
Evaluation of a Novel Oncolytic Adenovirus Silencing SYVN1

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
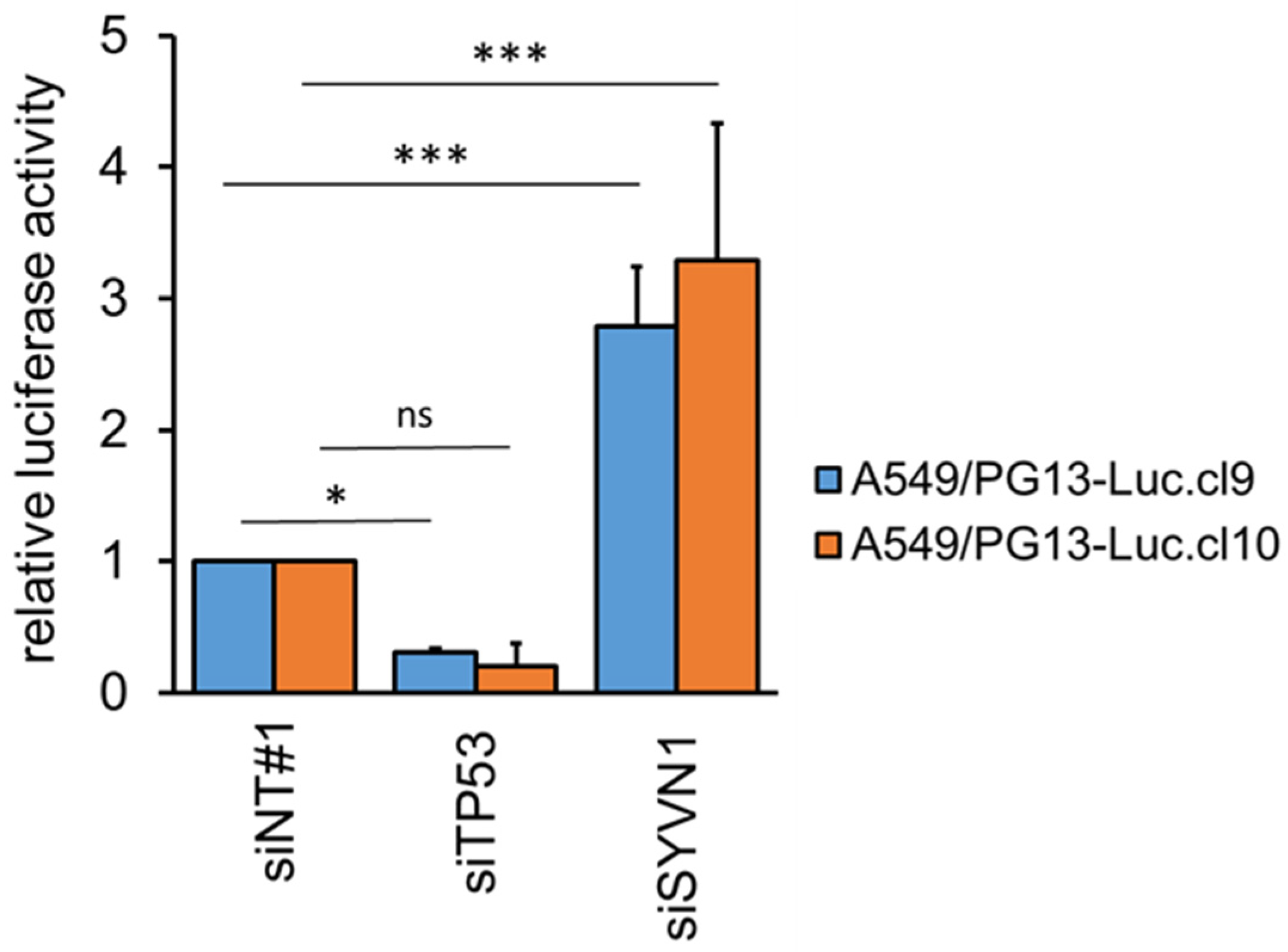
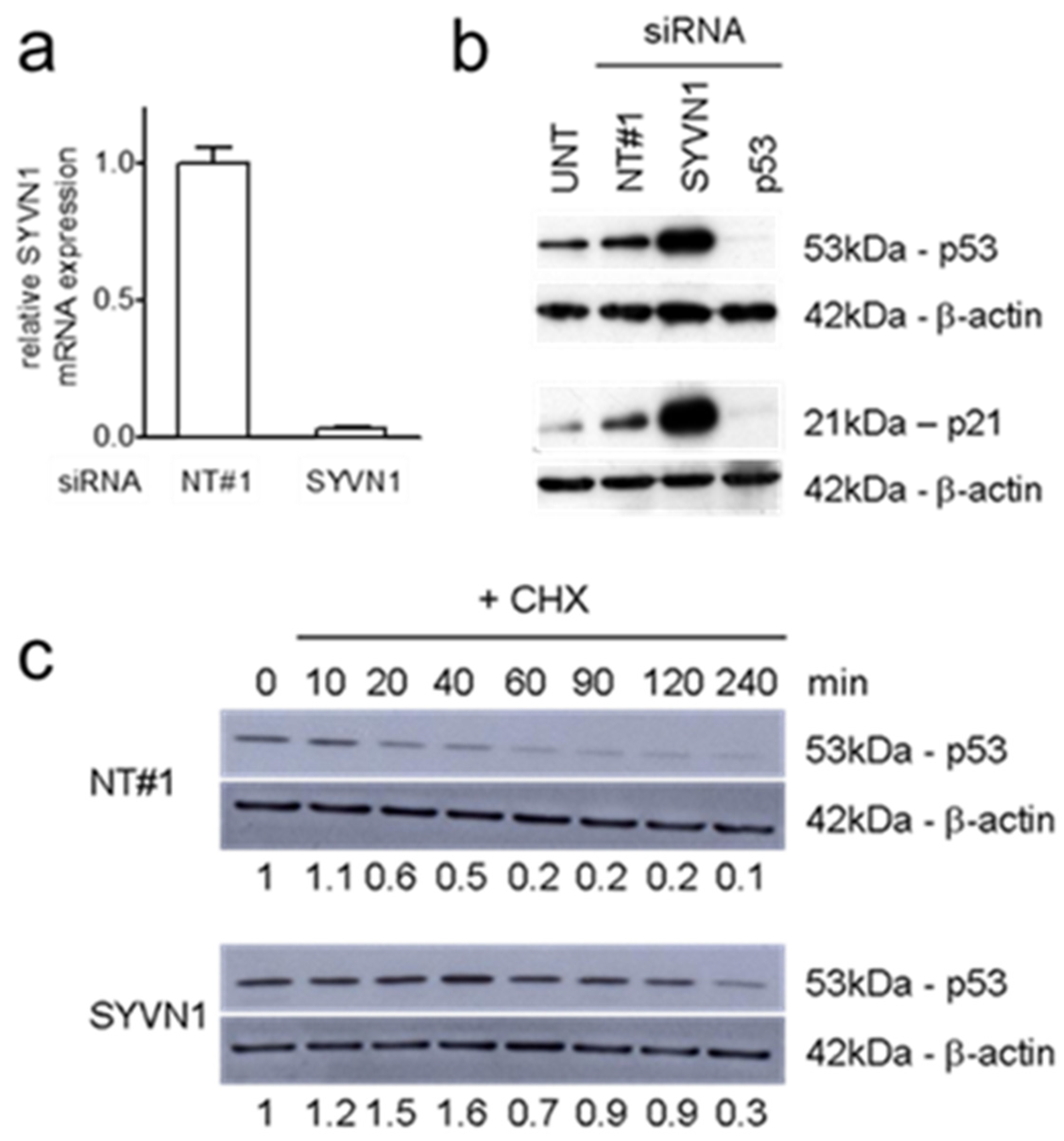
2.1. Synoviolin Silencing Induces p53 Activity in A549 Lung Cancer Cells
2.2. Silencing Synoviolin Increases Oncolysis of Adenovirus-Infected A549 Cells
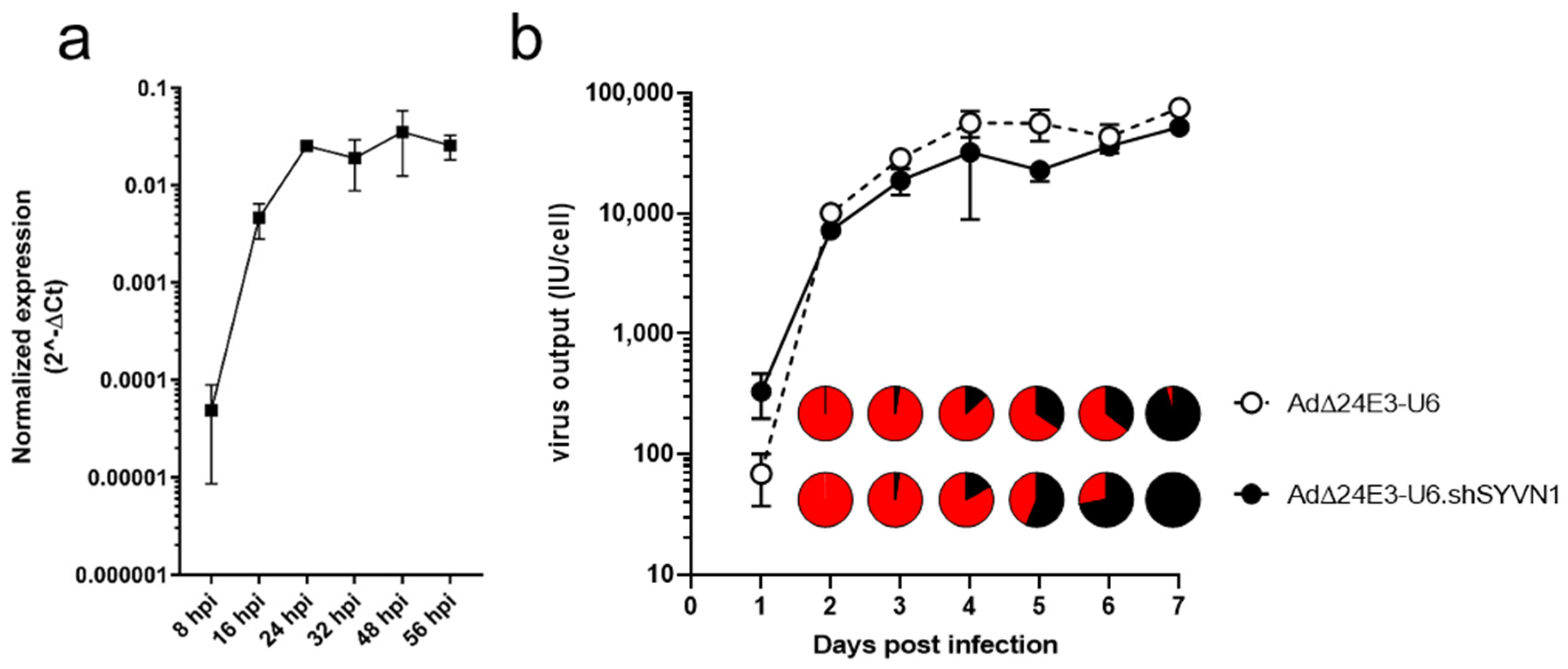
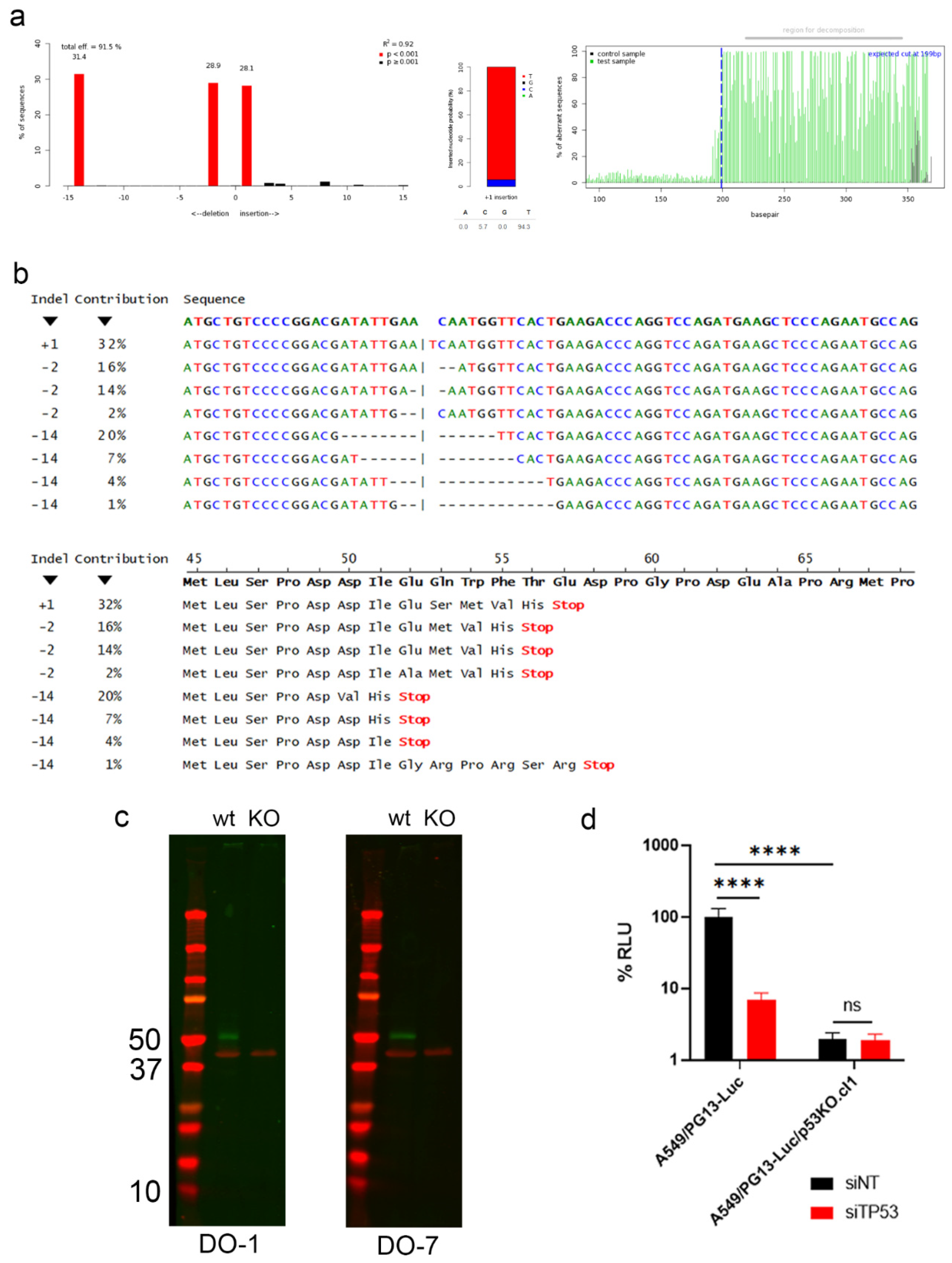
2.3. Construction and Characterization of an Oncolytic Adenovirus Expressing a Short Hairpin RNA against SYVN1
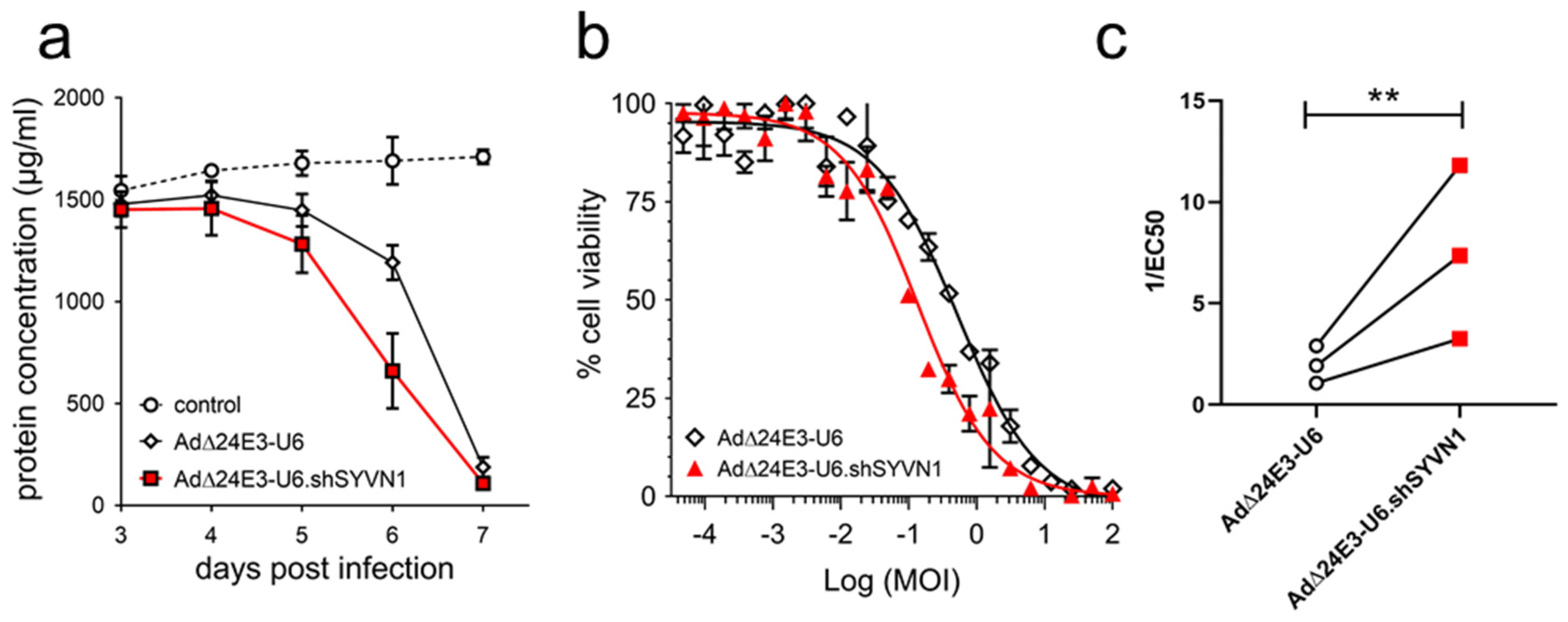
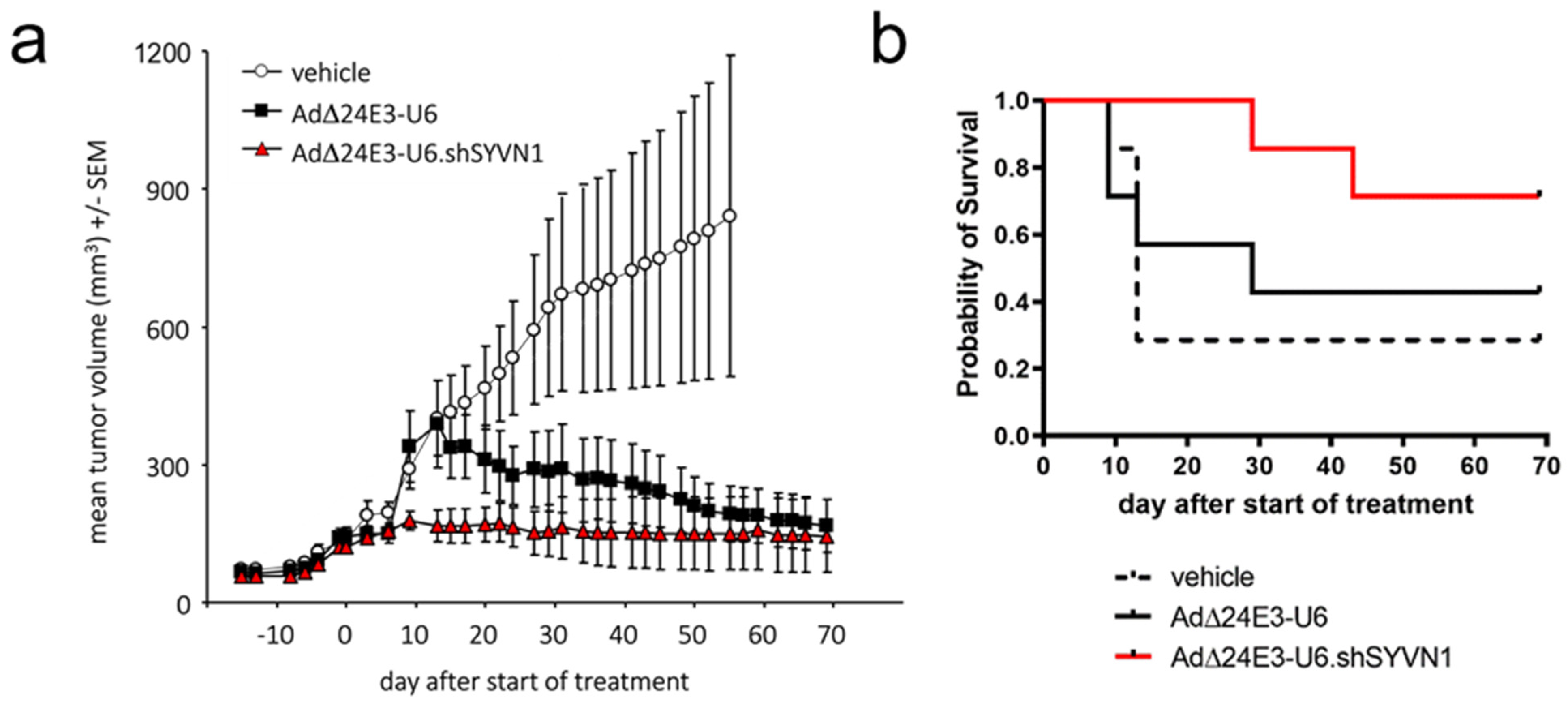
2.4. Oncolytic Adenovirus Expressing a Short Hairpin RNA against SYVN1 Exhibits Enhanced Oncolysis In Vitro and In Vivo
2.5. Enhanced Killing of Cancer Cells by SYVN1 Silencing Is p53-Independent
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Adenoviruses
4.3. P53 Reporter Assays
4.4. Quantitative Reverse Transcription Polymerase Chain Reaction
4.5. Western Blot Analysis
4.6. Cell Count Assay
4.7. BCA Protein Assay
4.8. Adenovirus Production Assay
4.9. Evaluation of Oncolytic Activity in Xenograft Tumors
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Oncolytic Virotherapy for Cancer: Clinical Experience. Biomedicines 2021, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef] [PubMed]
- Cody, J.J.; Douglas, J.T. Armed replicating adenoviruses for cancer virotherapy. Cancer Gene Ther. 2009, 16, 473–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gruijl, T.D.; Janssen, A.B.; van Beusechem, V.W. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin. Biol. Ther. 2015, 15, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Farrera-Sal, M.; Fillat, C.; Alemany, R. Effect of Transgene Location, Transcriptional Control Elements and Transgene Features in Armed Oncolytic Adenoviruses. Cancers 2020, 12, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauthoff, H.; Pipiya, T.; Heitner, S.; Chen, S.; Norman, R.G.; Rom, W.N.; Hay, J.G. Late expression of p53 from a replicating adenovirus improves tumor cell killing and is more tumor cell specific than expression of the adenoviral death protein. Hum. Gene Ther. 2002, 13, 1859–1871. [Google Scholar] [CrossRef]
- van Beusechem, V.W.; van den Doel, P.B.; Grill, J.; Pinedo, H.M.; Gerritsen, W.R. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res. 2002, 62, 6165–6171. [Google Scholar]
- Geoerger, B.; van Beusechem, V.W.; Opolon, P.; Morizet, J.; Laudani, L.; Lecluse, Y.; Barrois, M.; Idema, S.; Grill, J.; Gerritsen, W.R.; et al. Expression of p53, or targeting towards EGFR, enhances the oncolytic potency of conditionally replicative adenovirus against neuroblastoma. J. Gene Med. 2005, 7, 584–594. [Google Scholar] [CrossRef]
- Geoerger, B.; Vassal, G.; Opolon, P.; Dirven, C.M.; Morizet, J.; Laudani, L.; Grill, J.; Giaccone, G.; Vandertop, W.P.; Gerritsen, W.R.; et al. Oncolytic activity of p53-expressing conditionally replicative adenovirus AdDelta24-p53 against human malignant glioma. Cancer Res. 2004, 64, 5753–5759. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Lee, J.T.; Gu, W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010, 17, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Overmeer, R.M.; Schagen, F.H.; Alemany, R.; Barski, O.A.; Gerritsen, W.R.; Van Beusechem, V.W. Conditionally replicating adenoviruses expressing short hairpin RNAs silence the expression of a target gene in cancer cells. Cancer Res. 2004, 64, 2663–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebring-van Olst, E.; Blijlevens, M.; de Menezes, R.X.; van der Meulen-Muileman, I.H.; Smit, E.F.; van Beusechem, V.W. A genome-wide siRNA screen for regulators of tumor suppressor p53 activity in human non-small cell lung cancer cells identifies components of the RNA splicing machinery as targets for anticancer treatment. Mol. Oncol. 2018, 11, 534–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, T.; Yamasaki, S.; Yagishita, N.; Tsuchimochi, K.; Shin, H.; Kawahara, K.; Aratani, S.; Fujita, H.; Zhang, L.; Ikeda, R.; et al. Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 2003, 17, 2436–2449. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L.; et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J. 2007, 26, 113–122. [Google Scholar] [CrossRef]
- Siebring-van Olst, E.; Vermeulen, C.; de Menezes, R.X.; Howell, M.; Smit, E.F.; van Beusechem, V.W. Affordable luciferase reporter assay for cell-based high-throughput screening. J. Biomol. Screen 2013, 18, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Brachtlova, T.; van Ginkel, J.W.; Luinenburg, M.J.; de Menezes, R.X.; Koppers-Lalic, D.; Pegtel, D.M.; Dong, W.; de Gruijl, T.D.; van Beusechem, V.W. Expression of Oncolytic Adenovirus-Encoded RNAi Molecules Is Most Effective in a pri-miRNA Precursor Format. Mol. Ther. Oncolytics 2020, 19, 332–343. [Google Scholar] [CrossRef]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti- glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, C.; Cao, H.; Li, K.; Chen, J.; Jiang, L.; Zhang, Q.; Wu, X.; Jia, X.; Liu, Y.; et al. A novel triple-regulated oncolytic adenovirus carrying p53 gene exerts potent antitumor efficacy on common human solid cancers. Mol. Cancer Ther. 2008, 7, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Hasei, J.; Sasaki, T.; Tazawa, H.; Osaki, S.; Yamakawa, Y.; Kunisada, T.; Yoshida, A.; Hashimoto, Y.; Onishi, T.; Uno, F.; et al. Dual programmed cell death pathways induced by p53 transactivation overcome resistance to oncolytic adenovirus in human osteosarcoma cells. Mol. Cancer Ther. 2013, 12, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Bressy, C.; Hastie, E.; Grdzelishvili, V.Z. Combining Oncolytic Virotherapy with p53 Tumor Suppressor Gene Therapy. Mol. Ther. Oncolytics 2017, 5, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Graat, H.C.; Carette, J.E.; Schagen, F.H.; Vassilev, L.T.; Gerritsen, W.R.; Kaspers, G.J.; Wuisman, P.I.; van Beusechem, V.W. Enhanced tumor cell kill by combined treatment with a small-molecule antagonist of mouse double minute 2 and adenoviruses encoding p53. Mol. Cancer Ther. 2007, 6, 1552–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heideman, D.A.; Steenbergen, R.D.; van der Torre, J.; Scheffner, M.; Alemany, R.; Gerritsen, W.R.; Meijer, C.J.; Snijders, P.J.; van Beusechem, V.W. Oncolytic adenovirus expressing a p53 variant resistant to degradation by HPV E6 protein exhibits potent and selective replication in cervical cancer. Mol. Ther. 2005, 12, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Sauthoff, H.; Pipiya, T.; Chen, S.; Heitner, S.; Cheng, J.; Huang, Y.Q.; Rom, W.N.; Hay, J.G. Modification of the p53 transgene of a replication-competent adenovirus prevents mdm2- and E1b-55kD-mediated degradation of p53. Cancer Gene Ther. 2006, 13, 686–695. [Google Scholar] [CrossRef]
- van Beusechem, V.W.; van den Doel, P.B.; Gerritsen, W.R. Conditionally replicative adenovirus expressing degradation-resistant p53 for enhanced oncolysis of human cancer cells overexpressing murine double minute 2. Mol. Cancer Ther. 2005, 4, 1013–1018. [Google Scholar] [CrossRef] [Green Version]
- Brachtlova, T.; van Beusechem, V.W. Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens. Cells 2018, 7, 228. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Kim, J.H.; Kwon, Y.G.; Kim, E.C.; Kim, N.K.; Choi, H.J.; Yun, C.O. VEGF-specific short hairpin RNA-expressing oncolytic adenovirus elicits potent inhibition of angiogenesis and tumor growth. Mol. Ther. 2007, 15, 295–302. [Google Scholar] [CrossRef]
- Zhang, Y.A.; Nemunaitis, J.; Samuel, S.K.; Chen, P.; Shen, Y.; Tong, A.W. Antitumor activity of an oncolytic adenovirus-delivered oncogene small interfering RNA. Cancer Res. 2006, 66, 9736–9743. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.N.; Pei, D.S.; Sun, F.H.; Zhang, B.F.; Liu, X.Y.; Gu, J.F.; Liu, Y.H.; Hu, X.L.; Mao, L.J.; Wen, R.M.; et al. Inhibition of renal cancer cell growth by oncolytic adenovirus armed short hairpin RNA targeting hTERT gene. Cancer Biol. Ther. 2009, 8, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. p53 promotes adenoviral replication and increases late viral gene expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Yagishita, N.; Nishioka, K.; Nakajima, T. The roles of synoviolin in crosstalk between endoplasmic reticulum stress-induced apoptosis and p53 pathway. Cell Cycle 2007, 6, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Gronberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellmann, C.; Zuber, J.; McJunkin, K.; Chang, K.; Malone, C.D.; Dickins, R.A.; Xu, Q.; Hengartner, M.O.; Elledge, S.J.; Hannon, G.J.; et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol. Cell 2011, 41, 733–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Jin, L.; Zhang, Y.; Huang, Y.; Zhang, F.; Valdmanis, P.N.; Kay, M.A. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell 2012, 151, 900–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanas, A.; Cornelissen, L.A.M.; Kotsias, M.; van der Horst, J.C.; van de Vrugt, H.J.; Kalay, H.; Spencer, D.I.R.; Kozak, R.P.; van Vliet, S.J. Transcriptional activation of fucosyltransferase (FUT) genes using the CRISPR-dCas9-VPR technology reveals potent N-glycome alterations in colorectal cancer cells. Glycobiology 2019, 29, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Suzuki, K.; Alemany, R.; Yamamoto, M.; Curiel, D.T. The presence of the adenovirus E3 region improves the oncolytic potency of conditionally replicative adenoviruses. Clin. Cancer Res. 2002, 8, 3348–3359. [Google Scholar]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vermeulen, C.; Brachtlova, T.; Tol, N.; van der Meulen-Muileman, I.H.; Hodzic, J.; van de Vrugt, H.J.; van Beusechem, V.W. Evaluation of a Novel Oncolytic Adenovirus Silencing SYVN1. Int. J. Mol. Sci. 2022, 23, 15430. https://doi.org/10.3390/ijms232315430
Vermeulen C, Brachtlova T, Tol N, van der Meulen-Muileman IH, Hodzic J, van de Vrugt HJ, van Beusechem VW. Evaluation of a Novel Oncolytic Adenovirus Silencing SYVN1. International Journal of Molecular Sciences. 2022; 23(23):15430. https://doi.org/10.3390/ijms232315430
Chicago/Turabian StyleVermeulen, Christie, Tereza Brachtlova, Nikki Tol, Ida H. van der Meulen-Muileman, Jasmina Hodzic, Henri J. van de Vrugt, and Victor W. van Beusechem. 2022. "Evaluation of a Novel Oncolytic Adenovirus Silencing SYVN1" International Journal of Molecular Sciences 23, no. 23: 15430. https://doi.org/10.3390/ijms232315430
APA StyleVermeulen, C., Brachtlova, T., Tol, N., van der Meulen-Muileman, I. H., Hodzic, J., van de Vrugt, H. J., & van Beusechem, V. W. (2022). Evaluation of a Novel Oncolytic Adenovirus Silencing SYVN1. International Journal of Molecular Sciences, 23(23), 15430. https://doi.org/10.3390/ijms232315430