
Anti-Oxidant and Anti-Inflammatory Effects of Astaxanthin on Gastrointestinal Diseases



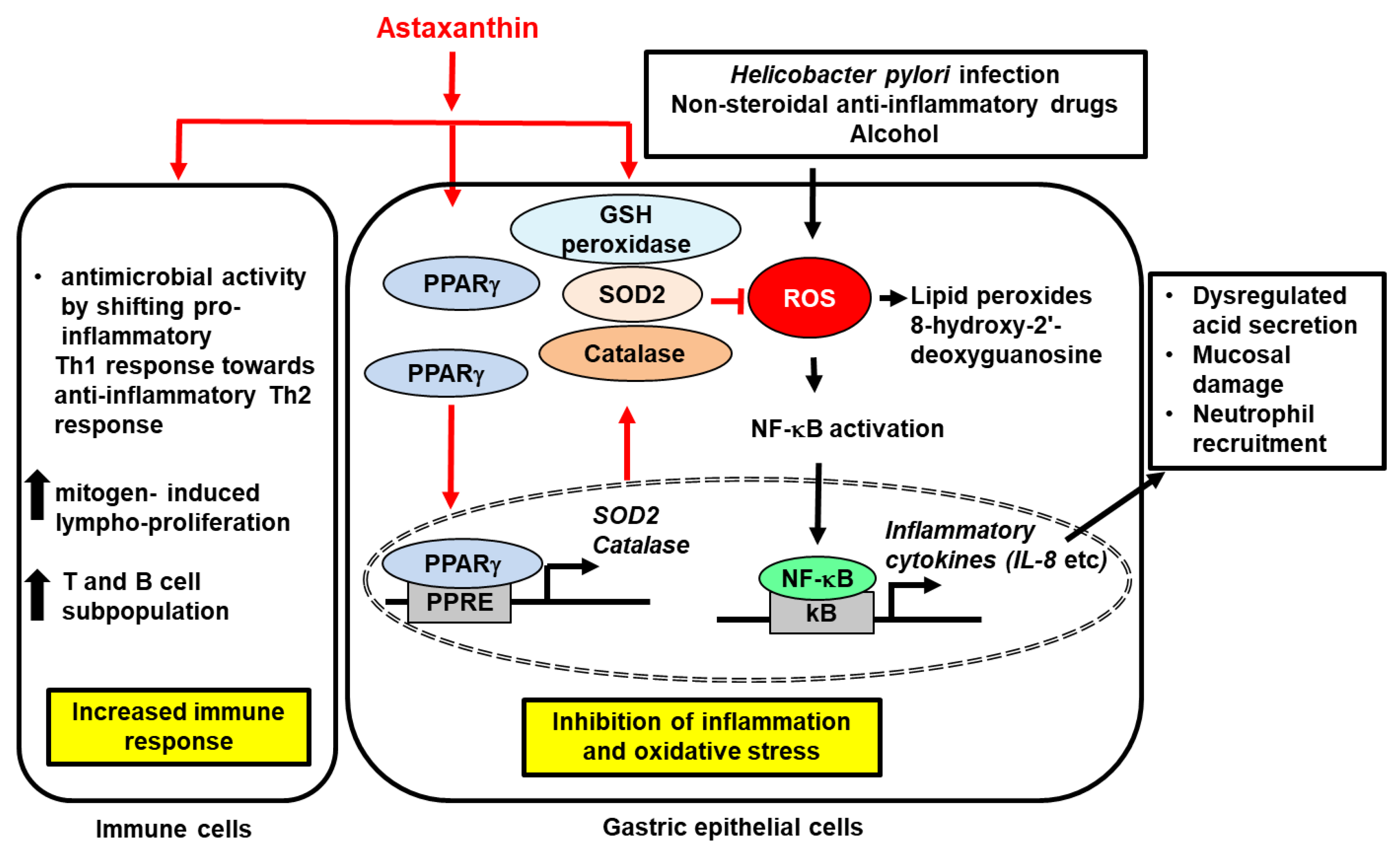{kind=link}
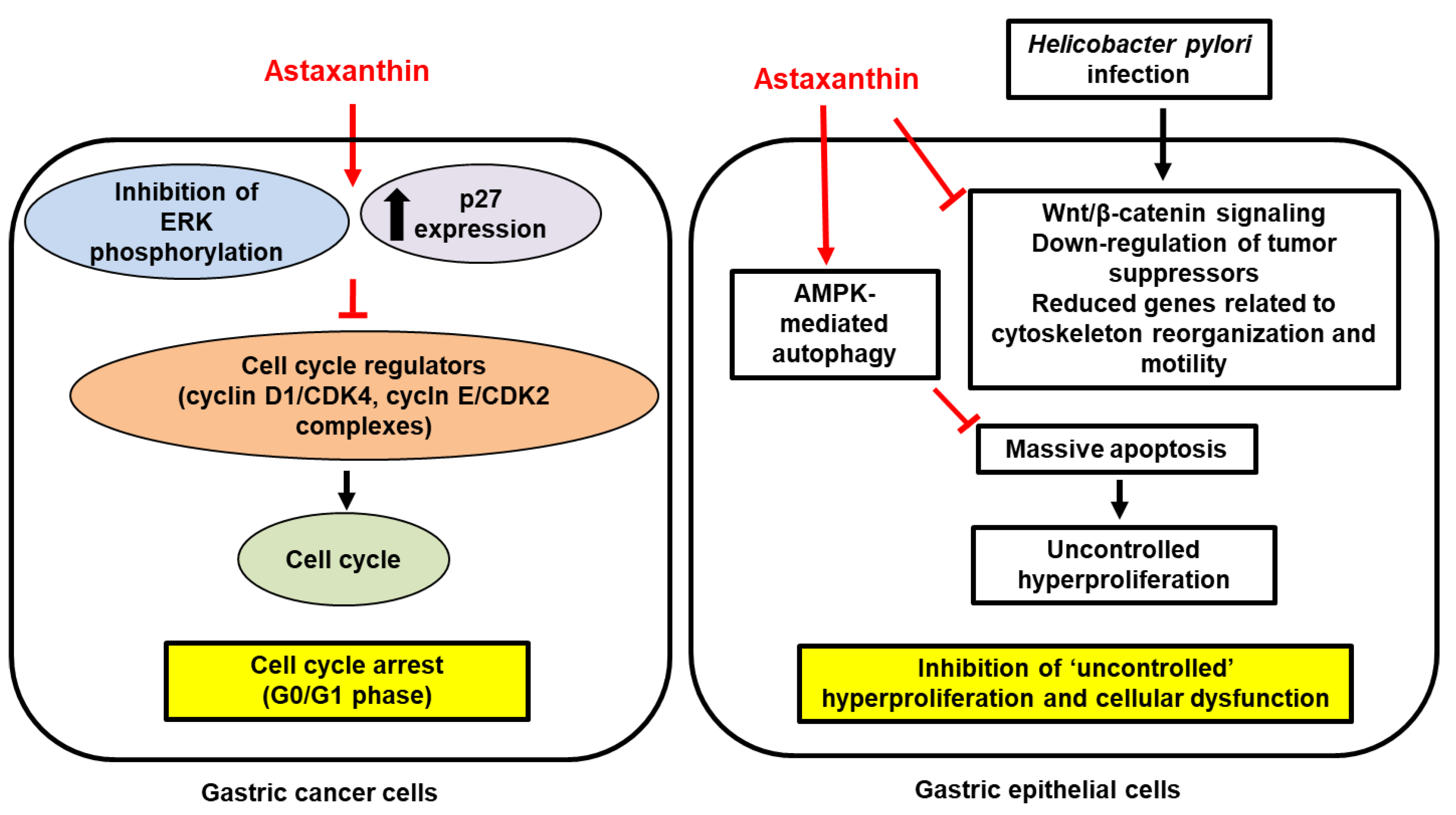{kind=link}
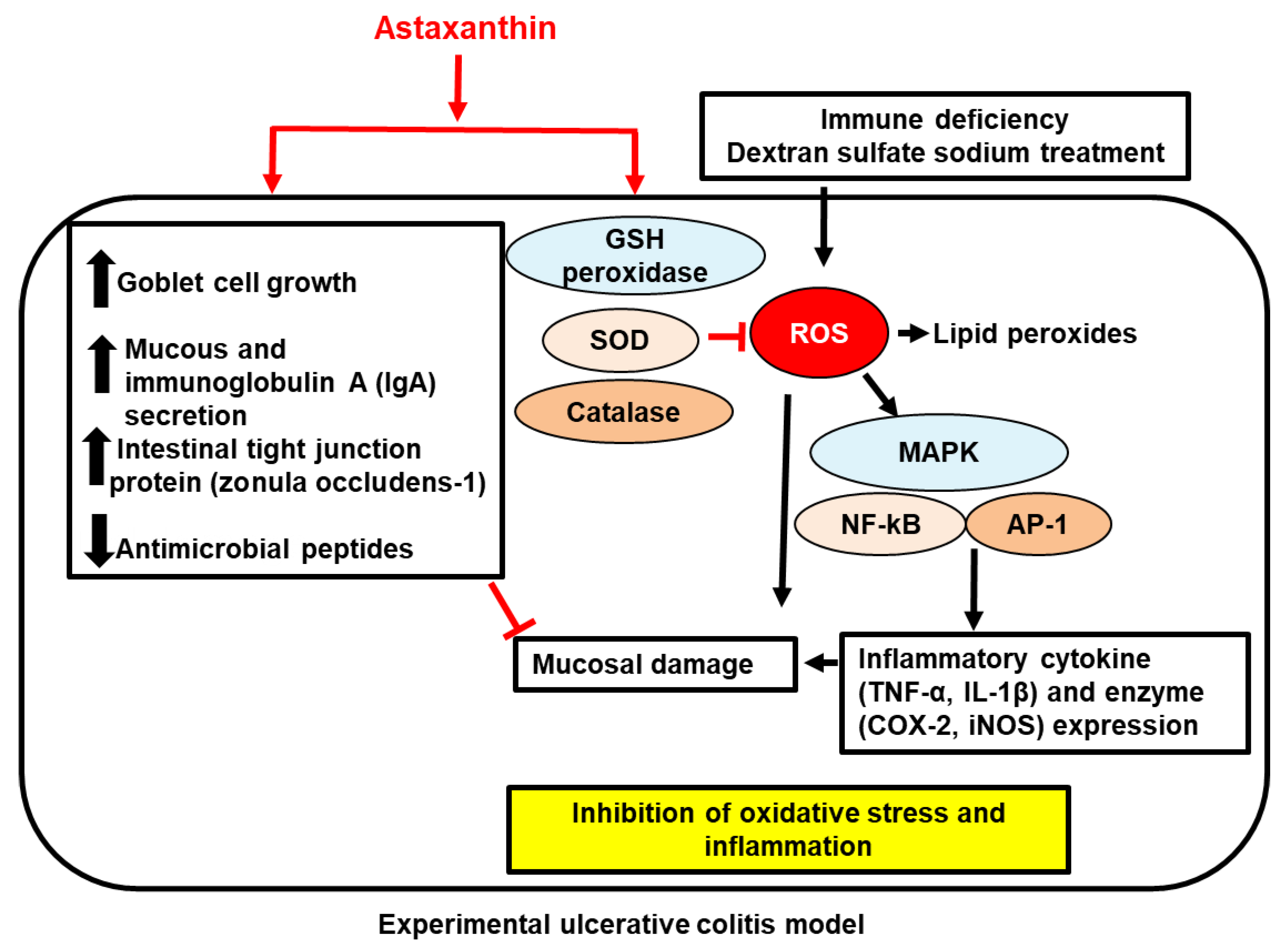{kind=link}
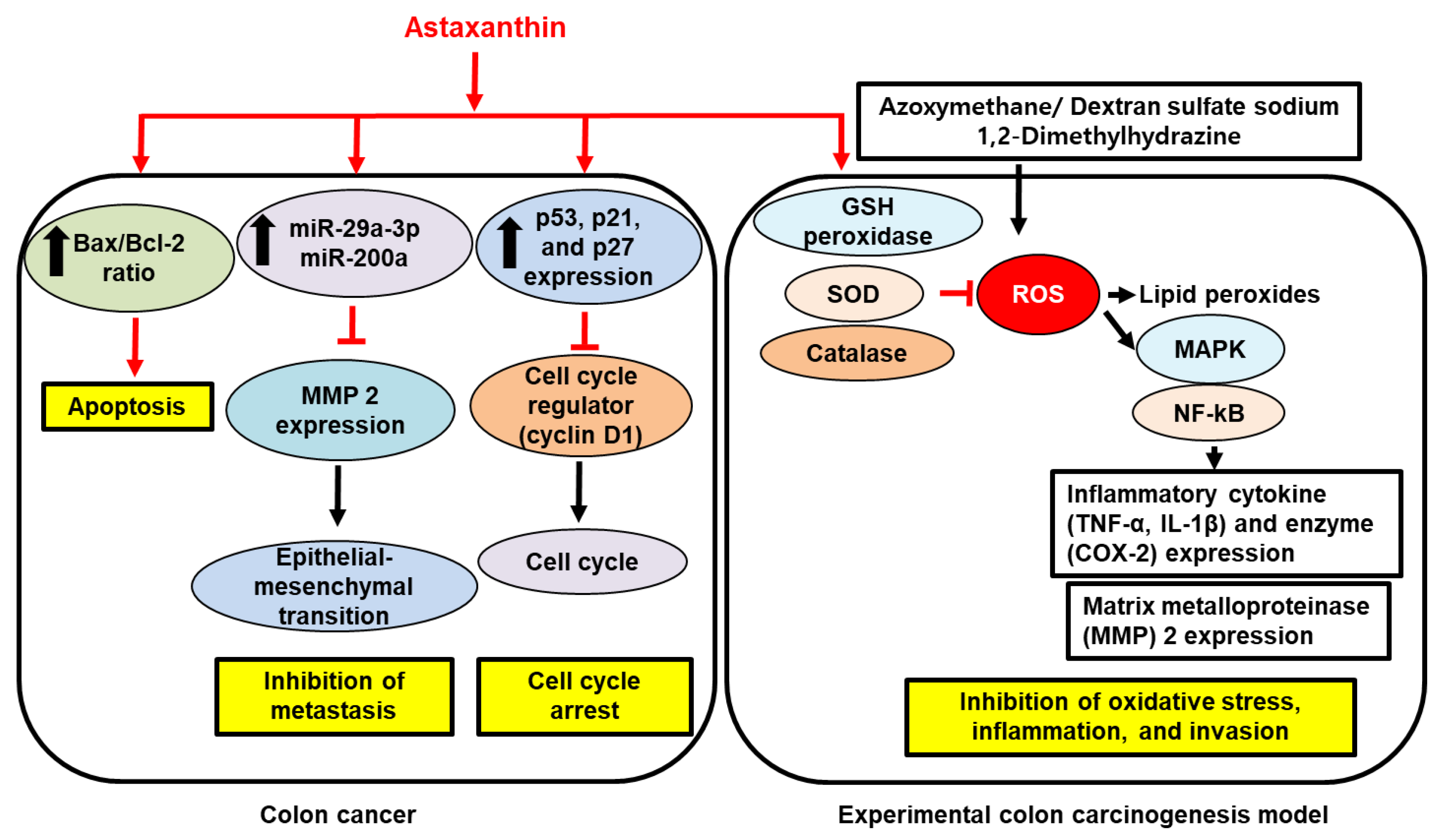{kind=link}
Abstract
1. Introduction
1.1. Anti-Oxidant and Anti-Inflammatory Mechanism of AST
1.2. Bioavailability of AST
2. Gastric Ulcer
2.1. The Effects of AST on H. pylori–Induced Gastric Ulcer
2.2. The Effects of AST on Other Gastric Ulcer Models
3. Gastric Cancer
The Effect of AST on Gastric Cancer
4. Ulcerative Colitis
The Effects of AST on Ulcerative Colitis
5. Colon Cancer
The Effects of AST on Colon Cancer
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nature Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Bayr, H. Reactive oxygen species. Critl. Care Med. 2005, 33, S498–S501. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.-H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Bondia-Pons, I.; Ryan, L.; Martinez, J.A. Oxidative stress and inflammation interactions in human obesity. J. Physiol. Biochem. 2012, 68, 701–711. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxid. Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Collins, T. Acute and chronic inflammation. In Robbins Pathologic Basis of Disease; Saunders: Philadelphia, PE, USA, 1999; pp. 83–84. [Google Scholar]
- Essick, E.E.; Sam, F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid. Med. Cell. Longev. 2010, 3, 168–177. [Google Scholar] [CrossRef]
- Soybel, D.I. Anatomy and physiology of the stomach. Surg. Clin. N. Am. 2005, 85, 875–894. [Google Scholar] [CrossRef]
- Suzuki, H.; Nishizawa, T.; Tsugawa, H.; Mogami, S.; Hibi, T. Roles of oxidative stress in stomach disorders. J. Clin. Biochem. Nutr. 2012, 50, 35–39. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Cordaro, M.; Fusco, R.; Peritore, A.F.; Siracusa, R.; Genovese, T.; D’Amico, R.; Impellizzeri, D.; Di Paola, R.; Cuzzocrea, S. Protective effect of snail secretion filtrate against ethanol-induced gastric ulcer in mice. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Jena, G.; Trivedi, P.P.; Sandala, B. Oxidative stress in ulcerative colitis: An old concept but a new concern. Free. Radic. Res. 2012, 46, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Perše, M. Oxidative stress in the pathogenesis of colorectal cancer: Cause or consequence? BioMed Res. Int. 2013, 2013, 725710. [Google Scholar] [CrossRef] [PubMed]
- Sørbye, H.; Svanes, K. The role of blood flow in gastric mucosal defence, damage and healing. Dig. Dis. 1994, 12, 305–317. [Google Scholar] [CrossRef]
- Warzecha, Z.; Dembiński, A.; Brzozowski, T.; Ceranowicz, P.; Pajdo, R.; Niemiec, J.; Drozdowicz, D.; Mitis-Musioł, M.; Konturek, S.J. Gastroprotective effect of histamine and acid secretion on ammonia-induced gastric lesions in rats. Scand J. Gastroenterol. 2000, 35, 916–924. [Google Scholar] [CrossRef]
- Warzecha, Z.; Dembiński, A.; Brzozowski, T.; Ceranowicz, P.; Dembiński, M.; Stachura, J.; Konturek, S.J. Histamine in stress ulcer prophylaxis in rats. J. Physiol. Pharmacol. 2001, 52, 407–421. [Google Scholar]
- Konarska, K.; Cieszkowski, J.; Warzecha, Z.; Ceranowicz, P.; Chmura, A.; Kuśnierz-Cabala, B.; Gałązka, K.; Kowalczyk, P.I.; Miskiewicz, A.; Konturek, T.J.; et al. Treatment with obestatin-a ghrelin gene-encoded peptide-reduces the severity of experimental colitis evoked by trinitrobenzene sulfonic acid. Int. J. Mol. Sci. 2018, 19, 1643. [Google Scholar] [CrossRef]
- Dembiński, A.; Warzecha, Z.; Ceranowicz, P.; Dembiński, M.; Cieszkowski, J.; Gosiewski, T.; Bulanda, M.; Kuśnierz-Cabala, B.; Gałązka, K.; Konturek, P.C. Synergic interaction of Rifaximin and Mutaflor (Escherichia coli Nissle 1917) in the treatment of acetic acid-induced colitis in rats. Gastroenterol. Res. Pract. 2016, 2016, 3126280. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [PubMed]
- Akki, R.; Raghay, K.; Errami, M. Potentiality of ghrelin as antioxidant and protective agent. Redox Rep. 2021, 26, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ginter, G.; Ceranowicz, P.; Warzecha, Z. Protective and healing effects of ghrelin and risk of cancer in the digestive system. Int. J. Mol. Sci. 2021, 22, 10571. [Google Scholar] [CrossRef]
- Warzecha, Z.; Ceranowicz, P.; Dembinski, M.; Cieszkowski, J.; Ginter, G.; Ptak-Belowska, A.; Dembinski, A. Involvement of cyclooxygenase-1 and cyclooxygenase-2 activity in the therapeutic effect of ghrelin in the course of ethanol-induced gastric ulcers in rats. J. Physiol. Pharmacol. 2014, 65, 95–106. [Google Scholar]
- Sibilia, V.; Rindi, G.; Pagani, F.; Rapetti, D.; Locatelli, V.; Torsello, A.; Campanini, N.; Deghenghi, R.; Netti, C. Ghrelin protects against ethanol-induced gastric ulcers in rats: Studies on the mechanisms of action. Endocrinology 2003, 144, 353–359. [Google Scholar] [CrossRef]
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A.; Sendur, R.; Cieszkowski, J.; Ceranowicz, D.; Pawlik, W.W.; Kuwahara, A.; Kato, I.; Konturek, P.C. Treatment with ghrelin accelerates the healing of acetic acid-induced gastric and duodenal ulcers in rats. J. Physiol. Pharmacol. 2009, 60, 87–98. [Google Scholar]
- Stempniewicz, A.; Ceranowicz, P.; Warzecha, Z. Potential therapeutic effects of gut hormones, ghrelin and obestatin in oral mucositis. Int. J. Mol. Sci. 2019, 20, 1534. [Google Scholar] [CrossRef]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Tomaszewska, R.; Stachura, J.; Konturek, S.J.; Konturek, P.C. Ghrelin attenuates the development of acute pancreatitis in rat. J. Physiol. Pharmacol. 2003, 54, 561–573. [Google Scholar]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Cieszkowski, J.; Pawlik, W.W.; Tomaszewska, P.; Kuśnierz-Cabala, B.; Naskalski, J.W.; Kuwahara, A.; Kato, I. Role of growth hormone and insulin-like growth factor-1 in the protective effect of ghrelin in ischemia/reperfusion-induced acute pancreatitis. Growth Horm. IGF Res. 2006, 16, 348–356. [Google Scholar] [CrossRef]
- Maduzia, D.; Matuszyk, A.; Ceranowicz, D.; Warzecha, Z.; Ceranowicz, P.; Fyderek, K.; Galazka, K.; Dembinski, A. The influence of pretreatment with ghrelin on the development of acetic-acid-induced colitis in rats. J. Physiol. Pharmacol. 2015, 66, 875–885. [Google Scholar]
- Matuszyk, A.; Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Gałązka, K.; Bonior, J.; Jaworek, J.; Bartuś, K.; Gil, K.; et al. Exogenous ghrelin accelerates the healing of acetic acid-induced colitis in rats. Int. J. Mol. Sci. 2016, 17, 1455. [Google Scholar] [CrossRef] [PubMed]
- 34 Bukowczan, J.; Warzecha, Z.; Ceranowicz, P.; Kusnierz-Cabala, B.; Tomaszewska, R.; Dembinski, A. Therapeutic effect of ghrelin in the course of ischemia/reperfusion-induced acute pancreatitis. Curr. Pharm. Des. 2015, 21, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, Z.; Ceranowicz, D.; Dembiński, A.; Ceranowicz, P.; Cieszkowski, J.; Kuwahara, A.; Kato, I.; Dembiński, M.; Konturek, P.C. Ghrelin accelerates the healing of cysteamine-induced duodenal ulcers in rats. Med. Sci. Monit. 2012, 18, BR181–BR187. [Google Scholar] [CrossRef] [PubMed]
- Matuszyk, A.; Ceranowicz, D.; Warzecha, Z.; Ceranowicz, P.; Fyderek, K.; Gałązka, K.; Cieszkowski, J.; Bonior, J.; Jaworek, J.; Pihut, M.; et al. The influence of ghrelin on the development of dextran sodium sulfate-induced colitis in rats. BioMed Res. Int. 2015, 2015, 718314. [Google Scholar] [CrossRef]
- Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Kuśnierz-Cabala, B.; Bonior, J.; Jaworek, J.; Ambroży, T.; Gil, K.; Olszanecki, R.; et al. Essential role of growth hormone and IGF-1 in therapeutic effect of ghrelin in the course of acetic acid-induced colitis. Int. J. Mol. Sci. 2017, 18, 1118. [Google Scholar] [CrossRef]
- Matuszyk, A.; Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Gałązka, K.; Bonior, J.; Jaworek, J.; Konturek, P.C.; Gil, K.; Dembinski, A. Pretreatment with obestatin inhibits the development of acetic acid-induced colitis in rats. Arch. Med. Sci. 2018, 14, 920–929. [Google Scholar] [CrossRef]
- Al-Bulishi, M.S.M.; Changhu, X.; Tang, Q.-J. Health aspects of astaxanthin: A review. Canad. J. Clin. Nutr. 2015, 3, 71–78. [Google Scholar] [CrossRef]
- Ushakumari, U.N.; Ramanujan, R. Isolation of astaxanthin from marine yeast and study of its pharmacological activity. Int. Curr. Pharm. J. 2013, 2, 67–69. [Google Scholar] [CrossRef]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Bennedsen, M.; Wang, X.; Willén, R.; Wadström, T.; Andersen, L.P. Treatment of H. pylori infected mice with antioxidant astaxanthin reduces gastric inflammation, bacterial load and modulates cytokine release by splenocytes. Immunol. Lett. 2000, 70, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Hosokawa, M.; Mikami, N.; Miyashita, K.; Tanaka, T. Dietary astaxanthin inhibits colitis and colitis-associated colon carcinogenesis in mice via modulation of the inflammatory cytokines. Chem. Biol. Interact. 2011, 193, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Naguib, Y.M. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.R.; Baskaran, V.; Sarada, R.; Ravishankar, G.A. In vivo bioavailability and antioxidant activity of carotenoids from microalgal biomass—A repeated dose study. Food Res. Int. 2013, 54, 711–717. [Google Scholar]
- Stahl, W.; Sies, H. Antioxidant activity of carotenoids. Mol. Asp. Med. 2003, 24, 345–351. [Google Scholar] [CrossRef]
- McNulty, H.; Jacob, R.F.; Mason, R.P. Biologic activity of carotenoids related to distinct membrane physicochemical interactions. Am. J. Cardiol. 2008, 101, S20–S29. [Google Scholar] [CrossRef]
- Goto, S.; Kogure, K.; Abe, K.; Kimata, Y.; Kitahama, K.; Yamashita, E.; Terada, H. Efficient radical trapping at the surface and inside the phospholipid membrane is responsible for highly potent antiperoxidative activity of the carotenoid astaxanthin. Bioch. Biophys. Acta (BBA)-Biomembr. 2001, 1512, 251–258. [Google Scholar] [CrossRef]
- Pereira, C.P.M.; Souza, A.C.R.; Vasconcelos, A.R.; Prado, P.S. Antioxidant and anti-inflammatory mechanisms of action of astaxanthin in cardiovascular diseases. Int. J. Mol. Med. 2021, 47, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.P.; Pinto, E.; Colepicolo, P.; Pedersén, M. Astaxanthin and peridinin inhibit oxidative damage in Fe2+-loaded liposomes: Scavenging oxyradicals or changing membrane permeability? Biochem. Biophys. Res. Commun. 2001, 288, 225–232. [Google Scholar] [CrossRef]
- McNulty, H.P.; Byun, J.; Lockwood, S.F.; Jacob, R.F.; Mason, R.P. Differential effects of carotenoids on lipid peroxidation due to membrane interactions: X-ray diffraction analysis. Bioch. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Pashkow, F.J.; Watumull, D.G.; Campbell, C.L. Astaxanthin: A novel potential treatment for oxidative stress and inflammation in cardiovascular disease. Am. J. Cardiol. 2008, 101, S58–S68. [Google Scholar] [CrossRef] [PubMed]
- May, J.M. Is ascorbic acid an antioxidant for the plasma membrane? FASEB J. 1999, 13, 995–1006. [Google Scholar] [CrossRef]
- Dose, J.; Matsugo, S.; Yokokawa, H.; Koshida, Y.; Okazaki, S.; Seidel, U.; Eggersdorfer, M.; Rimbach, G.; Esatbeyoglu, T. Free radical scavenging and cellular antioxidant properties of astaxanthin. Int. J. Mol. Sci. 2016, 17, 103. [Google Scholar] [CrossRef] [PubMed]
- Kohandel, Z.; Farkhondeh, T.; Aschner, M.; Pourbagher-Shahri, A.M.; Samarghandian, S. Anti-inflammatory action of astaxanthin and its use in the treatment of various diseases. Biomed. Pharmacother. 2022, 145, 112179. [Google Scholar] [CrossRef]
- Speranza, L.; Pesce, M.; Patruno, A.; Franceschelli, S.; De Lutiis, M.A.; Grilli, A.; Felaco, M. Astaxanthin treatment reduced oxidative induced pro-inflammatory cytokines secretion in U937: SHP-1 as a novel biological target. Mar. Drugs 2012, 10, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ohgami, K.; Shiratori, K.; Jin, X.-H.; Ilieva, I.; Koyama, Y.; Yazawa, K.; Yoshida, K.; Kase, S.; Ohno, S. Suppressive effects of astaxanthin against rat endotoxin-induced uveitis by inhibiting the NF-κB signaling pathway. Exp. Eye Res. 2006, 82, 275–281. [Google Scholar] [CrossRef]
- Li, J.; Wang, F.; Xia, Y.; Dai, W.; Chen, K.; Li, S.; Liu, T.; Zheng, Y.; Wang, J.; Lu, W. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar. Drugs 2015, 13, 3368–3387. [Google Scholar] [CrossRef]
- Yang, X.; Guo, A.-L.; Pang, Y.-P.; Cheng, X.-J.; Xu, T.; Li, X.-R.; Liu, J.; Zhang, Y.-Y.; Liu, Y. Astaxanthin attenuates environmental tobacco smoke-induced cognitive deficits: A critical role of p38 MAPK. Mar. Drugs 2019, 17, 24. [Google Scholar] [CrossRef]
- Liu, G.; Shi, Y.; Peng, X.; Liu, H.; Peng, Y.; He, L. Astaxanthin attenuates adriamycin-induced focal segmental glomerulosclerosis. Pharmacology 2015, 95, 193–200. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, J.; Yin, W.; Ding, X. Astaxanthin improves cognitive deficits from oxidative stress, nitric oxide synthase and inflammation through upregulation of PI3K/Akt in diabetes rat. Int. J. Clin. Exp. Pathol. 2015, 8, 6083. [Google Scholar] [PubMed]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Ambati, R.R.; Siew Moi, P.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Sy, C.; Gleize, B.; Dangles, O.; Landrier, J.F.; Veyrat, C.C.; Borel, P. Effects of physicochemical properties of carotenoids on their bioaccessibility, intestinal cell uptake, and blood and tissue concentrations. Mol. Nutr. Food Res. 2012, 56, 1385–1397. [Google Scholar] [CrossRef]
- Reboul, E.; Abou, L.; Mikail, C.; Ghiringhelli, O.; André, M.; Portugal, H.; Jourdheuil-Rahmani, D.; Amiot, M.-J.; Lairon, D.; Borel, P. Lutein transport by Caco-2 TC-7 cells occurs partly by a facilitated process involving the scavenger receptor class B type I (SR-BI). Biochem. J. 2005, 387, 455–461. [Google Scholar] [CrossRef]
- Kiefer, C.; Sumser, E.; Wernet, M.F.; Von Lintig, J. A class B scavenger receptor mediates the cellular uptake of carotenoids in Drosophila. Proc. Nat. Acad. Sci. USA 2002, 99, 10581–10586. [Google Scholar] [CrossRef]
- During, A.; Dawson, H.D.; Harrison, E.H. Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is downregulated in Caco-2 cells treated with ezetimibe. J. Nutr. 2005, 135, 2305–2312. [Google Scholar] [CrossRef]
- Zhou, Q.; Xu, J.; Yang, L.; Gu, C.; Xue, C. Thermal stability and oral absorbability of astaxanthin esters from Haematococcus pluvialis in Balb/c mice. J. Sci. Food Agric. 2019, 99, 3662–3671. [Google Scholar] [CrossRef]
- Petri, D.; Lundebye, A.-K. Tissue distribution of astaxanthin in rats following exposure to graded levels in the feed. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 145, 202–209. [Google Scholar] [CrossRef]
- Singh, G.S.; Ismail, M.; Zulkefli, N.; Affandi, M.; Meor, M. Tissue distribution of astaxanthin formulation in rats. Curr. Nutr. Food Sci. 2018, 14, 329–334. [Google Scholar] [CrossRef]
- Østerlie, M.; Bjerkeng, B.; Liaaen-Jensen, S. Plasma appearance and distribution of astaxanthin E/Z and R/S isomers in plasma lipoproteins of men after single dose administration of astaxanthin. J. Nutr. Biochem. 2000, 11, 482–490. [Google Scholar] [CrossRef]
- Coral-Hinostroza, G.N.; Ytrestøyl, T.; Ruyter, B.; Bjerkeng, B. Plasma appearance of unesterified astaxanthin geometrical E/Z and optical R/S isomers in men given single doses of a mixture of optical 3 and 3′ R/S isomers of astaxanthin fatty acyl diesters. Comp. Biochem. Physiol. C Toxicol. Pharmacol 2004, 139, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Chan, F.K.; McColl, K.E. Peptic ulcer disease. Lancet 2009, 374, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Lanas, A.; Chan, F.K. Peptic ulcer disease. Lancet 2017, 390, 613–624. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-κB and AP-1 connection: Mechanism of NF-κB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Helicobacter pylori: A ROS-inducing bacterial species in the stomach. Inflamm. Res. 2010, 59, 997–1003. [Google Scholar] [CrossRef]
- Iwamoto, T.; Hosoda, K.; Hirano, R.; Kurata, H.; Matsumoto, A.; Miki, W.; Kamiyama, M.; Itakura, H.; Yamamoto, S.; Kondo, K. Inhibition of low-density lipoprotein oxidation by astaxanthin. J. Atheroscler. Thromb. 2000, 7, 216–222. [Google Scholar] [CrossRef]
- Davies, G.; Simmonds, N.; Stevens, T.; Sheaff, M.; Banatvala, N.; Laurenson, I.; Blake, D.; Rampton, D. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 1994, 35, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Khoi, P.N.; Xia, Y.; Park, J.S.; Joo, Y.E.; Kim, K.K.; Choi, S.Y.; Do Jung, Y. Helicobacter pylori and interleukin-8 in gastric cancer. World J. Gastroenterol. 2013, 19, 8192. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Jones, K.R.; Olsen, C.H.; Joo, Y.M.; Yoo, Y.-J.; Chung, I.-S.; Cha, J.-H.; Merrell, D.S. Epidemiological link between gastric disease and polymorphisms in VacA and CagA. J. Clin. Microbiol. 2010, 48, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Nat. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef]
- Aihara, M.; Tsuchimoto, D.; Takizawa, H.; Azuma, A.; Wakebe, H.; Ohmoto, Y.; Imagawa, K.; Kikuchi, M.; Mukaida, N.; Matsushima, K. Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect Immun. 1997, 65, 3218–3224. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kikuchi, S.; El–Zimaity, H.M.; Gutierrez, O.; Osato, M.S.; Graham, D.Y. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 2002, 123, 414–424. [Google Scholar] [CrossRef]
- Crabtree, J.; Covacci, A.; Farmery, S.; Xiang, Z.; Tompkins, D.; Perry, S.; Lindley, I.; Rappuoli, R. Helicobacter pylori induced interleukin-8 expression in gastric epithelial cells is associated with CagA positive phenotype. J. Clin. Pathol. 1995, 48, 41–45. [Google Scholar] [CrossRef]
- Epplein, M.; Xiang, Y.-B.; Cai, Q.; Peek, R.M.; Li, H.; Correa, P.; Gao, J.; Wu, J.; Michel, A.; Pawlita, M. Circulating cytokines and gastric cancer risk. Cancer Causes Control 2013, 24, 2245–2250. [Google Scholar] [CrossRef]
- Ma, J.; Wu, D.; Hu, X.; Li, J.; Cao, M.; Dong, W. Associations between cytokine gene polymorphisms and susceptibility to Helicobacter pylori infection and Helicobacter pylori related gastric cancer, peptic ulcer disease: A meta-analysis. PLoS ONE 2017, 12, e0176463. [Google Scholar] [CrossRef]
- Atherton, J.C.; Blaser, M.J. Coadaptation of Helicobacter pylori and humans: Ancient history, modern implications. J. Clin. Investig. 2009, 119, 2475–2487. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin inhibits mitochondrial dysfunction and interleukin-8 expression in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2018, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin prevents decreases in superoxide dismutase 2 level and superoxide dismutase activity in helicobacter pylori-infected gastric epithelial cells. J. Cancer Preven. 2019, 24, 54. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Lim, J.W.; Kim, H. Astaxanthin inhibits Helicobacter pylori-induced inflammatory and oncogenic responses in gastric mucosal tissues of mice. J. Cancer Preven. 2020, 25, 244. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-J.; Du, Y.; Zhao, X.; Ma, L.-Y.; Cao, G.-W. Inflammation-related factors predicting prognosis of gastric cancer. World J. Gastroenterol. 2014, 20, 4586. [Google Scholar] [CrossRef]
- Knutson, K.L.; Disis, M. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef]
- Whary, M.; Morgan, T.; Dangler, C.; Gaudes, K.; Taylor, N.; Fox, J. Chronic active hepatitis induced by Helicobacter hepaticus in the A/JCr mouse is associated with a Th1 cell-mediated immune response. Infect Immun. 1998, 66, 3142–3148. [Google Scholar] [CrossRef]
- Davinelli, S.; Melvang, H.M.; Andersen, L.P.; Scapagnini, G.; Nielsen, M.E. Astaxanthin from shrimp cephalothorax stimulates the immune response by enhancing IFN-γ, IL-10, and IL-2 secretion in splenocytes of Helicobacter pylori-infected mice. Mar. Drugs 2019, 17, 382. [Google Scholar] [CrossRef]
- Andersen, L.P.; Holck, S.; Kupcinskas, L.; Kiudelis, G.; Jonaitis, L.; Janciauskas, D.; Permin, H.; Wadström, T. Gastric inflammatory markers and interleukins in patients with functional dyspepsia treated with astaxanthin. FEMS Immunol. Med. Microbiol. 2007, 50, 244–248. [Google Scholar] [CrossRef]
- Park, J.S.; Chyun, J.H.; Kim, Y.K.; Line, L.L.; Chew, B.P. Astaxanthin decreased oxidative stress and inflammation and enhanced immune response in humans. Nutr. Metabol. 2010, 7, 1–10. [Google Scholar] [CrossRef]
- Cashman, J.N. The mechanisms of action of NSAIDs in analgesia. Drugs 1996, 52, 13–23. [Google Scholar] [CrossRef]
- Tenenbaum, J. The epidemiology of nonsteroidal anti-inflammatory drugs. Canad. J. Gastroenterol. 1999, 13, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, W.; Gilman, S.; Datko, L.; Copenhaver, T.; Carlson, R. Interaction studies of tilomisole, aspirin, and naproxen in acute and chronic inflammation with assessment of gastrointestinal irritancy in the rat. Agents Actions 1992, 36, 99–106. [Google Scholar] [CrossRef]
- Graham, D.Y.; Malaty, H.M. Alendronate and naproxen are synergistic for development of gastric ulcers. Arch Intern. Med. 2001, 161, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Kim, Y.-S.; Song, G.-G.; Park, J.-J.; Chang, H.-I. Protective effect of astaxanthin on naproxen-induced gastric antral ulceration in rats. Eur. J. Pharmacol. 2005, 514, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Choi, S.-K.; Lim, W.-J.; Chang, H.-I. Protective effect of astaxanthin produced by Xanthophyllomyces dendrorhous mutant on indomethacin-induced gastric mucosal injury in rats. J. Microbiol. Biotechnol. 2004, 14, 996–1003. [Google Scholar]
- Laksitorini, M.D.; Yathindranath, V.; Xiong, W.; Parkinson, F.E.; Thliveris, J.A.; Miller, D.W. Impact of Wnt/β-catenin signaling on ethanol-induced changes in brain endothelial cell permeability. J. Neurochem. 2021, 157, 1118–1137. [Google Scholar] [CrossRef] [PubMed]
- Brătucu, M.N.; Prunoiu, V.-M.; Strâmbu, V.; Brătucu, E.; Răvaş, M.-M.; Simion, L.; Petre, R. Unusual Complicated Gastric Ulcers. Medicina 2021, 57, 1345. [Google Scholar] [CrossRef]
- Kim, J.-H.; Choi, S.-K.; Choi, S.-Y.; Kim, H.-K.; Chang, H.-I. Suppressive effect of astaxanthin isolated from the Xanthophyllomyces dendrorhous mutant on ethanol-induced gastric mucosal injury in rats. Biosci. Biotechnol. Biochem. 2005, 69, 1300–1305. [Google Scholar] [CrossRef]
- Kamath, B.S.; Srikanta, B.M.; Dharmesh, S.M.; Sarada, R.; Ravishankar, G.A. Ulcer preventive and antioxidative properties of astaxanthin from Haematococcus pluvialis. Eur. J. Pharmacol. 2008, 590, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Oyagi, A.; Takahira, D.; Tsuruma, K.; Shimazawa, M.; Ishibashi, T.; Hara, H. Protective effects of astaxanthin from Paracoccus carotinifaciens on murine gastric ulcer models. Phytother. Res. 2012, 26, 1126–1132. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Minenaka, Y.; Ichimura, M.; Tatsumi, K.; Nadamoto, T.; Urabe, K. Effects of astaxanthin and vitamin C on the prevention of gastric ulcerations in stressed rats. J. Nutr. Sci. Vitaminol. 2005, 51, 135–141. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Lian, S.; Li, S.; Zhu, J.; Xia, Y.; Do Jung, Y. Nicotine stimulates IL-8 expression via ROS/NF-κB and ROS/MAPK/AP-1 axis in human gastric cancer cells. Toxicology 2022, 466, 153062. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, S.; Mahadevan, N.; Balakumar, P. Possible involvement of PPARγ-associated eNOS signaling activation in rosuvastatin-mediated prevention of nicotine-induced experimental vascular endothelial abnormalities. Mol. Cell. Biochem. 2013, 374, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Hamraz, M.; Salimi, M.; Esfahani, M.; Sedaghati, B.; Aslani, H.R.; Bazarchi, A. Nicotine effect on P-ERK, COX-2, PGE2 and VEGF expression in oral squamous cancer. J. Biotechnol. 2010, 150, 442. [Google Scholar] [CrossRef]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef]
- Ma, K.; Baloch, Z.; He, T.-T.; Xia, X. Alcohol consumption and gastric cancer risk: A meta-analysis. Med. Sci. Monit. 2017, 23, 238. [Google Scholar] [CrossRef]
- Farinati, F.; Cardin, R.; Cassaro, M.; Bortolami, M.; Nitti, D.; Tieppo, C.; Zaninotto, G.; Rugge, M. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: A morphological, biological and molecular pathway. Eur. J. Cancer Prev. 2008, 17, 195–200. [Google Scholar] [CrossRef]
- Ladelfa, M.F.; Toledo, M.F.; Laiseca, J.E.; Monte, M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid. Redox Signal. 2011, 15, 1749–1761. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, G.Y.; Cao, J.L.; Li, Y.N.; Xue, H.; Wu, H.T.; Jin, C.H. Peimine-induced apoptosis and inhibition of migration by regulating reactive oxygen species-mediated MAPK/STAT3/NF-κB and Wnt/β-catenin signaling pathways in gastric cancer MKN-45 cells. Drug Develop. Res. 2022, 83, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhou, Y.; Wang, W.; Li, J.; Xie, G.; Zhao, Y.; Xu, D.; Shen, L. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013, 4, e794. [Google Scholar] [CrossRef] [PubMed]
- Hesari, A.; Ghasemi, F.; Cicero, A.F.; Mohajeri, M.; Rezaei, O.; Hayat, S.M.G.; Sahebkar, A. Berberine: A potential adjunct for the treatment of gastrointestinal cancers? J. Cell Biochem. 2018, 119, 9655–9663. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, J.-J.; Lee, B.J.; Joo, M.K.; Chun, H.J.; Lee, S.W.; Bak, Y.-T. Astaxanthin inhibits proliferation of human gastric cancer cell lines by interrupting cell cycle progression. Gut Liver 2016, 10, 369. [Google Scholar] [CrossRef] [PubMed]
- McCall, B.; McPartland, C.K.; Moore, R.; Frank-Kamenetskii, A.; Booth, B.W. Effects of astaxanthin on the proliferation and migration of breast cancer cells in vitro. Antioxid 2018, 7, 135. [Google Scholar] [CrossRef]
- Liu, X.; Song, M.; Gao, Z.; Cai, X.; Dixon, W.; Chen, X.; Cao, Y.; Xiao, H. Stereoisomers of astaxanthin inhibit human colon cancer cell growth by inducing G2/M cell cycle arrest and apoptosis. J. Agricul. Food Chem. 2016, 64, 7750–7759. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Transcriptome Analysis of the Inhibitory Effect of Astaxanthin on Helicobacter pylori-Induced Gastric Carcinoma Cell Motility. Mar. Drugs 2020, 18, 365. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Inhibitory Effect of Astaxanthin on Gene Expression Changes in Helicobacter pylori-Infected Human Gastric Epithelial Cells. Nutrients 2021, 13, 4281. [Google Scholar] [CrossRef]
- Moss, S.; Calam, J.; Agarwal, B.; Wang, S.; Holt, P. Induction of gastric epithelial apoptosis by Helicobacter pylori. Gut 1996, 38, 498–501. [Google Scholar] [CrossRef]
- Wang, H.; Liu, M.; Zeng, X.; Zheng, Y.; Wang, Y.; Zhou, Y. Cell death affecting the progression of gastric cancer. Cell Death Discov. 2022, 8, 377. [Google Scholar] [CrossRef]
- Zhao, H.; Lvauthor, G. Compound 13, an alpha1-selective small molecule activator of AMPK, inhibits Helicobacter pylori-induced oxidative stresses and gastric epithelial cell apoptosis. Biochem. Biophys. Res. Commun. 2015, 463, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Zhu, H.; Zhou, F.; Lin, Z.; Lin, G.; Li, C. AMP-activated protein kinase activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Biochem. Biophys. Res. Commun. 2014, 453, 13–18. [Google Scholar] [CrossRef]
- Lee, H.; Lim, J.W.; Kim, H. Effect of astaxanthin on activation of autophagy and inhibition of apoptosis in Helicobacter pylori-infected gastric epithelial cell line AGS. Nutrients 2020, 12, 1750. [Google Scholar] [CrossRef]
- Langan, R.C.; Gotsch, P.B.; Krafczyk, M.A.; Skillinge, D.D. Ulcerative colitis: Diagnosis and treatment. Am. Fam. Physician 2007, 76, 1323–1330. [Google Scholar] [PubMed]
- Gajendran, M.; Loganathan, P.; Jimenez, G.; Catinella, A.P.; Ng, N.; Umapathy, C.; Ziade, N.; Hashash, J.G. A comprehensive review and update on ulcerative colitis. Dis Month 2019, 65, 100851. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, F. Pathogenesis of ulcerative colitis. Lancet 1993, 342, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Yoshikawa, T. Neutrophil-dependent oxidative stress in ulcerative colitis. J. Clin. Biochem. Nutr. 2007, 41, 18–26. [Google Scholar] [CrossRef]
- Kandhare, A.D.; Raygude, K.S.; Ghosh, P.; Ghule, A.E.; Gosavi, T.P.; Badole, S.L.; Bodhankar, S.L. Effect of hydroalcoholic extract of Hibiscus rosa sinensis Linn. leaves in experimental colitis in rats. Asian Pac. J. Trop. Biomed. 2012, 2, 337–344. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, W.; Gao, Y.; Yang, R.; Zhang, X.; Xu, J.; Tang, Q. Astaxanthin (ATX) enhances the intestinal mucosal functions in immunodeficient mice. Food Funct. 2020, 11, 3371–3381. [Google Scholar] [CrossRef]
- Nagayama, T.; Sugimoto, M.; Ikeda, S.; Kume, S. Effects of astaxanthin-enriched yeast on mucosal IgA induction in the jejunum and ileum of weanling mice. Anim. Sci. J. 2014, 85, 449–453. [Google Scholar] [CrossRef]
- Lycke, N.; Bemark, M. The regulation of gut mucosal IgA B-cell responses: Recent developments. Mucosal Immunol. 2017, 10, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Akduman, H.; Tayman, C.; Korkmaz, V.; Akduman, F.; Fettah, N.D.; Gürsoy, B.K.; Turkmenoglu, T.T.; Çağlayan, M. Astaxanthin reduces the severity of intestinal damage in a neonatal rat model of necrotizing enterocolitis. Am. J. Perinatol. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Nishida, A.; Ohno, M.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; Andoh, A. Astaxanthin, a xanthophyll carotenoid, prevents development of dextran sulphate sodium-induced murine colitis. J. Clin. Biochem. Nutr. 2019, 64, 66–72. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Y.; Wu, S.; Zheng, W.; Song, S.; Ai, C. Fabrication of astaxanthin-enriched colon-targeted alginate microspheres and its beneficial effect on dextran sulfate sodium-induced ulcerative colitis in mice. Int. J. Biol. Macromol. 2022, 205, 396–409. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, X.; Tie, S.; Li, J.; Su, W.; Tan, M. A smart cauliflower-like carrier for astaxanthin delivery to relieve colon inflammation. J. Control Release 2022, 342, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Neu, J.; Walker, W.A. Necrotizing enterocolitis. N. Engl. J. Med. 2011, 364, 255–264. [Google Scholar] [CrossRef]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Stewart, B.W.; Kleihues, P. World Cancer Report; IARC pupblication: Lyon, France, 2003. [Google Scholar]
- Hagemann, T.; Balkwill, F.; Lawrence, T. Inflammation and cancer: A double-edged sword. Cancer cell 2007, 12, 300–301. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar] [CrossRef]
- Surh, Y.-J.; Kundu, J.K.; Na, H.-K.; Lee, J.-S. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J. Nutr. 2005, 135, 2993S–3001S. [Google Scholar] [CrossRef]
- Sun, S.-Q.; Zhao, Y.-X.; Li, S.-Y.; Qiang, J.-W.; Ji, Y.-Z. Anti-tumor effects of astaxanthin by inhibition of the expression of STAT3 in prostate cancer. Mar. Drugs 2020, 18, 415. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Ahn, Y.T.; Lee, C.W.; Kim, H.; An, W.G. Astaxanthin modulates apoptotic molecules to induce death of skbr3 breast cancer cells. Mar. Drugs 2020, 18, 266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H. Multiple mechanisms of anti-cancer effects exerted by astaxanthin. Mar Drugs 2015, 13, 4310–4330. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Demir, E.A.; Aliyazicioglu, Y. Selective cytotoxic effect of Astaxanthin on human lung and colon cancer cells. KSU J. Agric Nat. 2020, 23, 1489–1494. [Google Scholar]
- Palozza, P.; Torelli, C.; Boninsegna, A.; Simone, R.; Catalano, A.; Mele, M.C.; Picci, N. Growth-inhibitory effects of the astaxanthin-rich alga Haematococcus pluvialis in human colon cancer cells. Cancer Lett. 2009, 283, 108–117. [Google Scholar] [CrossRef]
- Tanaka, T.; Kawamori, T.; Ohnishi, M.; Makita, H.; Mori, H.; Satoh, K.; Hara, A. Suppression of azoxymethane-induced rat colon carcinogenesis by dietary administration of naturally occurring xanthophylls astaxanthin and canthaxanthin during the postinitiation phase. Carcinogenesis 1995, 16, 2957–2963. [Google Scholar] [CrossRef]
- Prabhu, P.N.; Ashokkumar, P.; Sudhandiran, G. Antioxidative and antiproliferative effects of astaxanthin during the initiation stages of 1, 2-dimethyl hydrazine-induced experimental colon carcinogenesis. Fundam. Clin. Pharmacol. 2009, 23, 225–234. [Google Scholar] [CrossRef]
- Nagendraprabhu, P.; Sudhandiran, G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFkB and COX-2. Investig. New Drug 2011, 29, 207–224. [Google Scholar] [CrossRef]
- Kochi, T.; Shimizu, M.; Sumi, T.; Kubota, M.; Shirakami, Y.; Tanaka, T.; Moriwaki, H. Inhibitory effects of astaxanthin on azoxymethane-induced colonic preneoplastic lesions in C57/BL/KsJ-db/dbmice. BMC Gastroenterol. 2014, 14, 212. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Kim, Y.-M.; Hong, S. Astaxanthin suppresses the metastasis of colon cancer by inhibiting the MYC-mediated downregulation of microRNA-29a-3p and microRNA-200a. Sci. Rep. 2019, 9, 9457. [Google Scholar] [CrossRef]
- Brendler, T.; Williamson, E.M. Astaxanthin: How much is too much? A safety review. Phytother. Res. 2019, 33, 3090–3111. [Google Scholar] [CrossRef] [PubMed]
 ; increase,
; increase,  . Red arrows represent the effects of astaxanthin.
; increase, . Red arrows represent the effects of astaxanthin.
. Red arrows represent the effects of astaxanthin.
; increase, . Red arrows represent the effects of astaxanthin.
 ; increase,
; increase,  . Red arrows represent the effects of astaxanthin.
; increase, . Red arrows represent the effects of astaxanthin.
. Red arrows represent the effects of astaxanthin.
; increase, . Red arrows represent the effects of astaxanthin.
 ; increase,
; increase,  ; decrease,
; decrease,  . Red arrows represent the effects of astaxanthin.
; increase, ; decrease, . Red arrows represent the effects of astaxanthin.
. Red arrows represent the effects of astaxanthin.
; increase, ; decrease, . Red arrows represent the effects of astaxanthin.
 ; increase,
; increase,  . Red arrows represent the effects of astaxanthin.
; increase, . Red arrows represent the effects of astaxanthin.
. Red arrows represent the effects of astaxanthin.
; increase, . Red arrows represent the effects of astaxanthin.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kim, M.-H.; Kim, H. Anti-Oxidant and Anti-Inflammatory Effects of Astaxanthin on Gastrointestinal Diseases. Int. J. Mol. Sci. 2022, 23, 15471. https://doi.org/10.3390/ijms232415471
Lee J, Kim M-H, Kim H. Anti-Oxidant and Anti-Inflammatory Effects of Astaxanthin on Gastrointestinal Diseases. International Journal of Molecular Sciences. 2022; 23(24):15471. https://doi.org/10.3390/ijms232415471
Chicago/Turabian StyleLee, Jaeeun, Min-Hyun Kim, and Hyeyoung Kim. 2022. "Anti-Oxidant and Anti-Inflammatory Effects of Astaxanthin on Gastrointestinal Diseases" International Journal of Molecular Sciences 23, no. 24: 15471. https://doi.org/10.3390/ijms232415471
APA StyleLee, J., Kim, M.-H., & Kim, H. (2022). Anti-Oxidant and Anti-Inflammatory Effects of Astaxanthin on Gastrointestinal Diseases. International Journal of Molecular Sciences, 23(24), 15471. https://doi.org/10.3390/ijms232415471