

Butyrate Lowers Cellular Cholesterol through HDAC Inhibition and Impaired SREBP-2 Signalling



Abstract
1. Introduction
2. Results
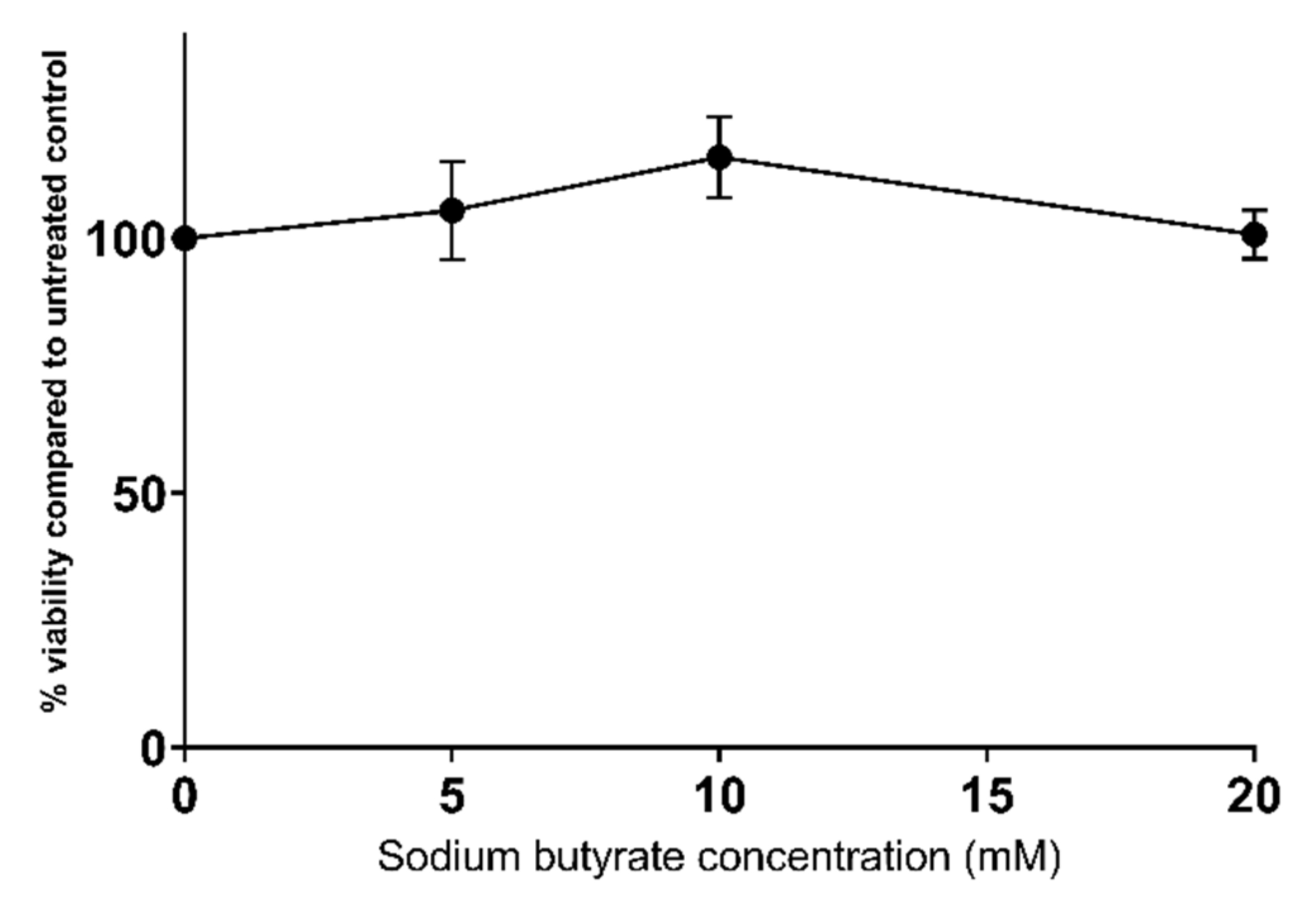
2.1. Sodium Butyrate Is Non-Toxic to HepG2 Cells
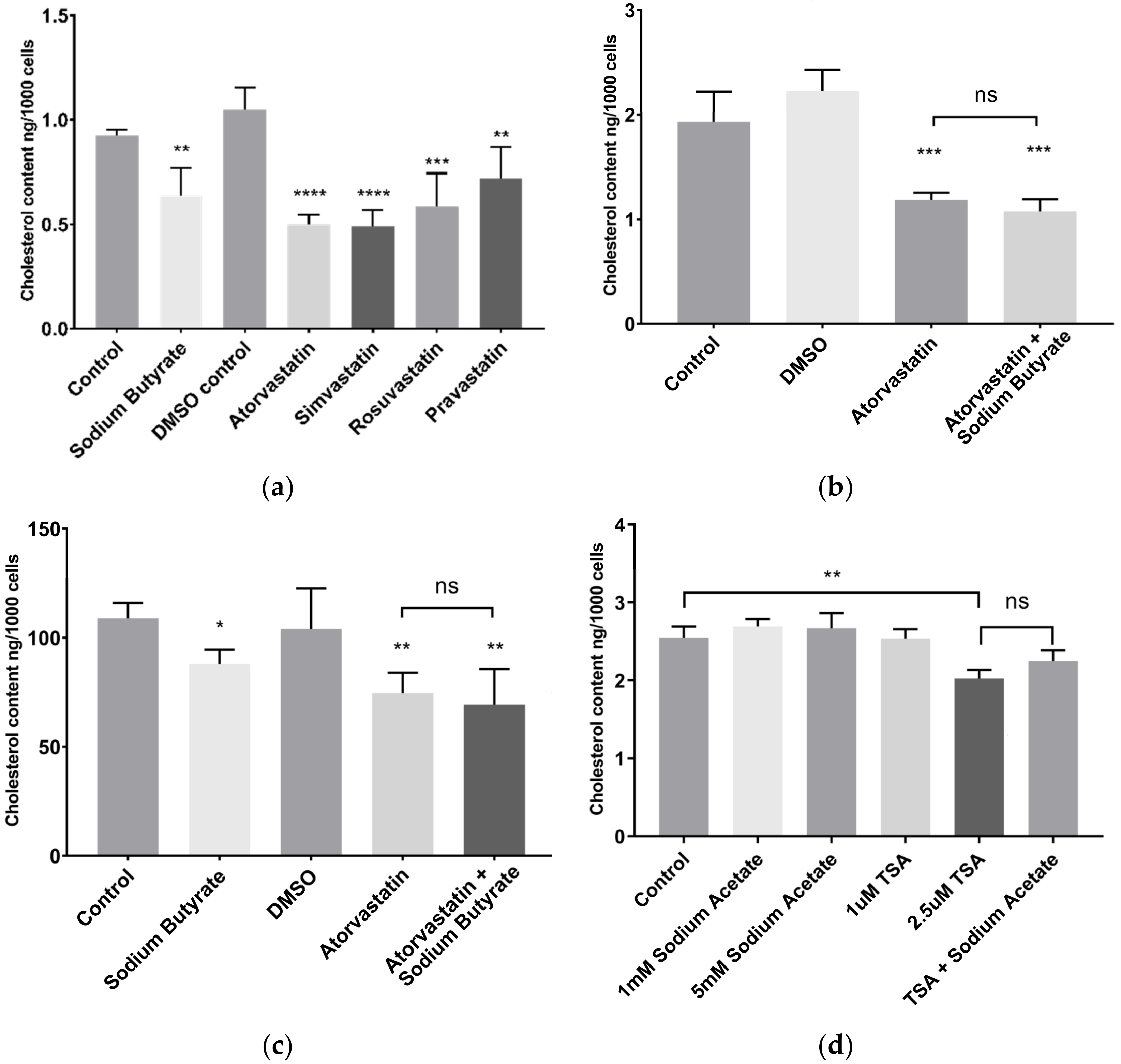
2.2. Sodium Butyrate Reduces Cellular Cholesterol
2.3. Sodium Butyrate Does Not Alter Cellular Triglyceride Content
2.4. Sodium Butyrate Affects Neither Cholesterol Uptake nor Export
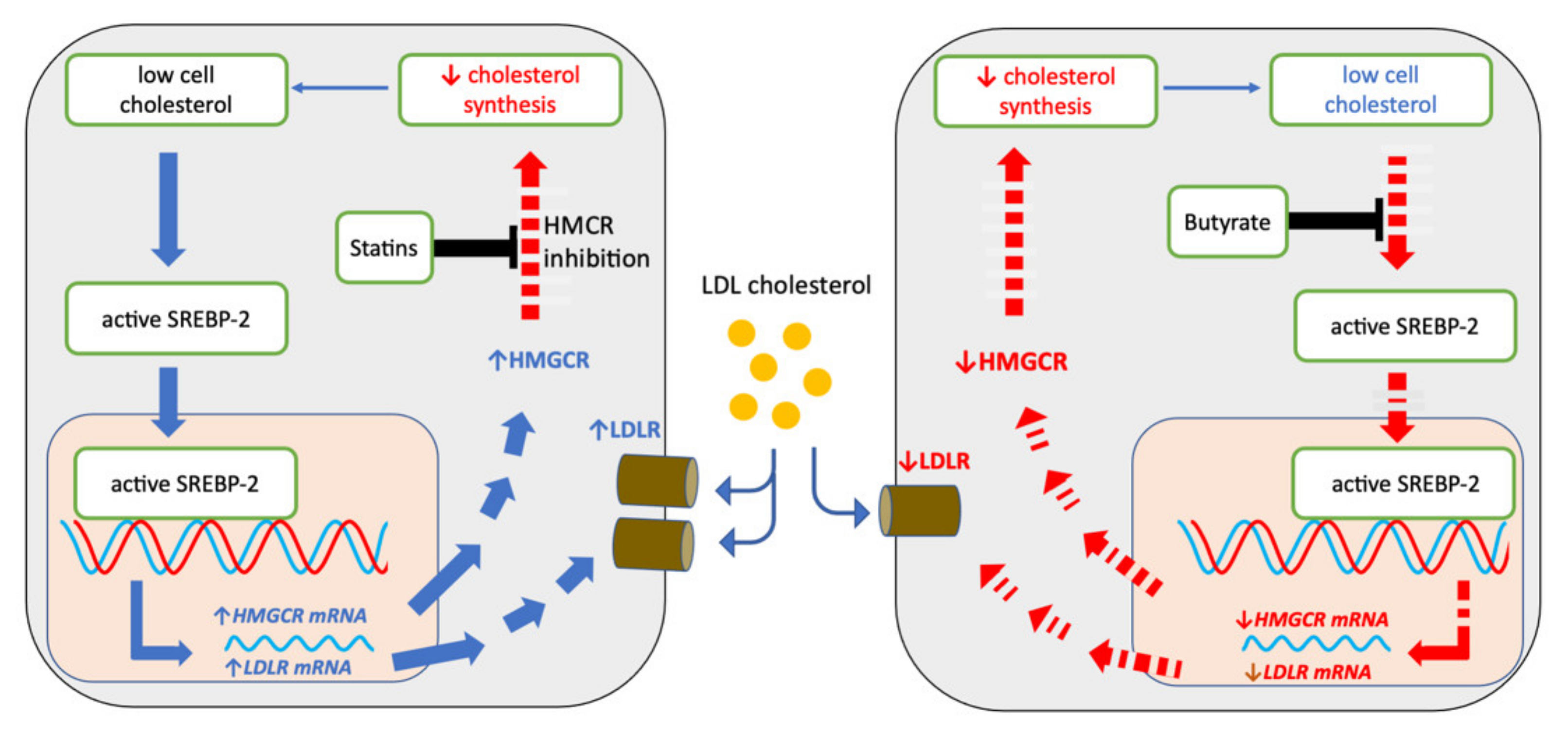
2.5. Sodium Butyrate and Statins Have Opposite Effects on SREBP-2 Signalling
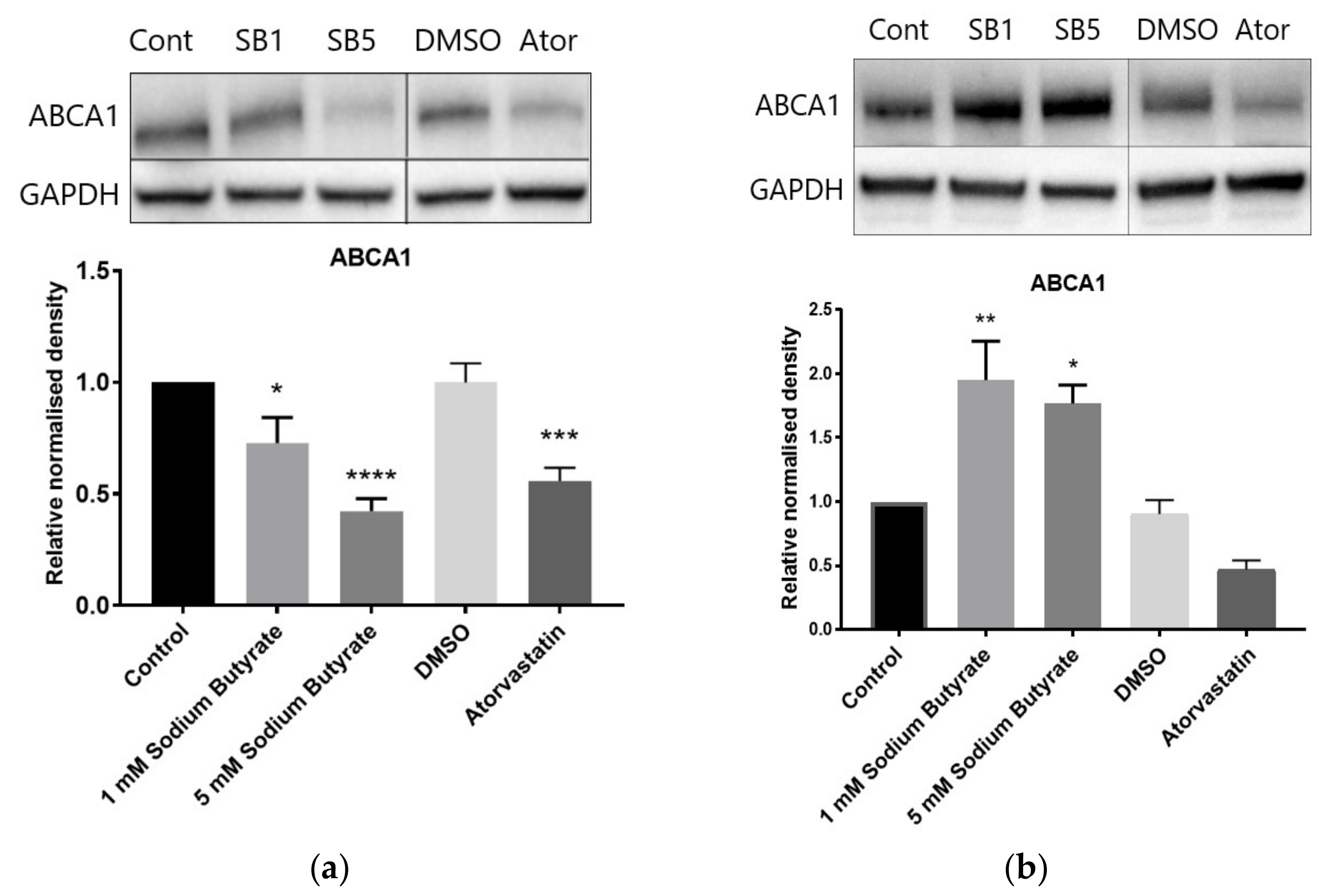
2.6. The Effect of Sodium Butyrate and Statins on Proteins Involved in Reverse Cholesterol Transport Depends on Cell Type and Media Lipoproteins
2.7. Sodium Butyrate Alters the Expression of Multiple Cholesterol Related Genes
3. Discussion
4. Materials and Methods
4.1. Preparation of Stock Solutions
4.2. Cell Culture and Treatment Conditions
4.3. Viability Assays
4.4. Lipid Quantification
4.5. Cholesterol Uptake and Export
4.6. Immunoblotting and Reverse Transcription Quantitative PCR
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organisation. Global Health Estimates 2016: Estimated Deaths by Cause and Region, 2000 and 2016. Available online: https://www.who.int/healthinfo/global_burden_disease/estimates/en/ (accessed on 6 September 2022).
- Johansen, M.E.; Green, L.A.; Sen, A.; Kircher, S.; Richardson, C.R. Cardiovascular risk and statin use in the United States. Ann. Fam. Med. 2014, 12, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.; Herrett, E.; Gasparrini, A.; Van Staa, T.; Goldacre, B.; Smeeth, L.; Bhaskaran, K. Impact of statin related media coverage on use of statins: Interrupted time series analysis with UK primary care data. BMJ 2016, 353, i3283. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.M.; Burke, M.; Davey Smith, G.; Ward, K.; Ebrahim, S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 2013, CD004816. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.S. Hepatotoxicity of statins and other lipid-lowering agents. Liver Int. 2017, 37, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Yiew, K.H.; Chatterjee, T.K.; Hui, D.Y.; Weintraub, N.L. Histone deacetylases and cardiometabolic diseases. Arter. Throm. Vasc. Biol. 2015, 35, 1914–1919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, J.; Yano, N.; Wang, H.; Zhao, Y.T.; Dubielecka, P.M.; Zhuang, S.; Chin, Y.E.; Qin, G.; Zhao, T.C. Sodium butyrate protects against high fat diet-induced cardiac dysfunction and metabolic disorders in type II diabetic mice. J. Cell. Biochem. 2017, 118, 2395–2408. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Marcil, V.; Delvin, E.; Garofalo, C.; Levy, E. Research Communication: Butyrate Impairs Lipid Transport by Inhibiting Microsomal Triglyceride Transfer Protein in Caco-2 Cells. J. Nutr. 2003, 133, 2180–2183. [Google Scholar] [CrossRef]
- Hao, M.; Head, W.S.; Gunawardana, S.C.; Hasty, A.H.; Piston, D.W. Direct effect of cholesterol on insulin secretion: A novel mechanism for pancreatic beta-cell dysfunction. Diabetes 2007, 56, 2328–2338. [Google Scholar] [CrossRef]
- Bridgeman, S.; Northrop, W.; Ellison, G.; Sabapathy, T.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Statins Do Not Directly Inhibit the Activity of Major Epigenetic Modifying Enzymes. Cancers 2019, 11, 516. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Cenedella, R.J. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 2009, 44, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef]
- Ji, A.; Meyer, J.M.; Cai, L.; Akinmusire, A.; de Beer, M.C.; Webb, N.R.; van der Westhuyzen, D.R. Scavenger receptor SR-BI in macrophage lipid metabolism. Atherosclerosis 2011, 217, 106–112. [Google Scholar] [CrossRef]
- Alvaro, A.; Sola, R.; Rosales, R.; Ribalta, J.; Anguera, A.; Masana, L.; Vallve, J.C. Gene expression analysis of a human enterocyte cell line reveals downregulation of cholesterol biosynthesis in response to short-chain fatty acids. IUBMB Life 2008, 60, 757–764. [Google Scholar] [CrossRef]
- Nunes, M.J.; Moutinho, M.; Gama, M.J.; Rodrigues, C.M.P.; Rodrigues, E. Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PLoS ONE 2013, 8, e53394. [Google Scholar] [CrossRef]
- Chittur, S.V.; Sangster-Guity, N.; McCormick, P.J. Histone deacetylase inhibitors: A new mode for inhibition of cholesterol metabolism. BMC Genom. 2008, 9, 507. [Google Scholar] [CrossRef]
- Giandomenico, V.; Simonsson, M.; Gronroos, E.; Ericsson, J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol. Cell. Biol. 2003, 23, 2587–2599. [Google Scholar] [CrossRef]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.-Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef] [PubMed]
- Howe, V.; Sharpe, L.J.; Prabhu, A.V.; Brown, A.J. New insights into cellular cholesterol acquisition: Promoter analysis of human HMGCR and SQLE, two key control enzymes in cholesterol synthesis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.; Ellison, G.; Newsholme, P.; Mamotte, C. The HDAC Inhibitor Butyrate Impairs beta Cell Function and Activates the Disallowed Gene Hexokinase I. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.C.; Northrop, W.; Melton, P.E.; Ellison, G.C.; Newsholme, P.; Mamotte, C.D.S. Butyrate generated by gut microbiota and its therapeutic role in metabolic syndrome. Pharm. Res. 2020, 160, 105174. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Hori, M.; Nakagami, T.; Harada-Shiba, M.; Uchigata, Y. Glucagon-like peptide-1 receptor agonists reduced the low-density lipoprotein cholesterol in Japanese patients with type 2 diabetes mellitus treated with statins. J. Clin. Lipidol. 2018, 12, 62–69.e61. [Google Scholar] [CrossRef]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. A Significant Effect of Oral Semaglutide on Cardiovascular Risk Factors in Patients With Type 2 Diabetes. Cardiol. Res. 2022, 13, 303–308. [Google Scholar] [CrossRef]
- Khan, S.; Jena, G. Sodium butyrate reduces insulin-resistance, fat accumulation and dyslipidemia in type-2 diabetic rat: A comparative study with metformin. Chem. Biol. Interact. 2016, 254, 124–134. [Google Scholar] [CrossRef]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE 2013, 8, e68626. [Google Scholar] [CrossRef]
- Hong, J.; Jia, Y.; Pan, S.; Jia, L.; Li, H.; Han, Z.; Cai, D.; Zhao, R. Butyrate alleviates high fat diet-induced obesity through activation of adiponectin-mediated pathway and stimulation of mitochondrial function in the skeletal muscle of mice. Oncotarget 2016, 7, 56071–56082. [Google Scholar] [CrossRef]
- Roshanravan, N.; Mahdavi, R.; Alizadeh, E.; Jafarabadi, M.A.; Hedayati, M.; Ghavami, A.; Alipour, S.; Alamdari, N.M.; Barati, M.; Ostadrahimi, A. Effect of Butyrate and Inulin Supplementation on Glycemic Status, Lipid Profile and Glucagon-Like Peptide 1 Level in Patients with Type 2 Diabetes: A Randomized Double-Blind, Placebo-Controlled Trial. Horm. Metab. Res. 2017, 49, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Bouter, K.E.C.; Bakker, G.J.; Levin, E.; Hartstra, A.V.; Kootte, R.S.; Udayappan, S.D.; Katiraei, S.; Bahler, L.; Gilijamse, P.W.; Tremaroli, V.; et al. Differential metabolic effects of oral butyrate treatment in lean versus metabolic syndrome subjects. Clin. Transl. Gastroenterol. 2018, 9, 155. [Google Scholar] [CrossRef]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef]
- Arguello, G.; Balboa, E.; Arrese, M.; Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. BBA Mol. Basis Dis. 2015, 1852, 1765–1778. [Google Scholar] [CrossRef]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef]
- Zhou, D.; Pan, Q.; Xin, F.-Z.; Zhang, R.-N.; He, C.-X.; Chen, G.-Y.; Liu, C.; Chen, Y.-W.; Fan, J.-G. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 2017, 23, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Tomioka, M.; Ueda, K. Function and regulation of ABCA1-membrane meso-domain organization and reorganization. FEBS J. 2011, 278, 3190–3203. [Google Scholar] [CrossRef]
- Li, A.C.; Glass, C.K. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 2004, 45, 2161–2173. [Google Scholar] [CrossRef]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. SR-B1: A unique multifunctional receptor for cholesterol influx and efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; McLean, M.P. Sterol regulatory element-binding protein-1a binds to cis elements in the promoter of the rat high density lipoprotein receptor SR-BI gene. Endocrinology 1999, 140, 5669–5681. [Google Scholar] [CrossRef] [PubMed]
- Malerod, L.; Juvet, L.K.; Hanssen-Bauer, A.; Eskild, W.; Berg, T. Oxysterol-activated LXRalpha/RXR induces hSR-BI-promoter activity in hepatoma cells and preadipocytes. Biochem. Biophys. Res. Commun. 2002, 299, 916–923. [Google Scholar] [CrossRef]
- Yu, L.; Cao, G.; Repa, J.; Stangl, H. Sterol regulation of scavenger receptor class B type I in macrophages. J. Lipid Res. 2004, 45, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Arai, T.; Ji, Y.; Rinninger, F.; Tall, A.R. Liver-specific overexpression of scavenger receptor BI decreases levels of very low density lipoprotein ApoB, low density lipoprotein ApoB, and high density lipoprotein in transgenic mice. J. Biol. Chem. 1998, 273, 32920–32926. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, I.M.; Svenson, K.L.; VanOrman, S.K.; Langle, Y.; Millar, J.S.; Paigen, B.; Rader, D.J. Novel ENU-induced point mutation in scavenger receptor class B, member 1, results in liver specific loss of SCARB1 protein. PLoS ONE 2009, 4, e6521. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | HepG2 (LPDS) | HepG2 (HLPS) | TPH-1 (LPDS) | BRIN-BD11 (LPDS) |
|---|---|---|---|---|
| Butyrate | ↓ **** | ↓ ** | ↓ ** | ↑ * |
| Atorvastatin | ↓ *** | ↑ * | ↓ ** | ↓ (p = 0.17) |
| Target | Type | Product Code | Company |
|---|---|---|---|
| GAPDH | Mouse monoclonal | ab8245 | abcam |
| HMGCR | Rabbit monoclonal | ab174830 | abcam |
| LDLR | Rabbit polyclonal | ab30532 | abcam |
| SREBP-2 | Rabbit polyclonal | ab28482 | abcam |
| APO-A1 | Mouse monoclonal | sc376818 | Santa Cruz Biotechnology |
| APO-B | Mouse monoclonal | sc-13538 | Santa Cruz Biotechnology |
| ABCA1 | Mouse monoclonal | ab66217 | abcam |
| ABCG1 | Rabbit monoclonal | ab52617 | abcam |
| SRB1 | Rabbit monoclonal | ab52629 | abcam |
| Caveolin-1 | Rabbit polyclonal | ab18199 | abcam |
| Goat Anti-Mouse | IgG H&L (HRP) | ab6789 | abcam |
| Goat Anti-Rabbit | IgG H&L (HRP) | ab6721 | abcam |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bridgeman, S.; Woo, H.C.; Newsholme, P.; Mamotte, C. Butyrate Lowers Cellular Cholesterol through HDAC Inhibition and Impaired SREBP-2 Signalling. Int. J. Mol. Sci. 2022, 23, 15506. https://doi.org/10.3390/ijms232415506
Bridgeman S, Woo HC, Newsholme P, Mamotte C. Butyrate Lowers Cellular Cholesterol through HDAC Inhibition and Impaired SREBP-2 Signalling. International Journal of Molecular Sciences. 2022; 23(24):15506. https://doi.org/10.3390/ijms232415506
Chicago/Turabian StyleBridgeman, Stephanie, Hon Chiu Woo, Philip Newsholme, and Cyril Mamotte. 2022. "Butyrate Lowers Cellular Cholesterol through HDAC Inhibition and Impaired SREBP-2 Signalling" International Journal of Molecular Sciences 23, no. 24: 15506. https://doi.org/10.3390/ijms232415506
APA StyleBridgeman, S., Woo, H. C., Newsholme, P., & Mamotte, C. (2022). Butyrate Lowers Cellular Cholesterol through HDAC Inhibition and Impaired SREBP-2 Signalling. International Journal of Molecular Sciences, 23(24), 15506. https://doi.org/10.3390/ijms232415506