

A Review of Genetic Polymorphisms and Susceptibilities to Complications after Aneurysmal Subarachnoid Hemorrhage
 ,
,  , and
, and
Abstract
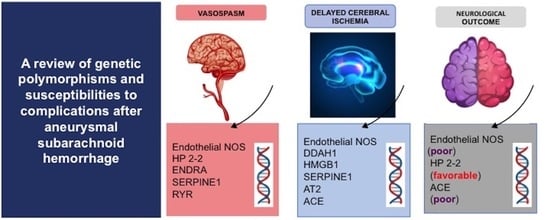
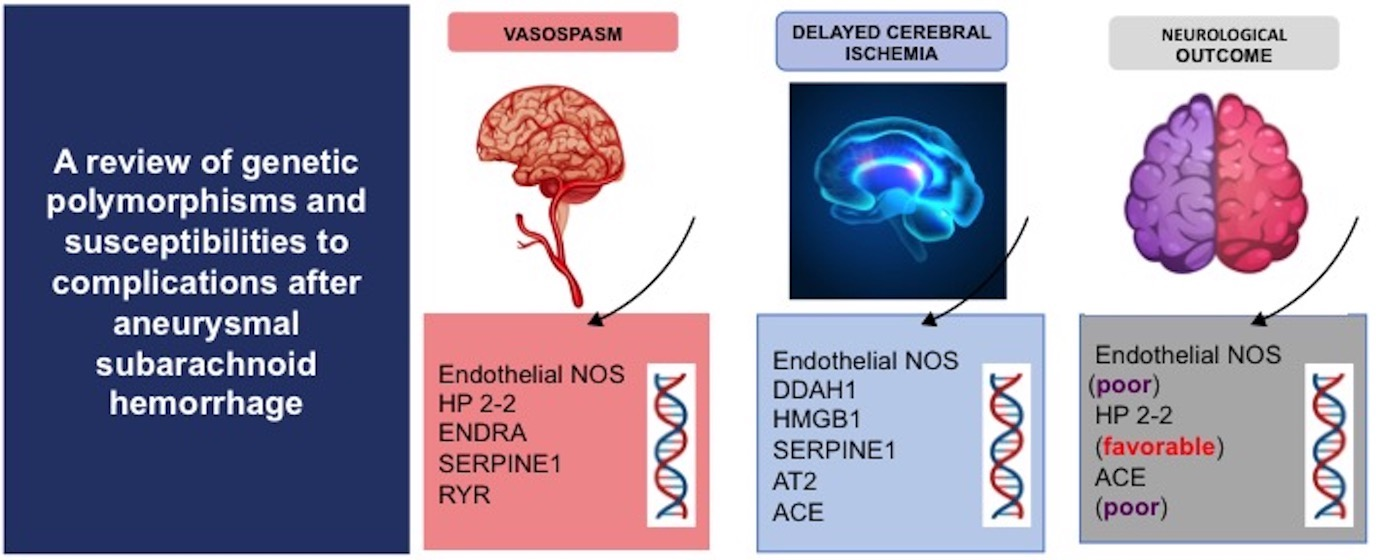
:
1. Introduction
2. Genetic Polymorphisms
2.1. Nitric Oxide Synthases
2.2. Haptoglobin
2.3. Endothelin-1
2.4. High-Mobility Group Box 1
2.5. Serpin Family E Member 1
2.6. Renin–Angiotensin–Aldosterone System
2.7. Ryanodine Receptor
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geraghty, J.; Testai, F. Delayed cerebral ischemia after subrachnoid hemorrhage: Beyond vasospasm and towards a multifactorial pathophysiology. Curr. Atheroscler. Rep. 2017, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Alimohammadi, E.; Ahadi, P.; Karbasforoushan, A.; Rahmani, S.; Bagheri, S.R.; Abdi, A. Nontraumatic nonaneurysmal subarachnoid hemorrhage: Risk factors, complications, and clinical outcomes. Indian J. Neurosurg. 2021, 1, 37–41. [Google Scholar] [CrossRef]
- Kole, M.; Shea, P.; Albrecht, J.; Cannarsa, G.; Wessel, A.; Miller, T.; Simard, J.M. Utility of the Hijdra Sum Score in predicting risk of aneurysm in patients with subarachnoid hemorrhage: A single-center experience with 550 patients. Neurosurgery 2009, 86, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Sarrafzadeh, A.; Vajkoczy, P.; Bijlenga, P.; Schaller, K. Monitoring in neurointensive care—The challenge to detect delayed cerebral ischemia in high-grade aneurysmal SAH. Front. Neurol. 2014, 5, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Carlson, A.P.; Hänggi, D.; Macdonald, R.L.; Shuttleworth, C.W. Nimodipine reappraised: An old drug with a future. Curr. Neuropharmacol. 2020, 18, 65–82. [Google Scholar] [CrossRef]
- Van der Steen, W.; Leemans, E.; van den Berg, R.; Roos, Y.; Marquering, H.; Verbaan, D.; Majoie, C.B. Radiological scales predicting delayed cerebral ischemia in subarachnoid hemorrhage: Systematic review and meta-analysis. Neuroradiology 2019, 61, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Ezra, M.; Garry, P.; Rowland, M.; Mitsis, G.; Pattison, K. Phase dynamics of cerebral flow in subarachnoid haemorrhage in response to sodium nitrite infusion. Nitric Oxide 2021, 106, 55–65. [Google Scholar] [CrossRef]
- Sabri, M.; Ai, J.; Lakovic, K.; D’abbondanza, J.; Ilodigwe, D.; Macdonald, R. Mechanisms of microthrombosis formation after experimental subarachnoid hemorrhage. Neuroscience 2012, 224, 26–37. [Google Scholar] [CrossRef]
- Crowley, R.; Medel, R.; Dumont, A.; Ilodigwe, D.; Kassell, N.; Mayer, S.; Macdonald, R.L. Angiographic vasospasm is strongly correlated with cerebral infarctation after subarachnoid hemorrhage. Stroke 2011, 42, 919–923. [Google Scholar] [CrossRef]
- Dreier, J.; Manning, A.; Woizik, J.; Drenckahn, C.; Steinbrink, J.; Tolias, C.; COSBID Study Group. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009, 132, 1866–1881. [Google Scholar] [CrossRef]
- Vergouwen, M.; Vermeulen, M.; Coert, B.; Stroes, E.; Roos, Y. Microthrombosis after aneurysmal subarachnoid hemorrhage: An additional explanation for delayed cerebral ischemia. Br. J. Pharmacol. 2008, 28, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.; Hopkins, S.; Vail, A.; King, A.; Smith, D.; Illingworth, K.; Tyrrell, P.J. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J. Neurointerv. Surg. 2013, 5, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, R.; Schweizer, T. Spontaneous subarachnoid haemorrhage. Lancet 2017, 389, 655–656. [Google Scholar] [CrossRef]
- Rass, V.; Helbok, R. How to diagnose delayed cerebral ischemia and symptomatic vasospasm and prevent cerebral infarction in patients with subarachnoid hemorrhage. Curr. Opin. Crit. Care 2021, 27, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.; Ahn, H.; Preziosi, T.; Battye, R.; Boone, S.; Chou, S.; Transou, C.R. Cerebral arterial spasm—A controlled trial of nimodipine in patients with subarachnoid hemorrhage. N. Engl. J. Med. 1983, 308, 619–624. [Google Scholar] [CrossRef]
- Endo, H.; Hagihara, Y.; Kimura, N.; Takizawa, K.; Niizuma, K.; Togo, O.; Tominaga, T. Effects of clazosentan on cerebral vapospasm-related morbidity and all-cause mortality after aneurysmal subarachnoid hemorrhage: Two randomized phase 3 trials in Japanese patients. J. Neurosurg. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Mahmoud, S.; Ji, X.; Isse, F. Nimodipine pharmacokinetic variability in various patient populations. Drugs R D 2020, 20, 307–318. [Google Scholar] [CrossRef]
- Contestabile, A.; Monti, B.; Polazzi, E. Neuronal-glial interactions define the role of nitric oxide in neural functional processes. Curr. Neuropharmacol. 2012, 10, 303–310. [Google Scholar] [CrossRef]
- Ebner, J.; Cagalinec, M.; Kubista, H.; Todt, H.; Szabo, P.; Kiss, A.; Koenig, X. Neuronal nitric oxide synthase regulation of calcium cycling in ventricular cardiomyocytes is independent of Cav1.2 channel modulation under basal conditions. Eur. J. Physiol. 2020, 472, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Becker-Barroso, E. Calcium, channels, and brain damage. Lancet Neurol. 2006, 5, 26–27. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; García-Buendía, J.; Muñoz, F. Functions and dysfunctions of nitric oxide in brain. BBA—Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Song, W.; Li, L.; Fan, X. Endothelial nitric oxide synthase: A potential therapeutic target for cerebrovascular diseases. Mol. Brain 2016, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, V.; Sohni, Y.; Mangrun, W.; McClelland, R.; O’Kane, D.; Meyer, F.; Meissner, I. Endothelial nitric oxide synthase T-786C single nucleotide polymorphism. A putative genetic marker differentiating small versus large ruptured intracraneal aneurysms. Stroke 2003, 34, 2555–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, B.; Guo, Q.L.; He, Z.J.; Yuan, Y.J.; Wang, N.; Zhou, J. Remote ischemic postconditioning protects the brain from global cerebral ischemia/reperfusion injury by up-regulating endothelial nitric acyd synthase through the PI3K/Akt pathway. Brain Res. 2012, 1445, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.; Ezra, M.; Rowland, M.; Westbrook, J.; Pattison, T. The role of the nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athiraman, U.; Jayaraman, K.; Meizi, L.; Tusar, G.; Yuan, J.; Zipfel, G.J. Role of endothelial nitric oxide synthase in isoflurane conditioning-induced neurovascular protection in subarachnoid hemorrhage. J. Am. Heart Assoc. 2020, 9, e017477. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jayamaran, K.; Giri, T.; Zipfel, G.; Athiraman, U. Role of SIRT1 in isoflurane conditioning-induced neurovascular protection against delayed cerebral ischemia secondary to subarachnoid hemorrhage. Int. J. Mol. Sci. 2021, 22, 4291. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.; Sohni, Y.; Mangrun, W.; McClelland, R.; O’Kane, D.; Meyer, F.; Meissner, I. Endothelial nitric oxide synthase gene polymorphisms predict susceptibility to aneurysmal subarachnoid hemorrhage and cerebral vasospasm. J. Cereb. Blood Flow Metab. 2004, 24, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, P.; Foreman, P.; Harrigan, M.; Fisher, W.; Vyas, N.; Lipsky, R.; Lin, M. Endothelial nitric oxide synthase polymorphism is associated with delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017, 101, 514–519. [Google Scholar] [CrossRef]
- Lai, P.; Du, R. Role of genetic polymorphism in predicting delayed cerebral ischemia and radiographic vasospasm after aneurysmal subarachnoid hemorrhage: A meta-analysis. World Neurosurg. 2015, 84, 933–941. [Google Scholar] [CrossRef]
- Foreman, P.; Starke, R.; Hendrix, P.; Harrigan, M.; Fisher, W.; Vyas, N.; Griessenauer, C.J. Endothelin polymorphisms as a risk factor for cerebral aneurysm rebleeding following aneurysmal subarachnoid hemorrhage. Clin. Neurol. Neurosurg. 2017, 157, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Grissenauer, C.; Tubbs, R.; Foreman, P.; Chua, M.; Vyas, N.; Lipsky, R.; Shoja, M.M. Associations of renin-angiotensin system genetic polymorphisms and clinical course after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2017, 126, 1585–1597. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Grissenauer, C.; Starke, R.; Tubbs, R.; Shoja, M.; Foreman, P.; Lipsky, R.H. Haplotype analysis of SERPINE1 gene: Risk for aneurysmal subarachnoid hemorrhage and clinical outcomes. Mol. Genet. Genom. Med. 2019, 7, e737. [Google Scholar] [CrossRef] [Green Version]
- Song, M.K.; Myeong-Kyu, K.; Kim, T.S.; Joo, S.P.; Park, M.S.; Kim, B.C.; Cho, K.H. Endothelial nitric oxide gene T-786C polymorphism and subarachnoid hemorrhage in Korean population. J. Korean Med. Sci. 2006, 21, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, N.; Rajendran, P.; Kim, H.; Rutkowski, M.; Pawlikowska, L.; Kwok, P.Y.; Young, W.L. Endothelial nitric oxide synthase polymorphism (−7867->C) and increased risk of angiographic vasospasm after aneurysmal subarachnoid hemorrhage. Stroke 2008, 39, 1103–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.; Kim, G.; Komotar, R.; Hickman, Z.; Black, E.; Rosales, M.; Kellner, C.P. Endothelial nitric oxide synthase gene signle-nucleotide polymorphism predicts cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2008, 28, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Wispel, C.; Zweckberger, K.; Beynon, C.; Hertle, D.; Sakowitz, O.; Unterberg, A.W. Endogenous Nitric-Oxide Synthase Inhibitor ADMA after acute brain injury. Int. J. Mol. Sci. 2014, 15, 4088–4103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlert, A.; Starekova, J.; Manthei, G.; Ehlert-Gamm, A.; Flack, J.; Gessert, M.; Hesselmann, V. Nitric oxide-based treatment of poor-grade patients after severe aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2020, 32, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Budohoski, K.P.; Guilfoyle, M.; Helmy, A.; Huuskonen, T.; Czosnyka, M.; Kirollos, R.; Kirkpatrick, P.J. The pathophysiology and treatment of delayed cerebral ischemia following subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1343–1353. [Google Scholar] [CrossRef]
- Appel, D.; Seerberger, M.; Schwedhelm, E.; Czorlich, P.; Goetz, A.; Böger, R.; Hannemann, J. Assymetric and symmetric dimethylarginines are markers of delayed cerebral ischemia and neurological outcome in patients with subarachnoide hemorrhage. Neurocrit. Care 2018, 29, 84–93. [Google Scholar] [CrossRef]
- Bergström, A.; Staalso, J.; Romner, B.; Olsen, N. Impaired endothelial function after aneurysmal subarachnoid haemorrhage correlates with arginine: Asymmetric dimethylarginine ratio. Br. J. Anaesth. 2014, 112, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanneman, J.; Appel, D.; Seerberger-Steinmeister, M.; Brüning, T.; Zummack, J.; Böger, R. Sequence variation in the DDAH1 gene predisposes for delayed cerebral ischemia in subarachnoidal hemorrhage. J. Clin. Med. 2020, 9, 3900. [Google Scholar] [CrossRef]
- Hugelshofer, M.; Buzzi, R.; Schaer, C.; Richter, H.; Akeret, K.; Anagnostaku, V.; Schaer, D.J. Haptoglobin administration into the subarachnoid space prevents hemoglobin-induced cerebral vasospasm. J. Clin. Investig. 2019, 129, 5219–5235. [Google Scholar] [CrossRef] [Green Version]
- Sabri, M.; Jinglu, A.; Lass, E.; D’abbondanza, J.; Macdonald, R. Genetic elimination of eNOS reduces secondary complications of experimental subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2013, 33, 1008–1014. [Google Scholar] [CrossRef] [Green Version]
- Hugelshofer, M.; Sikorski, C.; Seule, M.; Deuel, J.; Muroi, C.; Seboek, M.; Keller, E. Cell-free oxyhemoglobin in cerebrospinal fluid after aneurysmal subarachnoid hemorrhage: Biomarker and potential therapeutic target. World Neurosurg. 2018, 120, e660–e666. [Google Scholar] [CrossRef]
- Morton, M.; Hostetler, I.; Kazmi, N.; Alg, V.; Borner, S.; Brown, M.; Galea, I. Haptoglobin genotype and outcome after aneurysmal subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 2020, 91, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Ciurea, A.; Palade, C.; Voinescu, D.; Nica, D. Subarachnoid hemorrhage and cerebral vasospasm—Literature review. J. Med. Life 2013, 6, 120–125. [Google Scholar]
- Grissenauer, C.; Starke, R.; Foreman, P.; Hendrix, P.; Harrigan, M.; Fisher, W.; Mathru, M. Associations between endothelin polymorphisms and aneurysmal subarachnoid hemorrhage, clinical vasospasm, delayed cerebral ischemia, and functional outcome. J. Neurosurg. 2018, 128, 1311–1317. [Google Scholar] [CrossRef]
- Stein, S.; Browne, K.; Chen, X.; Smith, D.; Graham, D. Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: An autopsy study. Neurosurgery 2006, 59, 781–787. [Google Scholar] [CrossRef]
- Maksoud, M.; Tellios, V.; Xiang, Y.Y.; Lu, W.Y. Nitric oxide displays a biphasic effect on calcium dynamics in microglia. Nitric Oxide 2021, 108, 28–39. [Google Scholar] [CrossRef]
- Laskowitz, D.; Kolls, B. Neuroprotection in subarachnoid hemorrhage. Stroke 2010, 41, 79–84. [Google Scholar] [CrossRef]
- Francoeur, C.; Mayer, S. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit. Care Med. 2016, 20, 277. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, M.; LaRusso, N.; Burgart, L.; Gores, G. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000, 60, 184–190. [Google Scholar]
- Li, Y.; Ghen, G.; Zhao, J.; Nie, X.; Wan, C.; Liu, J.; Xu, G. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces microgial nitric oxide production and subsequent rat primary cortical neuron apoptosis through p38/JNK MAPK pathway. Toxicology 2013, 312, 132–141. [Google Scholar] [CrossRef]
- Green, J.; Rolfe, M.; Smith, L. Transcriptional regulation of bacterial virulence gene expression by molecular oxygen and nitric oxide. Virulence 2014, 5, 794–809. [Google Scholar] [CrossRef]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Bosch-Morató, M.; Valls-Comamala, V.; Munoz, F.J. Methylglyoxal reduces mithocondrial potential and activates Bax and caspase-3 in neurons: Implications for Alzheimer’s disease. Neurosci. Lett. 2014, 580, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Guix, F.X. Methylglyoxal produced by amyloid-B peptide-induced nitrotyrosination of triosephosphate isomerase triggers neuronal death in Alzheimer’s disease. J. Alzheimers Dis. 2014, 41, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Chaudry, S.; Güresir, A.; Stoffel-Wagner, B.; Fimmers, R.; Kinfe, T.; Dietrich, D.; Muhammad, S. Systemic high-mobility group box-1: A novel predictive biomarker for cerebral vasospasm in aneurysmal subarachnoid hemorrhage. Crit. Care Med. 2018, 46, e1023–e1028. [Google Scholar] [CrossRef]
- Hemmer, S.; Senger, S.; Griessenauer, C.; Simgen, A.; Oertel, J.; Geisel, J.; Hendrix, P. Admission serum high mobility group box 1 (HMGB1) protein predicts delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 2022, 45, 807–817. [Google Scholar] [CrossRef]
- Hendrix, P.; Foreman, P.; Fisher, W.; Harrigan, M.; Vyas, N.; Lipsky, R.; Griessenauer, C.J. Impact of high-mobility group box 1 polymorphism on delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017, 101, 325–330. [Google Scholar] [CrossRef]
- Ohkuma, H.; Suzuki, S.; Fujita, S.; Nakamura, W. Role of a decreased expression of the local renin-angiotensin system in the etiology of cerebral aneurysms. Circulation 2003, 108, 785–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueffert, H.; Gumplinger, A.; Renner, C.; Dengl, M.; Reske, A.; Kaisers, U.X.; Meixensberger, J. Search for genetic variants in the Ryanodine receptor 1 gene in patients with symptomatic cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2011, 15, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, P.; Foreman, P.; Harrigan, M.; Fisher, W.; Vyas, N.; Lipsky, R.; Griessenauer, C.J. Ryanodine Receptor 1 polymorphism is not associated with aneurysmal subarachnoid hemorrhage or its clinical sequelae. World Neurosurg. 2017, 100, 190–194. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
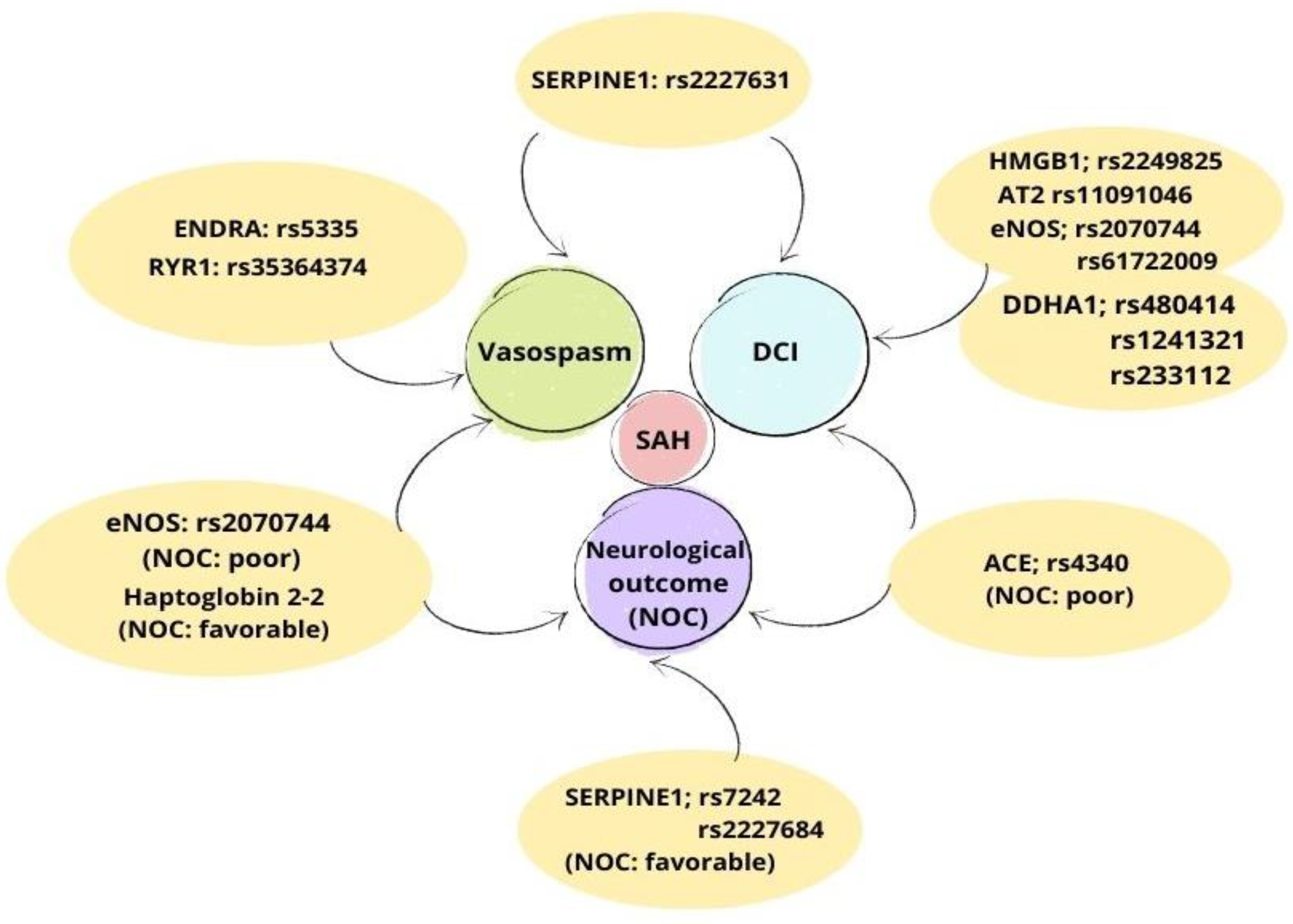
| Gene | SNP | NOC | DCI | Vasospasm |
|---|---|---|---|---|
| eNOS | rs2070744 | A (poor) | A | A |
| rs61722009 | NA | A | NA | |
| DDAH1 | rs480414 | NA | A | NA |
| rs1241321 | NA | A | NA | |
| rs233112 | NA | A | NA | |
| HP2-2 | - | A (favorable) | NA | A |
| ENDRA | rs5335 | NA | NA | A |
| HMGB1 | rs2249825 | NA | A | NA |
| SERPINE1 | rs2227631 | NA | A | A |
| AT2 | rs11092046 | NA | A | NA |
| ACE | rs4340 | A (poor) | A | NA |
| RYR | rs35364374 | NA | NA | A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Suárez, J.; Rodríguez-Esparragón, F.; Sosa-Pérez, C.; Cazorla-Rivero, S.; Torres-Mata, L.B.; Jiménez-O’Shanahan, A.; Clavo, B.; Morera-Molina, J. A Review of Genetic Polymorphisms and Susceptibilities to Complications after Aneurysmal Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2022, 23, 15427. https://doi.org/10.3390/ijms232315427
Medina-Suárez J, Rodríguez-Esparragón F, Sosa-Pérez C, Cazorla-Rivero S, Torres-Mata LB, Jiménez-O’Shanahan A, Clavo B, Morera-Molina J. A Review of Genetic Polymorphisms and Susceptibilities to Complications after Aneurysmal Subarachnoid Hemorrhage. International Journal of Molecular Sciences. 2022; 23(23):15427. https://doi.org/10.3390/ijms232315427
Chicago/Turabian StyleMedina-Suárez, Jose, Francisco Rodríguez-Esparragón, Coralia Sosa-Pérez, Sara Cazorla-Rivero, Laura B. Torres-Mata, Aruma Jiménez-O’Shanahan, Bernardino Clavo, and Jesús Morera-Molina. 2022. "A Review of Genetic Polymorphisms and Susceptibilities to Complications after Aneurysmal Subarachnoid Hemorrhage" International Journal of Molecular Sciences 23, no. 23: 15427. https://doi.org/10.3390/ijms232315427