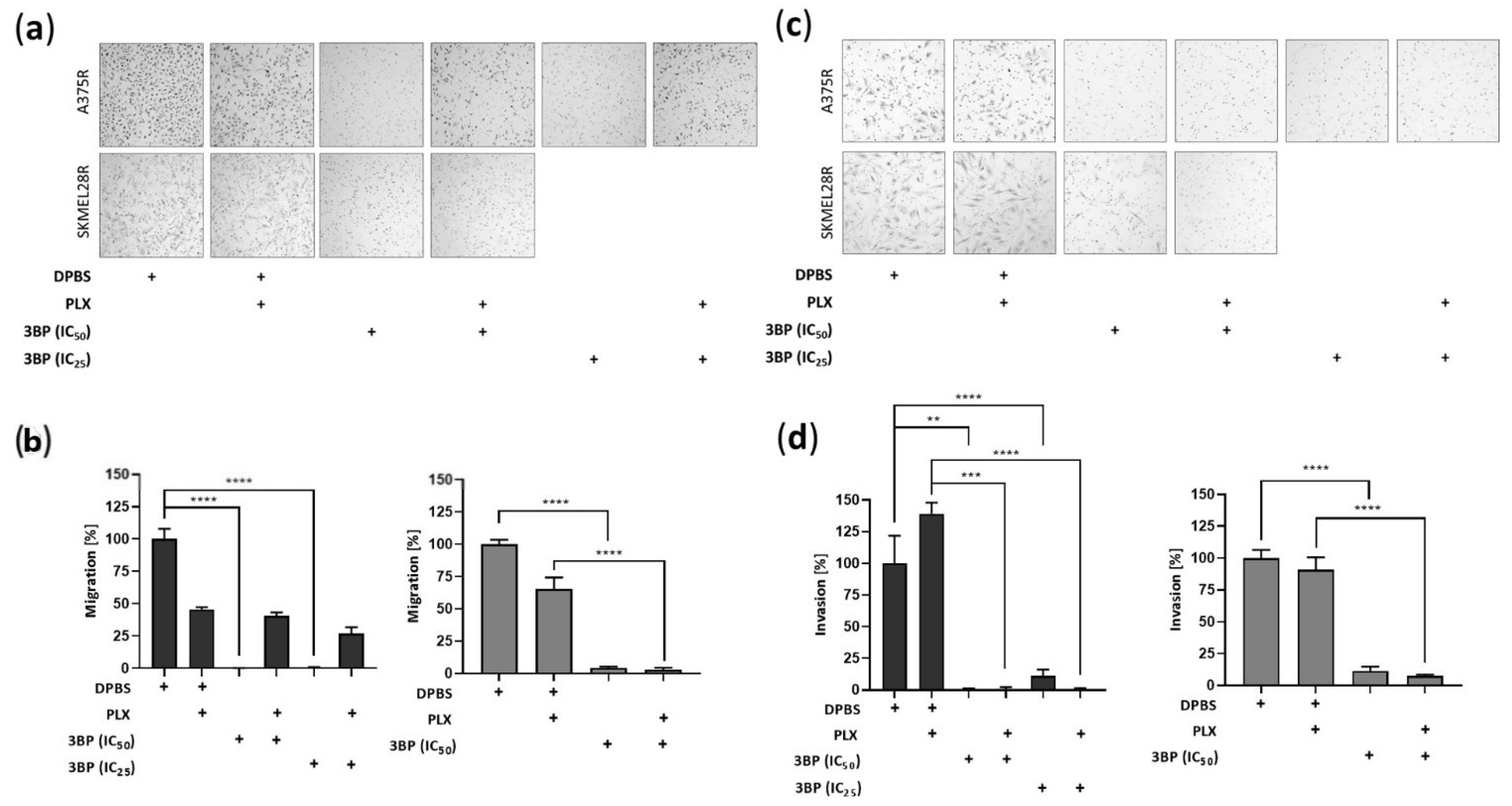
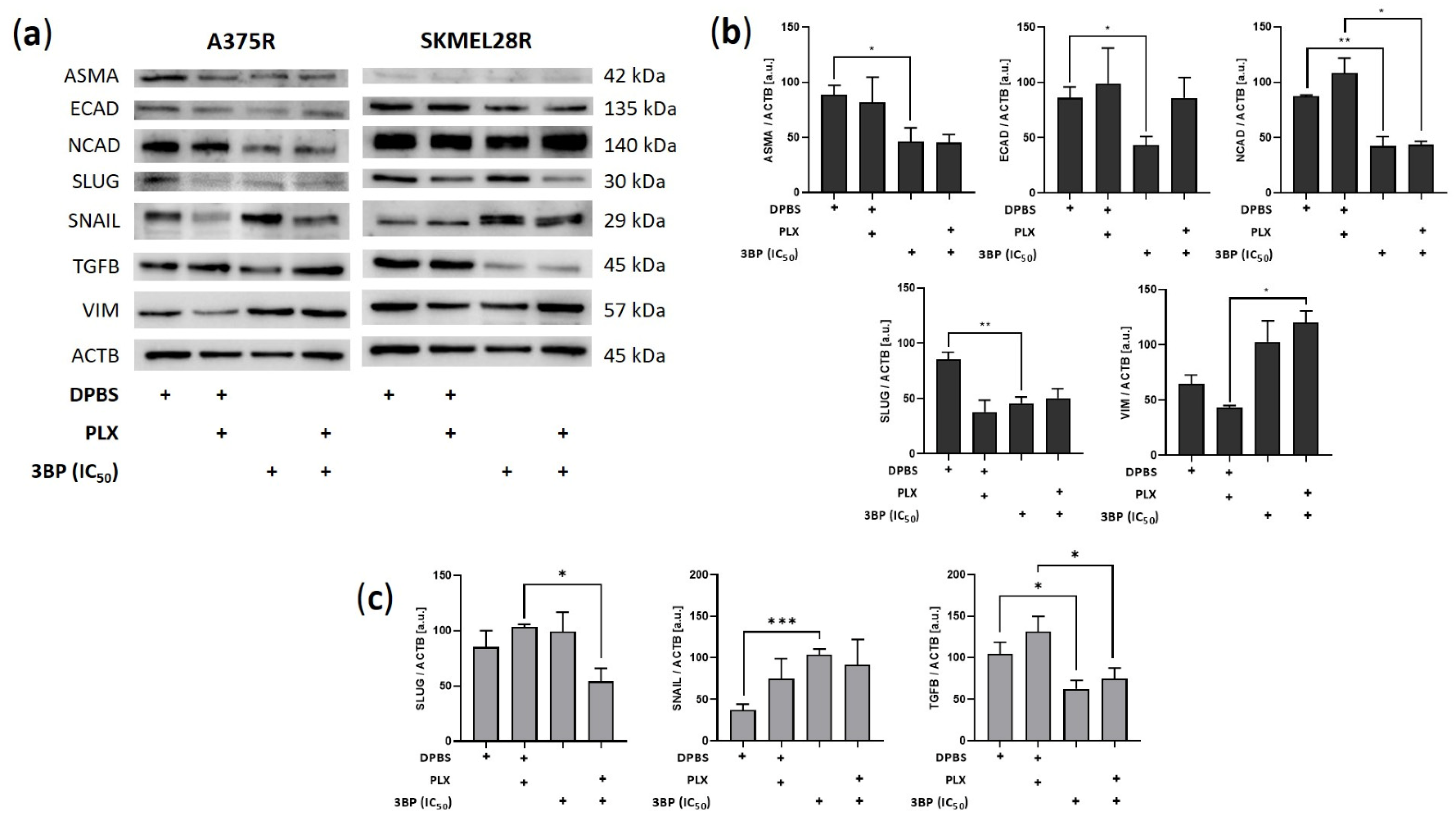
2.4. 3-Bromopyruvate Impairs the Migration and Invasion of Vemurafenib-Resistant Melanoma Cells and Inhibits the Expression of Epithelial-Mesenchymal Transition Markers
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Growth Conditions
4.2. Drugs
4.3. Cell Viability and IC50 and IC25 Determination
4.4. Drug Combined Effect
4.5. Cell Proliferation Assay
4.6. Colony Formation Assay
4.7. Cell Death Assay
4.8. Reactive Oxygen Species Quantification
4.9. Cell Migration and Invasion Assays
4.10. Western Blot
4.11. Extracellular Glucose and Lactate Quantification
4.12. Glucose Uptake Quantification
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erdmann, F.; Lortet-Tieulent, J.; Schüz, J.; Zeeb, H.; Greinert, R.; Breitbart, E.W.; Bray, F. International Trends in the Incidence of Malignant Melanoma 1953-2008-Are Recent Generations at Higher or Lower Risk? Int. J. Cancer 2013, 132, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma Treatment in Review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U. ESMO Guidelines Committee Electronic address: Clinicalguidelines@esmo.org Cutaneous Melanoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef]
- Guan, J.; Gupta, R.; Filipp, F.V. Cancer Systems Biology of TCGA SKCM: Efficient Detection of Genomic Drivers in Melanoma. Sci. Rep. 2015, 5, 7857. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF Kinases for Cancer Therapy: BRAF-Mutated Melanoma and Beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Amaria, R.N.; Rozeman, E.A.; Huang, A.C.; Tetzlaff, M.T.; van de Wiel, B.A.; Lo, S.; Tarhini, A.A.; Burton, E.M.; Pennington, T.E.; et al. Pathological Response and Survival with Neoadjuvant Therapy in Melanoma: A Pooled Analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat. Med. 2021, 27, 301–309. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK Reactivation in Early Resistance to Dabrafenib/Trametinib Combination Therapy of BRAF-Mutant Metastatic Melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting Cellular Metabolism to Improve Cancer Therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef]
- Pinheiro, C.; Miranda-Gonçalves, V.; Longatto-Filho, A.; Vicente, A.L.S.A.; Berardinelli, G.N.; Scapulatempo-Neto, C.; Costa, R.F.A.; Viana, C.R.; Reis, R.M.; Baltazar, F.; et al. The Metabolic Microenvironment of Melanomas: Prognostic Value of MCT1 and MCT4. Cell Cycle 2016, 15, 1462–1470. [Google Scholar] [CrossRef]
- Hardeman, K.N.; Peng, C.; Paudel, B.B.; Meyer, C.T.; Luong, T.; Tyson, D.R.; Young, J.D.; Quaranta, V.; Fessel, J.P. Dependence on Glycolysis Sensitizes BRAF-Mutated Melanomas for Increased Response to Targeted BRAF Inhibition. Sci. Rep. 2017, 7, srep42604. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Cancer Cell Oncogenic BRAF Regulates Oxidative Metabolism via PGC1a and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Davies, J.E.; Tran, T.Q.; Reid, M.A.; Rosales, K.R.; Lowman, X.H.; Pan, M.; Moriceau, G.; Yang, Y.; Wu, J.; Lo, R.S.; et al. Vemurafenib Resistance Reprograms Melanoma Cells towards Glutamine Dependence. J. Transl. Med. 2015, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Chaneton, B.; Smith, M.; van den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF Inhibitors Induces Glutamine Dependency in Melanoma Cells. Mol. Oncol. 2016, 10, 73–84. [Google Scholar] [CrossRef]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporters in Cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front. Oncol. 2020, 10, 231. [Google Scholar] [CrossRef]
- Baltazar, F.; Pinheiro, C.; Morais-Santos, F.; Azevedo-Silva, J.; Queirós, O.; Preto, A.; Casal, M. Monocarboxylate Transporters as Targets and Mediators in Cancer Therapy Response. Histol. Histopathol. 2014, 29, 1511–1524. [Google Scholar]
- Zhu, J.; Thompson, C.B. Metabolic Regulation of Cell Growth and Proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Muñoz-Torrero, D.; Rapposelli, S.; Gütschow, M.; Matos, M.J.; Emília De Sousa, M.; Saso, L.; Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; et al. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Possemato, R.; Yilmaz, O.H.; Koch, C.E.; Chen, W.W.; Hutchins, A.W.; Gultekin, Y.; Peterson, T.R.; Carette, J.E.; et al. MCT1-Mediated Transport of a Toxic Molecule Is an Effective Strategy for Targeting Glycolytic Tumors. Nat. Genet. 2013, 45, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 Bound to Mitochondria: Cancer’s Stygian Link to the “Warburg Effect” and a Pivotal Target for Effective Therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef]
- Kwiatkowska, E.; Wojtala, M.; Gajewska, A.; Soszyński, M.; Bartosz, G.; Sadowska-Bartosz, I. Effect of 3-Bromopyruvate Acid on the Redox Equilibrium in Non-Invasive MCF-7 and Invasive MDA-MB-231 Breast Cancer Cells. J. Bioenerg. Biomembr. 2016, 48, 23–32. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.A.; de Bari, L. 3-Bromopyruvate Induces Rapid Human Prostate Cancer Cell Death by Affecting Cell Energy Metabolism, GSH Pool and the Glyoxalase System. J. Bioenerg. Biomembr. 2015, 47, 493–506. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular Mechanisms of Drug Resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Wilson, T.R.; Longley, D.B.; Johnston, P.G. Chemoresistance in Solid Tumours. Ann. Oncol. 2006, 17, x315–x324. [Google Scholar] [CrossRef]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A Translational Study “Case Report” on the Small Molecule “Energy Blocker” 3-Bromopyruvate (3BP) as a Potent Anticancer Agent: From Bench Side to Bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef]
- Shoshan, M.C. 3-Bromopyruvate: Targets and Outcomes. J. Bioenerg. Biomembr. 2012, 44, 7–15. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.; Alfarouk, K. The Warburg Effect and the Hallmarks of Cancer. Anti-Cancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F.H. Tumor Glycolysis as a Target for Cancer Therapy: Progress and Prospects. Mol. Cancer 2013, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The First Drug Approved for BRAF-Mutant Cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK Pathways in the Regulation of Metabolic Reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Sinkala, M.; Mulder, N.; Patrick Martin, D. Metabolic Gene Alterations Impact the Clinical Aggressiveness and Drug Responses of 32 Human Cancers. Commun. Biol. 2019, 2, 1–14. [Google Scholar] [CrossRef]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic Heterogeneity Confers Differences in Melanoma Metastatic Potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef]
- Xu, D.Q.; Tan, X.Y.; Zhang, B.W.; Wu, T.; Liu, P.; Sun, S.J.; Cao, Y.G. 3-Bromopyruvate Inhibits Cell Proliferation and Induces Apoptosis in CD133+ Population in Human Glioma. Tumor Biol. 2016, 37, 3543–3548. [Google Scholar] [CrossRef]
- Wang, T.A.; Zhang, X.D.; Guo, X.Y.; Xian, S.L.; Lu, Y.F. 3-Bromopyruvate and Sodium Citrate Target Glycolysis, Suppress Survivin, and Induce Mitochondrial-Mediated Apoptosis in Gastric Cancer Cells and Inhibit Gastric Orthotopic Transplantation Tumor Growth. Oncol. Rep. 2016, 35, 1287–1296. [Google Scholar] [CrossRef]
- Gan, L.; Ren, Y.; Lu, J.; Ma, J.; Shen, X.; Zhuang, Z. Synergistic Effect of 3-Bromopyruvate in Combination with Rapamycin Impacted Neuroblastoma Metabolism by Inhibiting Autophagy. Onco Targets Ther. 2020, 13, 11125–11137. [Google Scholar] [CrossRef]
- Qin, J.Z.; Xin, H.; Nickoloff, B.J. 3-Bromopyruvate Induces Necrotic Cell Death in Sensitive Melanoma Cell Lines. Biochem. Biophys. Res. Commun. 2010, 396, 495–500. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Z.; Zou, X.; Lan, Y.; Sun, X.; Wang, X.; Zhao, S.; Jiang, C.; Liu, H. Mechanisms Underlying 3-Bromopyruvate-Induced Cell Death in Colon Cancer. J. Bioenerg. Biomembr. 2015, 47, 319–329. [Google Scholar] [CrossRef]
- Calviño, E.; Estañ, M.C.; Sánchez-Martín, C.; Brea, R.; de Blas, E.; del Carmen Boyano-Adánez, M.; Rial, E.; Aller, P. Regulation of Death Induction and Chemosensitizing Action of 3-Bromopyruvate in Myeloid Leukemia Cells: Energy Depletion, Oxidative Stress, and Protein Kinase Activity Modulations. J. Pharmacol. Exp. Ther. 2014, 348, 324–335. [Google Scholar] [CrossRef]
- Messner, K.R.; Imlay, J.A. Mechanism of Superoxide and Hydrogen Peroxide Formation by Fumarate Reductase, Succinate Dehydrogenase, and Aspartate Oxidase. J. Biol. Chem. 2002, 277, 42563–42571. [Google Scholar] [CrossRef]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef]
- Li, L.K.; Zhang, X.J.; Wang, L.M.; Hu, J.Y.; Cao, F.H. Inhibitory Effect of 3-Bromopyruvate on the Proliferation, Migration and Invasive Ability of Prostate Cancer PC-3 Cells. Zhonghua Nan Ke Xue 2020, 26, 17–23. [Google Scholar]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A Multi-Tool for Tumor Progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Børretzen, A.; Gravdal, K.; Haukaas, S.A.; Mannelqvist, M.; Beisland, C.; Akslen, L.A.; Halvorsen, O.J. The Epithelial-Mesenchymal Transition Regulators Twist, Slug, and Snail Are Associated with Aggressive Tumour Features and Poor Outcome in Prostate Cancer Patients. J. Pathol. Clin. Res. 2021, 7, 253–270. [Google Scholar] [CrossRef]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-Cadherin-Mediated Cell-Cell Adhesion Activates Akt and Mitogen Activated Protein Kinase via Phosphatidylinositol 3 Kinase and Ligand-Independent Activation of Epidermal Growth Factor Receptor in Ovarian Cancer Cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar] [CrossRef]
- Bussink, J.; Kaanders, J.H.A.M.; van der Kogel, A.J. Microenvironmental Transformations by VEGF- and EGF-Receptor Inhibition and Potential Implications for Responsiveness to Radiotherapy. Radiother. Oncol. 2007, 82, 10–17. [Google Scholar] [CrossRef]
- Hara, J.; Miyata, H.; Yamasaki, M.; Sugimura, K.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; Takiguchi, S.; Mori, M.; Doki, Y. Mesenchymal Phenotype after Chemotherapy Is Associated with Chemoresistance and Poor Clinical Outcome in Esophageal Cancer. Oncol. Rep. 2014, 31, 589–596. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual Breast Cancers after Conventional Therapy Display Mesenchymal as Well as Tumor-Initiating Features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- Ho, N.; Morrison, J.; Silva, A.; Coomber, B.L. The Effect of 3-Bromopyruvate on Human Colorectal Cancer Cells Is Dependent on Glucose Concentration but Not Hexokinase II Expression. Biosci. Rep. 2016, 36, e00299. [Google Scholar] [CrossRef]
- Da Silva, A.P.P.; El-Bacha, T.; Kyaw, N.; Dos Santos, R.S.; Da-Silva, W.S.; Almeida, F.C.L.; Da Poian, A.T.; Galina, A. Inhibition of Energy-Producing Pathways of HepG2 Cells by 3-Bromopyruvate. Biochem. J. 2009, 417, 717–726. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Fedorova, M.S.; Zhavoronkov, A.; Moskalev, A.A.; Zasedatelev, A.S.; Dmitriev, A.A.; Sadritdinova, A.F.; Karpova, I.Y.; Nyushko, K.M.; Kalinin, D.v.; et al. Effect of Lentivirus-Mediated ShRNA Inactivation of HK1, HK2, and HK3 Genes in Colorectal Cancer and Melanoma Cells. BMC Genet. 2016, 17, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic Treatment with the Antidiabetic Drug Metformin Selectively Impairs P53-Deficient Tumor Cell Growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef]
- Mashhedi, H.; Blouin, M.-J.; Zakikhani, M.; David, S.; Zhao, Y.; Bazile, M.; Birman, E.; Algire, C.; Aliaga, A.; Bedell, B.J.; et al. Metformin Abolishes Increased Tumor 18 F-2-Fluoro-2-Deoxy-D-Glucose Uptake Associated with a High Energy Diet. Cell Cycle 2011, 10, 2770–2778. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef]
- Swinson, D.E.; Jones, J.L.; Richardson, D.; Wykoff, C.; Turley, H.; Pastorek, J.; Taub, N.; Harris, A.L.; O’Byrne, K.J. Carbonic Anhydrase IX Expression, a Novel Surrogate Marker of Tumor Hypoxia, Is Associated with a Poor Prognosis in Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2003, 21, 473–482. [Google Scholar] [CrossRef]
- Swietach, P.; Wigfield, S.; Supuran, C.T.; Harris, A.L.; Vaughan-Jones, R.D. Cancer-Associated, Hypoxia-Inducible Carbonic Anhydrase IX Facilitates CO2 Diffusion. Br. J. Urol. 2008, 101, 22–24. [Google Scholar] [CrossRef]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling Vemurafenib Resistance in Melanoma Reveals a Strategy to Forestall Drug Resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Sargent, D.J.; Grothey, A.; Kerbel, R.S. Drug Rechallenge and Treatment beyond Progression-Implications for Drug Resistance. Nat. Rev. Clin. Oncol. 2013, 10, 571–587. [Google Scholar] [CrossRef]
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; de Lima Vazquez, V.; Reis, R.M.; et al. Vemurafenib Resistance Increases Melanoma Invasiveness and Modulates the Tumor Microenvironment by MMP-2 Upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef]
- Faião-Flores, F.; Alves-Fernandes, D.K.; Pennacchi, P.C.; Sandri, S.; Vicente, A.L.S.A.; Scapulatempo-Neto, C.; Vazquez, V.L.; Reis, R.M.; Chauhan, J.; Goding, C.R.; et al. Targeting the Hedgehog Transcription Factors GLI1 and GLI2 Restores Sensitivity to Vemurafenib-Resistant Human Melanoma Cells. Oncogene 2017, 36, 1849–1861. [Google Scholar] [CrossRef]
- Sandri, S.; Watanabe, L.R.M.; Oliveira, E.A.D.; Faião-Flores, F.; Migliorini, S.; Tiago, M.; Felipe-Silva, A.; Vazquez, V.D.L.; da Costa Souza, P.; Consolaro, M.E.L.; et al. Indoleamine 2,3-Dioxygenase in Melanoma Progression and BRAF Inhibitor Resistance. Pharmacol. Res. 2020, 159, 104998. [Google Scholar] [CrossRef] [PubMed]
- Dirks, W.G.; Fähnrich, S.; Estella, I.A.J.; Drexler, H.G. Short Tandem Repeat DNA Typing Provides an International Reference Standard for Authentication of Human Cell Lines. ALTEX-Altern. Anim. Exp. 2005, 22, 103–109. [Google Scholar]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vital, P.d.S.; Bonatelli, M.; Dias, M.P.; de Salis, L.V.V.; Pinto, M.T.; Baltazar, F.; Maria-Engler, S.S.; Pinheiro, C. 3-Bromopyruvate Suppresses the Malignant Phenotype of Vemurafenib-Resistant Melanoma Cells. Int. J. Mol. Sci. 2022, 23, 15650. https://doi.org/10.3390/ijms232415650
Vital PdS, Bonatelli M, Dias MP, de Salis LVV, Pinto MT, Baltazar F, Maria-Engler SS, Pinheiro C. 3-Bromopyruvate Suppresses the Malignant Phenotype of Vemurafenib-Resistant Melanoma Cells. International Journal of Molecular Sciences. 2022; 23(24):15650. https://doi.org/10.3390/ijms232415650
Chicago/Turabian StyleVital, Patrik da Silva, Murilo Bonatelli, Marina Pereira Dias, Larissa Vedovato Vilela de Salis, Mariana Tomazini Pinto, Fátima Baltazar, Silvya Stuchi Maria-Engler, and Céline Pinheiro. 2022. "3-Bromopyruvate Suppresses the Malignant Phenotype of Vemurafenib-Resistant Melanoma Cells" International Journal of Molecular Sciences 23, no. 24: 15650. https://doi.org/10.3390/ijms232415650
APA StyleVital, P. d. S., Bonatelli, M., Dias, M. P., de Salis, L. V. V., Pinto, M. T., Baltazar, F., Maria-Engler, S. S., & Pinheiro, C. (2022). 3-Bromopyruvate Suppresses the Malignant Phenotype of Vemurafenib-Resistant Melanoma Cells. International Journal of Molecular Sciences, 23(24), 15650. https://doi.org/10.3390/ijms232415650