
The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy



Abstract
1. Introduction
2. Vascular Endothelial Growth Factor in Physiology and Disease
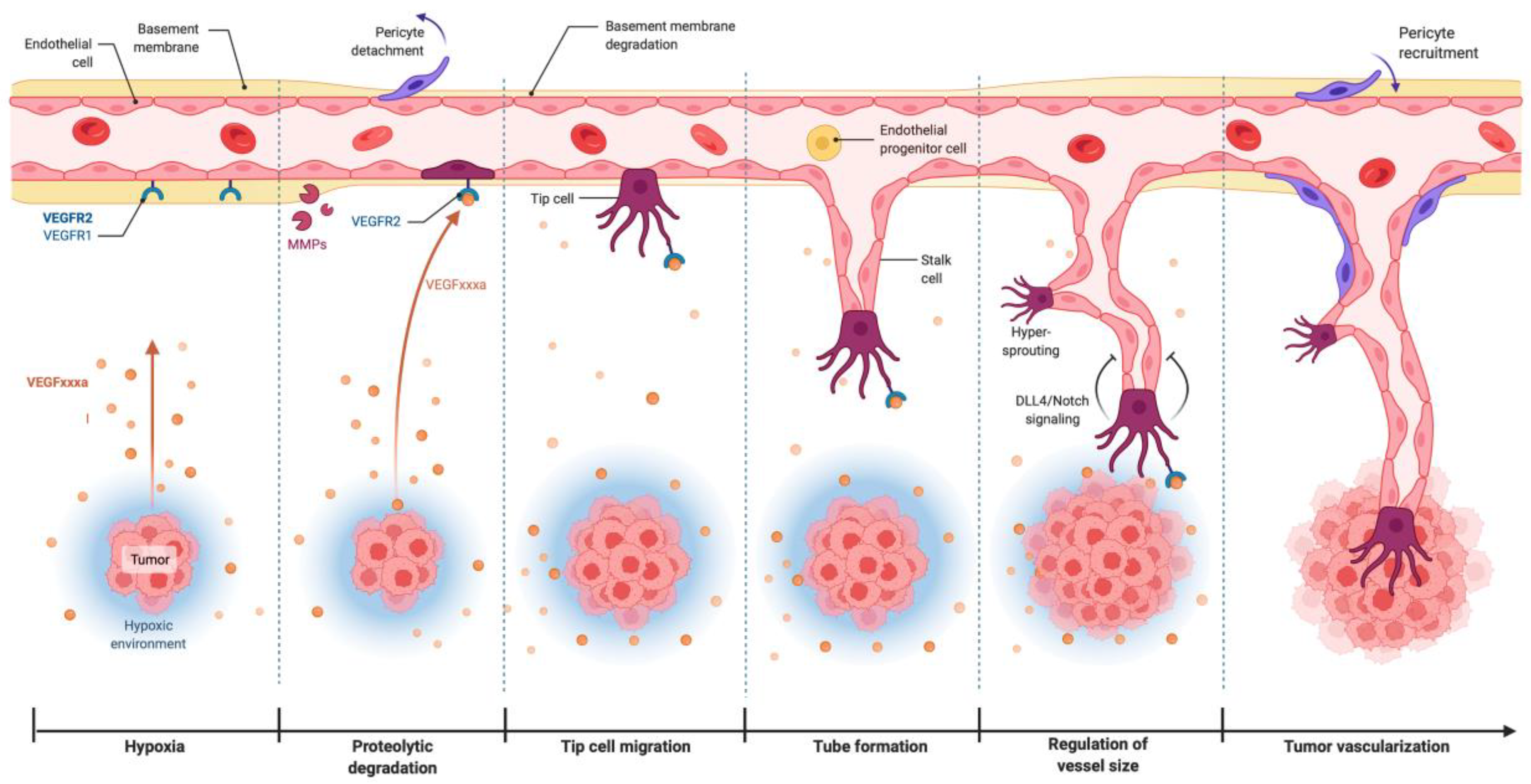
2.1. The VEGF in Physiological Angiogenesis
2.2. VEGF/VEGFR-2 Signaling in Tumor Angiogenesis
3. Alternative Splicing of VEGF and Angiogenesis
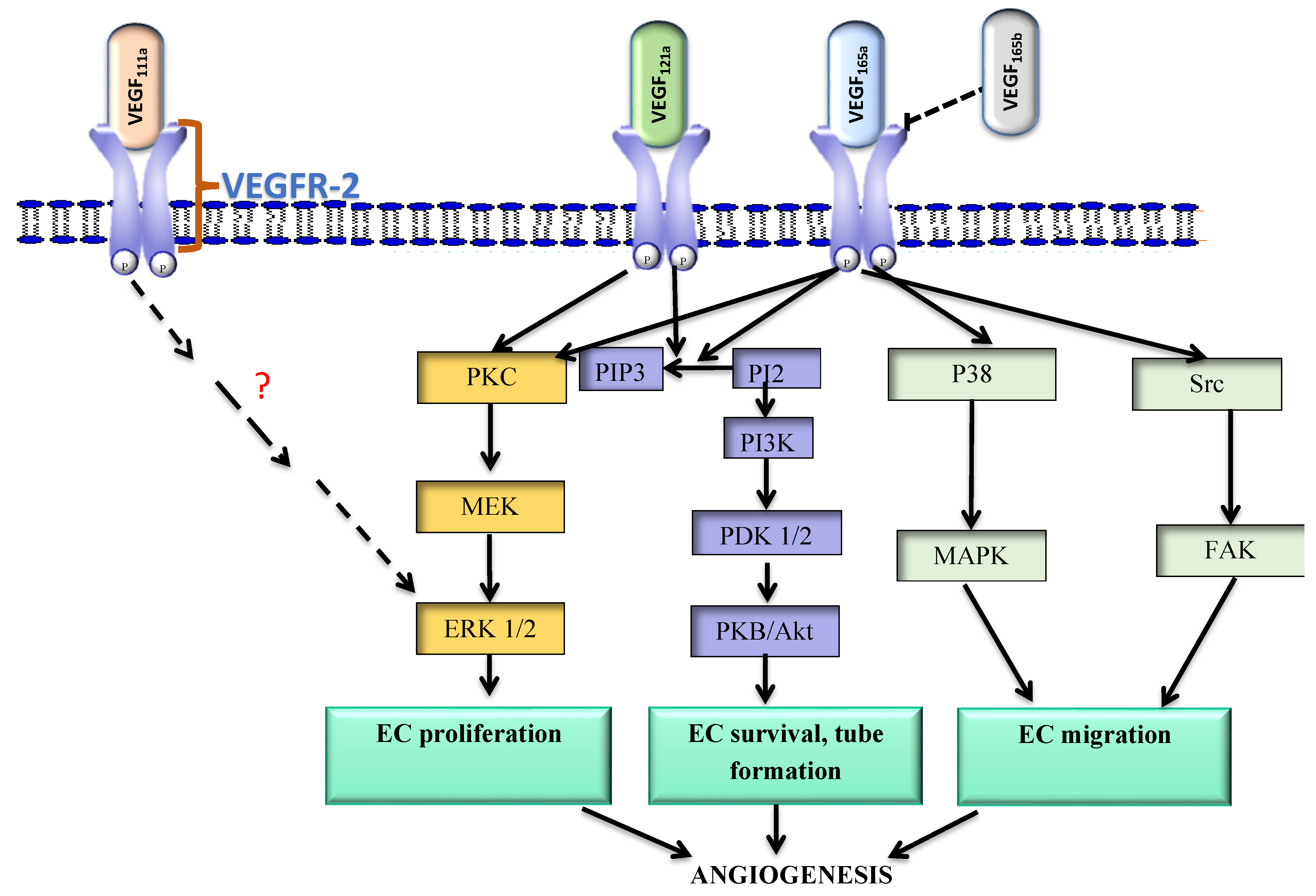
3.1. VEGF111a and VEGF121a
3.2. VEGF165a
3.3. VEGF165b
3.4. Alternative Splicing of VEGF Receptors
3.5. Regulation of VEGF Splicing
4. Clinical Implications of VEGF Splice Products
Improving VEGF-Targeting Approaches
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ide, A. Vascularization of the brown-pearce rabbit epithelioma transplant as seen in the transparent ear chamber. Am. J. Roentg. 1939, 42, 891. [Google Scholar]
- Folkman, J. Tumor angiogenesis: A possible control point in tumor growth. Ann. Inter. Med. 1975, 82, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P.; Pepper, M.S. Manipulating the tumor microenvironment: Opportunities for therapeutic targeting. Front. Anti. Cancer Drug Discov. 2017, 8, 46–71. [Google Scholar]
- Ribatti, D.; Vacca, A. New insights in anti-angiogenesis in multiple myeloma. Int. J. Mol. Sci. 2018, 19, 2031. [Google Scholar] [CrossRef]
- Makanya, A.N.; Hlushchuk, R.; Djonov, V.G. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis 2009, 12, 113–123. [Google Scholar] [CrossRef]
- Mabeta, P.; Hull, R.; Dlamini, Z. LncRNAs and the angiogenic switch in cancer: Clinical significance and therapeutic opportunities. Genes 2022, 13, 152. [Google Scholar] [CrossRef]
- Mabeta, P.; Pepper, M.S. A comparative study on the anti-angiogenic effects of DNA-damaging and cytoskeletal-disrupting agents. Angiogenesis 2009, 12, 81–90. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Orenstein, N.S.; Carvalho, A.C.; Churchill, W.H.; Dvorak, A.M.; Galli, S.J.; Feder, J.; Bitzer, A.M.; Rypysc, J.; Giovinco, P. Induction of a fibrin-gel investment: An early event in line 10 hepatocarcinoma growth mediated by tumor-secreted products. J. Immunol. 1979, 122, 166–174. [Google Scholar]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Ferrara, N. The role of the VEGF signaling pathway in tumor angiogenesis. In Tumor Angiogenesis: A Key Target for Cancer Therapy; Marmè, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 211–226. [Google Scholar]
- Ferrara, N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010, 21, 21–26. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Dakowicz, D.; Zajkowska, M.; Mroczko, B. Relationship between VEGF family members, their receptors and cell death in the neoplastic transformation of colorectal cancer. Int. J. Mol. Sci. 2022, 23, 3375. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [PubMed]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.-M.; Roman, M.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Huijbers, E.J.; Khan, K.A.; Kerbel, R.S.; Griffioen, A.W. Tumors resurrect an embryonic vascular program to escape immunity. Sci. Immunol. 2022, 7, eabm6388. [Google Scholar] [CrossRef] [PubMed]
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. Am. J. Physiol.-Cell Physiol. 2002, 282, C947–C970. [Google Scholar] [CrossRef]
- Weddell, J.C.; Chen, S.; Imoukhuede, P. VEGFR1 promotes cell migration and proliferation through PLCγ and PI3K pathways. NPJ Syst. Biol. Appl. 2018, 4, 1. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 signaling and its inhibition in modulating tumor invasion: Experimental evidence in different metastatic cancer models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef]
- Zachary, I. VEGF signalling: Integration and multi-tasking in endothelial cell biology. Biochem. Soc. Trans. 2003, 31, 1171–1177. [Google Scholar] [CrossRef]
- Lal, B.K.; Varma, S.; Pappas, P.J.; Hobson, R.W., II; Durán, W.N. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc. Res. 2001, 62, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; Potente, M. Regulation of angiogenesis by PI3K signaling networks. Exp. Cell Res. 2013, 319, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P. Inhibition of phosphoinositide 3-kinase is associated with reduced angiogenesis and an altered expression of angiogenic markers in endothelioma cells. Biomed. Pharmacother. 2014, 68, 611–617. [Google Scholar] [CrossRef]
- Qi, J.H.; Claesson-Welsh, L. VEGF-induced activation of phosphoinositide 3-kinase is dependent on focal adhesion kinase. Exp. Cell Res. 2001, 263, 173–182. [Google Scholar] [CrossRef]
- Mabeta, P.; Pepper, M.S. Inhibition of hemangioma development in a syngeneic mouse model correlates with bcl-2 suppression and the inhibition of Akt kinase activity. Angiogenesis 2012, 15, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Barak, V.; Pe’er, J.; Kalickman, I.; Frenkel, S. VEGF as a biomarker for metastatic uveal melanoma in humans. Curr. Eye Res. 2011, 36, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Alevizakos, M.; Kaltsas, S.; Syrigos, K.N. The VEGF pathway in lung cancer. Cancer Chemother. Pharmacol. 2013, 72, 1169–1181. [Google Scholar] [CrossRef]
- Ugurel, S.; Rappl, G.; Tilgen, W.; Reinhold, U. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J. Clin. Oncol. 2001, 19, 577–583. [Google Scholar] [CrossRef]
- Claffey, K.P.; Brown, L.F.; del Aguila, L.F.; Tognazzi, K.; Yeo, K.-T.; Manseau, E.J.; Dvorak, H.F. Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis, and experimental metastasis. Cancer Res. 1996, 56, 172–181. [Google Scholar]
- Jinnin, M.; Medici, D.; Park, L.; Limaye, N.; Liu, Y.; Boscolo, E.; Bischoff, J.; Vikkula, M.; Boye, E.; Olsen, B.R. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat. Med. 2008, 14, 1236–1246. [Google Scholar] [CrossRef]
- Marasco, L.E.; Kornblihtt, A.R. The physiology of alternative splicing. Nat. Rev. Mol. Cell Biol. 2022, 13, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-X.; Zhu, G.-Q.; Cai, J.-L.; Wang, B.; Luo, Y.-H.; Chen, C.; Cai, C.-Z.; Zhang, S.-J.; Zhou, J.; Fan, J.; et al. Splicing factors: Insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 2021, 501, 83–104. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Li, L.; Yang, L.; Wang, S.; Ning, X.; Gao, S.; Ren, L.; Chaulagain, A.; Tang, J.; et al. Alternative splicing: An important regulatory mechanism in colorectal carcinoma. Mol. Carcinog. 2021, 60, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Mehterov, N.; Kazakova, M.; Sbirkov, Y.; Vladimirov, B.; Belev, N.; Yaneva, G.; Todorova, K.; Hayrabedyan, S.; Sarafian, V. Alternative RNA splicing—The trojan horse of cancer cells in chemotherapy. Genes 2021, 12, 1085. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B. Targeting the spliceosome machinery: A new therapeutic axis in cancer? Biochem. Pharmacol. 2021, 189, 114039. [Google Scholar] [CrossRef]
- Eymin, B.; Boudria, A.; Abou-Faycal, C. VEGF-A splice variants: Do they play a role in tumor responses to anti-angiogenic therapies? In Molecular Mechanisms of Angiogenesis; Feige, J.-J., Pagès, G., Soncin, F., Eds.; Springer: Paris, France, 2014; pp. 421–442. [Google Scholar]
- Natua, S.; Ashok, C.; Shukla, S. Hypoxia-induced alternative splicing in human diseases: The pledge, the turn, and the prestige. Cell Mol. Life Sci. 2021, 78, 2729–2747. [Google Scholar] [CrossRef]
- Elias, A.P.; Dias, S. Microenvironment changes (in pH) affect VEGF alternative splicing. Cancer Microenviron. 2008, 1, 131–139. [Google Scholar] [CrossRef]
- Mamer, S.B.; Wittenkeller, A.; Imoukhuede, P. VEGF-A splice variants bind VEGFRs with differential affinities. Sci. Rep. 2020, 10, 14413. [Google Scholar] [CrossRef]
- Robinson, C.J.; Stringer, S.E. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 2001, 114, 853–865. [Google Scholar] [CrossRef]
- Woolard, J.; Bevan, H.S.; Harper, S.J.; Bates, D.O. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation 2009, 16, 572–592. [Google Scholar] [CrossRef]
- Mineur, P.; Colige, A.C.; Deroanne, C.F.; Dubail, J.; Kesteloot, F.; Habraken, Y.; Noël, A.; Vöö, S.; Waltenberger, J.; Lapière, C.M.; et al. Newly identified biologically active and proteolysis-resistant VEGF-A isoform VEGF111 is induced by genotoxic agents. J. Cell Biol. 2007, 179, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Chathery, Y.; Wu, Y.; Rathore, N.; Tong, R.K.; Peale, F.; Bagri, A.; Tessier-Lavigne, M.; Koch, A.W.; Watts, R.J. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J. Biol. Chem. 2007, 282, 24049–24056. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Minamiya, Y.; Ito, M.; Saito, H.; Ogawa, J. VEGF121 promotes lymphangiogenesis in the sentinel lymph nodes of non-small cell lung carcinoma patients. Lung Cancer 2008, 59, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.; Carrer, A.; Moimas, S.; Zandonà, L.; Bussani, R.; Casagranda, B.; Palmisano, S.; Prelazzi, P.; Giacca, M.; Zentilin, L.; et al. VEGF121 and VEGF165 differentially promote vessel maturation and tumor growth in mice and humans. Cancer Gene Ther. 2016, 23, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, C.; Guyot, M. VEGF spliced variants: Possible role of anti-angiogenesis therapy. J. Nucleic Acids 2012, 2012, 162692. [Google Scholar] [CrossRef]
- Patel, K.A.; Patel, B.M.; Thobias, A.R.; Gokani, R.A.; Chhikara, A.B.; Desai, A.D.; Patel, P.S. Overexpression of VEGF165 is associated with poor prognosis of cervical cancer. J. Obstet. Gynaecol. Res. 2020, 46, 2397–2406. [Google Scholar] [CrossRef]
- Mehedi, F. Vascular endothelial growth factor (VEGF) splice isoforms may hold the key to targeting tumour angiogenesis in oesophageal cancer. Ann. Oncol. 2018, 29, 11. [Google Scholar] [CrossRef]
- Catena, R.; Muniz-Medina, V.; Moralejo, B.; Javierre, B.; Best, C.J.; Emmert-Buck, M.R.; Green, J.E.; Baker, C.C.; Calvo, A. Increased expression of VEGF121/VEGF165–189 ratio results in a significant enhancement of human prostate tumor angiogenesis. Int. J. Cancer 2007, 120, 2096–2109. [Google Scholar] [CrossRef]
- Bates, D.O.; Cui, T.-G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillat, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar]
- Jones, R.P.; Rennel, E.; Harper, S.; Orlando, A.; Bates, D. The endogenous antiangiogenic molecule VEGF165b inhibits the proliferation of A375 melanoma in vitro. Melanoma Res. 2006, 16, S8–S9. [Google Scholar] [CrossRef]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef]
- Volpi, N.; Pecorelli, A.; Lorenzoni, P.; Di Lazzaro, F.; Belmonte, G.; Aglianò, M.; Cantarini, L.; Giannini, F.; Grasso, G.; Valacchi, G. Antiangiogenic VEGF isoform in inflammatory myopathies. Mediators Inflamm. 2013, 2013, 219313. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Nakamura, K.; MacLauchlan, S.; Ngo, D.T.-M.; Shimizu, I.; Fuster, J.J.; Katanasaka, Y.; Yoshida, S.; Qiu, Y.; Yamaguchi, T.P.; et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 2014, 20, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Wang, W.-Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.-G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: Mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Romano, E.; Ceccarelli, C.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ. Res. 2011, 109, e14–e26. [Google Scholar] [CrossRef]
- Catena, R.; Larzabal, L.; Larrayoz, M.; Molina, E.; Hermida, J.; Agorreta, J.; Montes, R.; Pio, R.; Montuenga, L.M.; Calvo, A. VEGF₁₆₅b are weakly angiogenic isoforms of VEGF-A. Mol. Cancer 2010, 9, 320. [Google Scholar] [CrossRef]
- Peiris-Pagès, M. The role of VEGF165b in pathophysiology. Cell Adh. Migr. 2012, 6, 561–568. [Google Scholar] [CrossRef]
- Kawamura, H.; Li, X.; Harper, S.J.; Bates, D.O.; Claesson-Welsh, L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008, 68, 4683–4692. [Google Scholar] [CrossRef]
- Pritchard-Jones, R.; Dunn, D.; Qiu, Y.; Varey, A.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGFxxxb, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230. [Google Scholar] [CrossRef]
- Karsten, M.M.; Beck, M.H.; Rademacher, A.; Knabl, J.; Blohmer, J.-U.; Jückstock, J.; Radosa, J.C.; Jank, P.; Rack, B.; Janni, W. VEGF-A165b levels are reduced in breast cancer patients at primary diagnosis but increase after completion of cancer treatment. Sci. Rep. 2020, 10, 3635. [Google Scholar] [CrossRef]
- Boudria, A.; Abou Faycal, C.; Jia, T.; Gout, S.; Keramidas, M.; Didier, C.; Lemaître, N.; Manet, S.; Coll, J.-L.; Toffart, A.-C.; et al. VEGF165b, a splice variant of VEGF-A, promotes lung tumor progression and escape from anti-angiogenic therapies through a β1 integrin/VEGFR autocrine loop. Oncogene 2019, 38, 1050–1066. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, M.N.; Sainson, R.C.; Pérez-del-Pulgar, S.; Aoto, J.N.; Aitkenhead, M.; Taylor, K.L.; Carpenter, P.M.; Hughes, C.C.W. VEGF121 and VEGF165 regulate blood vessel diameter through vascular endothelial growth factor receptor 2 in an in vitro angiogenesis model. Lab. Investig. 2003, 83, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Bowler, E.; Oltean, S. Alternative splicing in angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Furumura, M.; Morita, E. Distinct signaling pathways confer different vascular responses to VEGF 121 and VEGF 165. Growth Factors 2008, 26, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.; Waltenberger, J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR). Biochem. Biophys. Res. Commun. 1998, 252, 743–746. [Google Scholar] [CrossRef]
- Poltorak, Z.; Cohen, T.; Sivan, R.; Kandelis, Y.; Spira, G.; Vlodavsky, I.; Keshet, E.; Neufeld, G. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J. Biol. Chem. 1997, 272, 7151–7158. [Google Scholar] [CrossRef]
- Yuan, A.; Yu, C.-J.; Kuo, S.-H.; Chen, W.-J.; Lin, F.-Y.; Luh, K.-T.; Lee, Y.-C. Vascular endothelial growth factor 189 mRNA isoform expression specifically correlates with tumor angiogenesis, patient survival, and postoperative relapse in non–small-cell lung cancer. J. Clin. Oncol. 2001, 19, 432–441. [Google Scholar] [CrossRef]
- Ng, I.O.; Poon, R.T.; Lee, J.M.; Fan, S.T.; Ng, M.; Tso, W.K. Microvessel density, vascular endothelial growth factor and its receptors Flt-1 and Llk-1/KDR in hepatocellular carcinoma. Am. J. Clin. Pathol. 2001, 116, 838–845. [Google Scholar] [CrossRef]
- Hervé, M.-A.; Buteau-Lozano, H.; Mourah, S.; Calvo, F.; Perrot-Applanat, M. VEGF189 stimulates endothelial cells proliferation and migration in vitro and up-regulates the expression of Flk-1/KDR mRNA. Exp. Cell Res. 2005, 309, 24–31. [Google Scholar] [CrossRef]
- Failla, C.M.; Carbo, M.; Morea, V. Positive and negative regulation of angiogenesis by soluble vascular endothelial growth factor receptor-1. Int. J. Mol. Sci. 2018, 19, 1306. [Google Scholar] [CrossRef]
- Luttun, A.; Autiero, M.; Tjwa, M.; Carmeliet, P. Genetic dissection of tumor angiogenesis: Are PlGF and VEGFR-1 novel anti-cancer targets? Biochim. Biophys. Acta-Rev. Cancer 2004, 1654, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.-P.; Condorelli, F.; Park, J.; Ferrara, N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes: Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.T.; Stefanini, M.O.; Gabhann, F.M.; Kontos, C.D.; Annex, B.H.; Popel, A.S. A systems biology perspective on sVEGFR1: Its biological function, pathogenic role and therapeutic use. J. Cell Mol. Med. 2010, 14, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Pavlakovic, H.; Becker, J.; Albuquerque, R.; Wilting, J.; Ambati, J. Soluble VEGFR-2: An anti-lymphangiogenic variant of VEGF receptors. Ann. N. Y. Acad. Sci. 2010, 1207, E7–E15. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Kamal, M.A.; Akhtar, S. Tumor angiogenesis and VEGFR-2: Mechanism, pathways and current biological therapeutic interventions. Curr. Drug Metab. 2021, 22, 50–59. [Google Scholar]
- Bando, H.; Weich, H.A.; Brokelmann, M.; Horiguchi, S.; Funata, N.; Ogawa, T.; Toi, M. Association between intratumoral free and total VEGF, soluble VEGFR-1, VEGFR-2 and prognosis in breast cancer. Br. J. Cancer 2005, 92, 553–561. [Google Scholar] [CrossRef]
- Star, E.; Stevens, M.; Gooding, C.; Smith, C.W.; Li, L.; Ayine, M.L.; Harper, S.J.; Bates, D.O.; Oltean, S. A drug repositioning screen using splicing-sensitive fluorescent reporters identifies novel modulators of VEGF-A splicing with anti-angiogenic properties. Oncogenesis 2021, 10, 36. [Google Scholar] [CrossRef]
- Hulse, R.; Beazley-Long, N.; Hua, J.; Kennedy, H.; Prager, J.; Bevan, H.; Qiu, Y.; Fernandes, E.S.; Gammons, M.V.; Ballmer-Hofer, K.; et al. Regulation of alternative VEGF-A mRNA splicing is a therapeutic target for analgesia. Neurobiol. Dis. 2014, 71, 245–259. [Google Scholar] [CrossRef]
- Pan, X.W.; Xu, D.; Chen, W.J.; Chen, J.X.; Chen, W.J.; Ye, J.Q.; Gan, S.S.; Zhou, W.; Song, X.; Shi, L.; et al. USP39 promotes malignant proliferation and angiogenesis of renal cell carcinoma by inhibiting VEGF-A165b alternative splicing via regulating SRSF1 and SRPK1. Cancer Cell Int. 2021, 21, 486. [Google Scholar] [CrossRef]
- Stimpfl, M.; Tong, D.; Fasching, B.; Schuster, E.; Obermair, A.; Leodolter, S.; Zeillinger, R. Vascular endothelial growth factor splice variants and their prognostic value in breast and ovarian cancer. Clin. Cancer Res. 2002, 8, 2253–2259. [Google Scholar]
- Dagmura, H.; Yigit, S.; Gumusay, O.; Nursal, A.F.; Daldal, E.; Karakus, N. eNOS and VEGF variants might increase the risk of pancreatic cancer. Cytol. Genet. 2021, 55, 177–182. [Google Scholar] [CrossRef]
- Bates, D.O.; Harper, S.J. Therapeutic potential of inhibitory VEGF splice variants. Future Med. 2005, 1, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.; Vimalraj, S.; Pavani, K.; Nikarika, R.; Sumantran, V.N. Intussusceptive angiogenesis as a key therapeutic target for cancer therapy. Life Sci. 2020, 252, 117670. [Google Scholar] [CrossRef]
- Djonov, V.G. VEGF withdrawal induces vascular remodelling and pruning via intussusception. FASEB J. 2006, 20, A440. [Google Scholar] [CrossRef]
- Crafts, T.D.; Jensen, A.R.; Blocher-Smith, E.C.; Markel, T.A. Vascular endothelial growth factor: Therapeutic possibilities and challenges for the treatment of ischemia. Cytokine 2015, 71, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Wilting, J.; Birkenhäger, R.; Eichmann, A.; Kurz, H.; Martiny-Baron, G.; Marmé, D.; McCarthy, J.E.; Christ, B.; Weich, H.A. VEGF121 induces proliferation of vascular endothelial cells and expression of flk-1 without affecting lymphatic vessels of the chorioallantoic membrane. Dev. Biol. 1996, 176, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Soker, S.; Miao, H.Q.; Nomi, M.; Takashima, S.; Klagsbrun, M. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J. Cell Biochem. 2002, 85, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Canavese, M.; Ngo, D.T.; Maddern, G.J.; Hardingham, J.E.; Price, T.J.; Hauben, E. Biology and therapeutic implications of VEGF-A splice isoforms and single-nucleotide polymorphisms in colorectal cancer. Int. J. Cancer 2017, 140, 2183–2191. [Google Scholar] [CrossRef]
- Carter, J.J.; Wheal, A.J.; Hill, S.J.; Woolard, J. Effects of receptor tyrosine kinase inhibitors on VEGF165a- and VEGF165b-stimulated gene transcription in HEK-293 cells expressing human VEGFR-2. Br. J. Pharmacol. 2015, 172, 3141–3150. [Google Scholar] [CrossRef]
- Rennel, E.; Waine, E.; Guan, H.; Schüler, Y.; Leenders, W.; Woolard, J.; Sugiono, M.; Gillatt, D.; Kleinerman, E.S.; Bates, D.O.; et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br. J. Cancer 2008, 98, 1250–1257. [Google Scholar] [CrossRef]
- Varey, A.; Rennel, E.; Qiu, Y.; Bevan, H.; Perrin, R.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; et al. VEGF165b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: Balance of pro-and antiangiogenic VEGF-A isoforms has implications for therapy. Br. J. Cancer 2008, 98, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Al Kawas, H.; Saaid, I.; Jank, P.; Westhoff, C.C.; Denkert, C.; Pross, T.; Weiler, K.B.S.; Karsten, M.M. How VEGF-A and its splice variants affect breast cancer development—Clinical implications. Cell Oncol. 2022, 45, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M.; Balasubramanian, S.; Vose, J.M.; Harb, W.; Gordon, L.I.; Langdon, R.; Sprague, J.; Sirisawad, M.; Mani, C.; Yue, J.; et al. Phase I/II multicenter, open-label study of the oral histone deacetylase inhibitor abexinostat in relapsed/refractory lymphoma abexinostat in mantle cell and follicular lymphoma. Clin. Cancer Res. 2016, 22, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Assouline, S.E.; Nielsen, T.H.; Yu, S.; Alcaide, M.; Chong, L.; MacDonald, D.; Tosikyan, A.; Kukreti, V.; Kezouh, A.; Petrogiannis-Haliotis, T.; et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood 2016, 128, 185–194. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variant | Receptor | Mechanism | Effect | Expression in Cancer | Reference |
|---|---|---|---|---|---|
| 111a | VEGFR-2 VEGFR-1 | VEGFR-2 phosphorylation; ERK 1/2 activation | Neovessel formation | ↑NSCLC ↑Breast cancer ↑Ovarian cancer | [40,42] |
| 121a | VEGFR-2 VEGFR-1 | VEGFR-2 phosphorylation at Y1175; P13k/p38, MEK1-ERK1/2 activation | EC proliferation, tube formation in matrigel plug; regulates vessel diameter; vessel maturation | ↑Prostate cancer ↑Colorectal cancer ↑Breast cancer | [43,64,65,66,67] |
| 145a | VEGFR-2 | EC mitogen; induces angiogenesis | [68] | ||
| 165a | VEGFR-2 VEGFR-1 | VEGFR-2 phosphorylation at Y1175 PIP2 hydrolysis and formation of IP3, activation of PKC | EC proliferation, migration, sprout formation; regulates vessel diameter; vessel maturation | ↑Colorectal cancer ↑Cervical cancer ↑Esophageal cancer | [40,41,42,43,49,67] |
| 165b | VEGFR-2 VEGFR-1 | Incomplete phosphorylation at Y1175; No PKC activation/ PIP2 hydrolysis | Weakly angiogenic; competitively inhibits VEGF- VEGFR-2 binding | ↓Breast cancer ↓RCC ↑Melanoma | [51,59,60] |
| 189a | VEGFR-2 (weak) | Binds NRP-1; upregulates Flk-1 | Cancer cell proliferation, EC proliferation, chemotaxis, tube formation | [69,70,71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. https://doi.org/10.3390/ijms232415585
Mabeta P, Steenkamp V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. International Journal of Molecular Sciences. 2022; 23(24):15585. https://doi.org/10.3390/ijms232415585
Chicago/Turabian StyleMabeta, Peace, and Vanessa Steenkamp. 2022. "The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy" International Journal of Molecular Sciences 23, no. 24: 15585. https://doi.org/10.3390/ijms232415585
APA StyleMabeta, P., & Steenkamp, V. (2022). The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. International Journal of Molecular Sciences, 23(24), 15585. https://doi.org/10.3390/ijms232415585