
Alterations in UPR Signaling by Methylmercury Trigger Neuronal Cell Death in the Mouse Brain

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
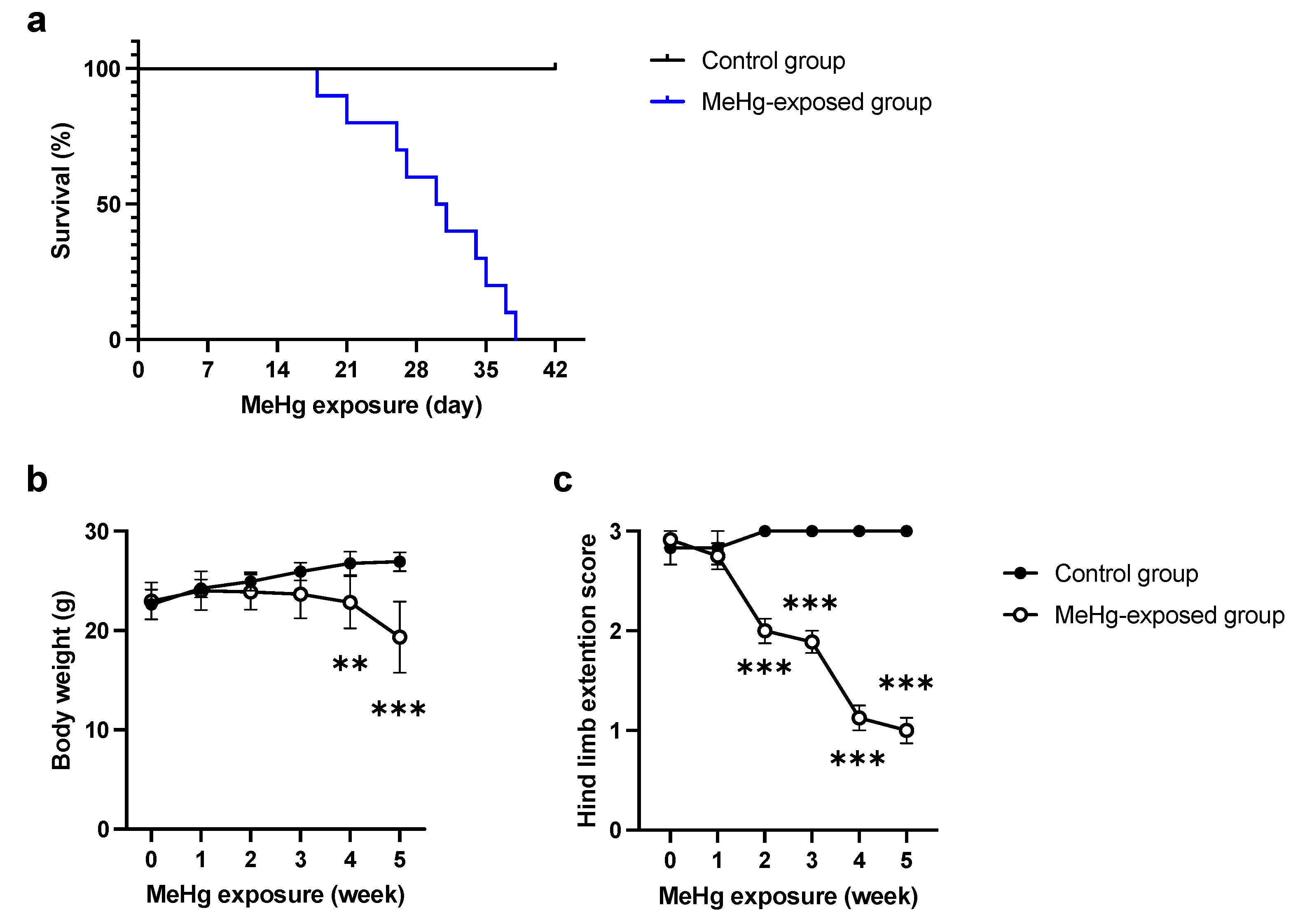
2.1. ERAI-Venus Mice Exhibit Neurological Symptoms Similar to Wild-Type Mice upon MeHg Exposure
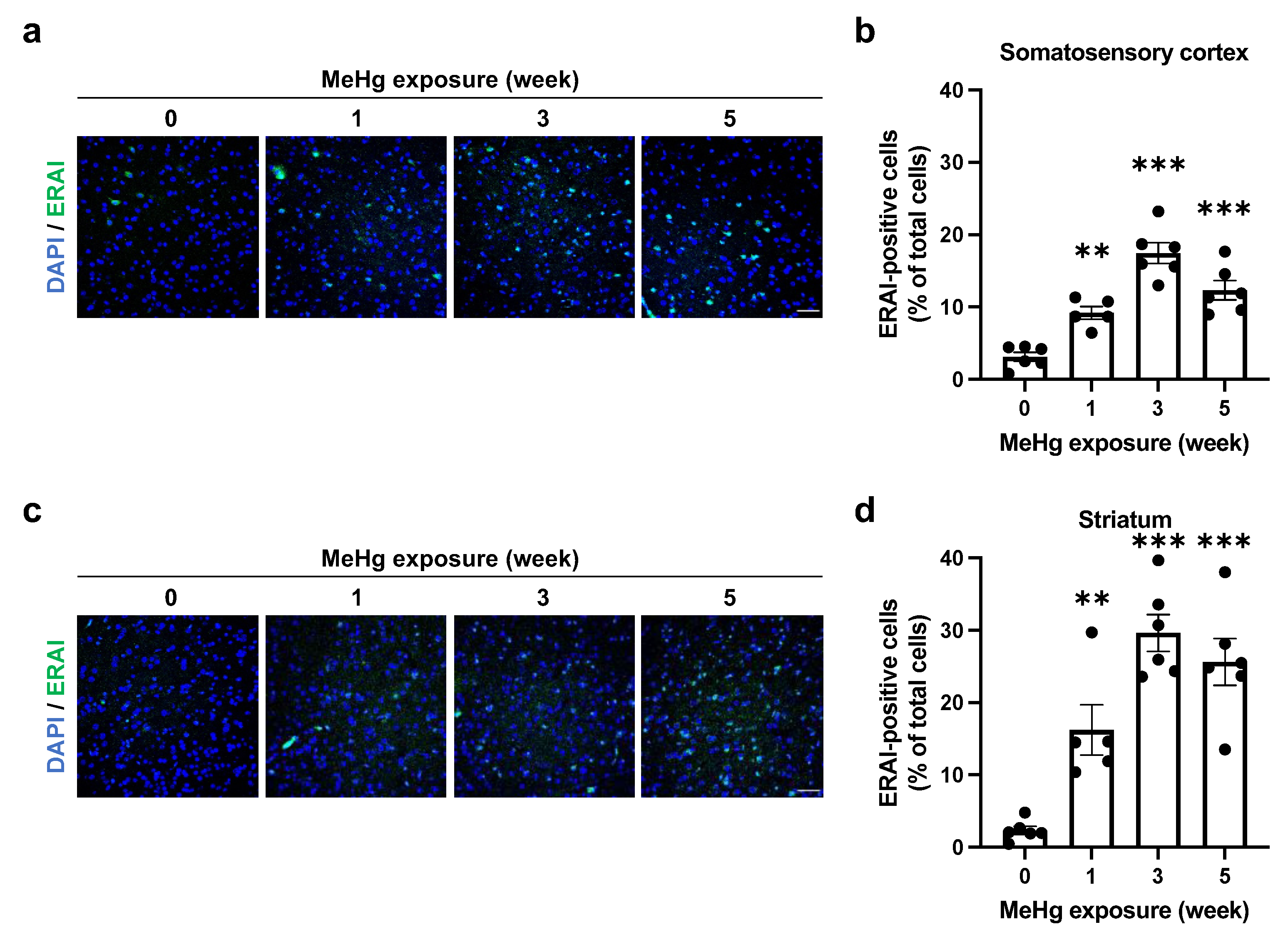
2.2. ER Stress Induction by MeHg Exposure in the Cerebral Cortex and Striatum
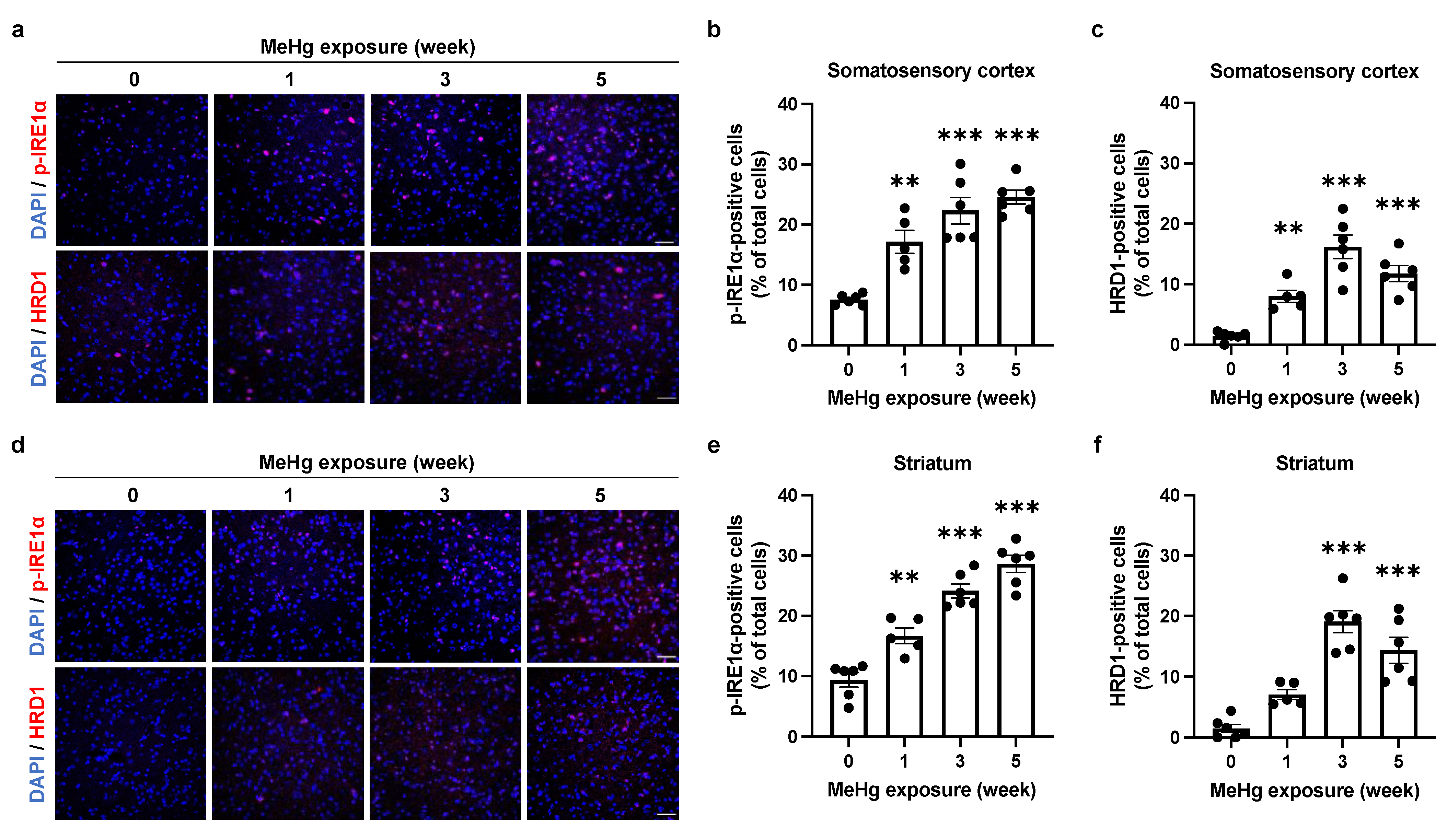
2.3. The UPR Signals Are Altered by MeHg Exposure in the Mouse Brain
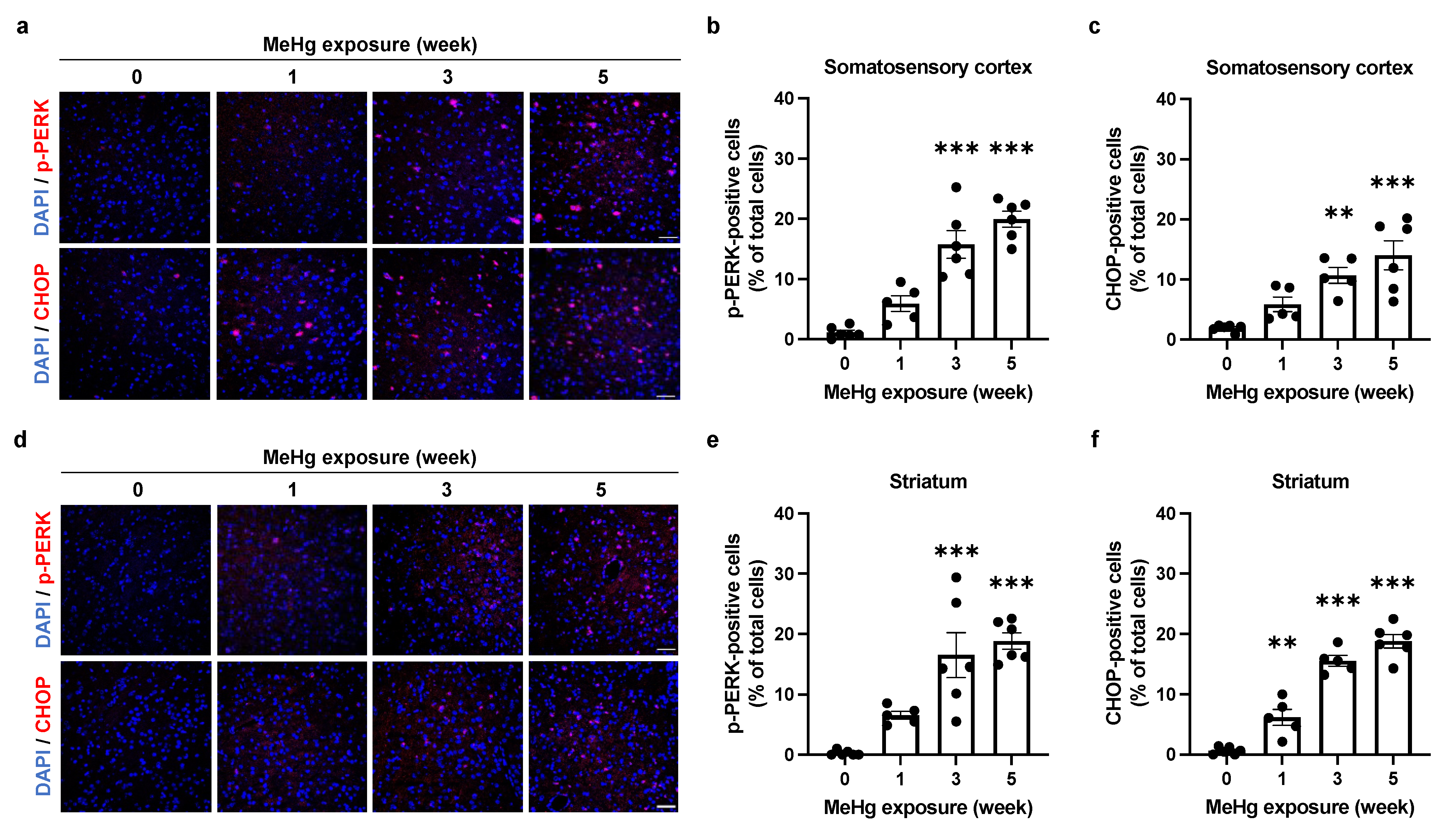
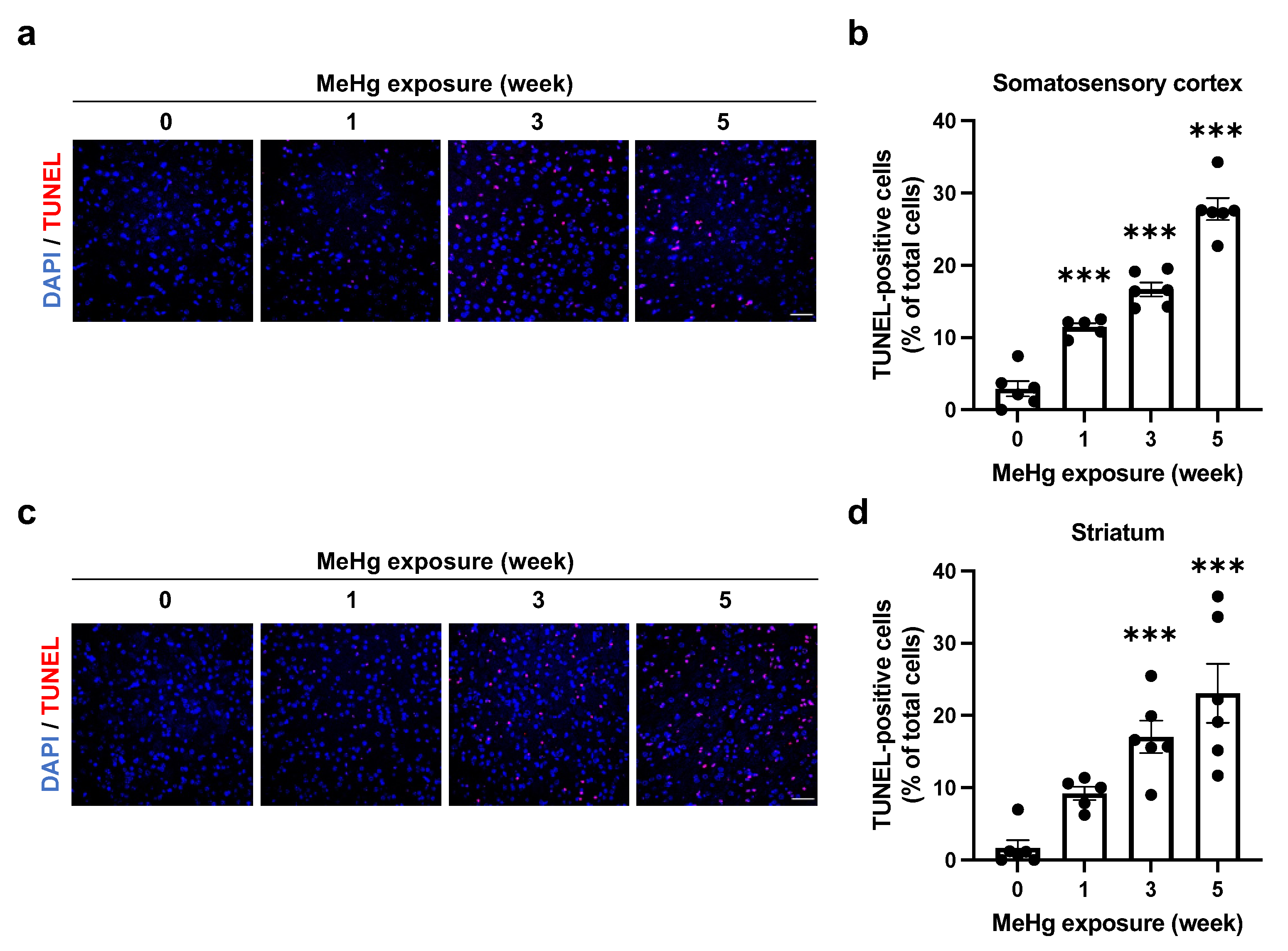
2.4. Alterations of UPR Signaling by MeHg Are Related to Cell Death
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. MeHg Administration
4.3. Hind Limb Extension Analysis
4.4. Measurement of Mercury Deposition
4.5. Immunohistochemistry
4.6. TUNEL Staining
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mahafey, K.R.; Clickner, R.P.; Bodurow, C.C. Blood organic mercury and dietary mercury intake: National Health and Nutrition Examination Survey, 1999 and 2000. Environ. Health Perspect. 2004, 112, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Kerper, L.E.; Ballatori, N.; Clarkson, T.W. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am. J. Physiol. 1992, 262, R761–R765. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Syversen, T. Methylmercury: Recent Advances in the Understanding of its Neurotoxicity. Ther. Drug Monit. 2005, 27, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Takeuchi, T. A pathological study of prolonged cases of Minamata disease. With particular reference to 83 autopsy cases. Acta Pathol. JPN 1978, 28, 565–584. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F.; Sawada, M.; Takashima, A. Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain. Neurotoxicology 2009, 30, 1000–1007. [Google Scholar] [CrossRef]
- Usuki, F.; Fujita, E.; Sasagawa, N. Methylmercury activates ASK1/JNK signaling pathways, leading to apoptosis due to both mitochondria- and endoplasmic reticulum (ER)-generated processes in myogenic cell lines. Neurotoxicology 2008, 29, 22–30. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, R.; Liu, W.; Wu, Y.; Qian, H.; Zhao, X.; Wang, S.; Xing, G. Hormetic effects of acute methylmercury exposure on GRP78 expression in rat brain cortex. Dose-Response 2013, 11, 109–120. [Google Scholar] [CrossRef]
- Liu, W.; Yang, T.; Xu, Z.; Xu, B.; Deng, Y. Methyl-mercury induces apoptosis through ROS-mediated endoplasmic reticulum stress and mitochondrial apoptosis pathways activation in rat cortical neurons. Free Radic. Res. 2019, 53, 26–44. [Google Scholar] [CrossRef]
- Chung, Y.; Yen, C.; Tang, F.; Lee, K.; Liu, S.; Wu, C.; Hsieh, S.; Su, C.; Kuo, C.; Chen, Y. Methylmercury exposure induces ROS/Akt inactivation-triggered endoplasmic reticulum stress-regulated neuronal cell apoptosis. Toxicology 2019, 425, 152245. [Google Scholar] [CrossRef]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2013, 14, 434–456. [Google Scholar] [CrossRef]
- Kanda, H.; Shinkai, Y.; Kumagai, Y. S-mercuration of cellular proteins by methylmercury and its toxicological implications. J. Toxicol. Sci. 2014, 39, 687–700. [Google Scholar] [CrossRef]
- Kumagai, Y.; Homma-Takeda, S.; Shinyashiki, M.; Shimojo, N. Alterations in superoxide dismutase isozymes by methylmercury. Appl. Organomet. Chem. 1997, 11, 635–643. [Google Scholar] [CrossRef]
- Mori, N.; Yasutake, A.; Hirayama, K. Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Arch. Toxicol. 2007, 81, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Okuda, K.; Sugino, E.; Nishiya, T.; Toyama, T.; Iwawaki, T.; Fujimura, M.; Kumagai, Y.; Uehara, T. Correlation between attenuation of protein disulfide isomerase activity through S-mercuration and neurotoxicity induced by methylmercury. Neurotox. Res. 2014, 27, 99–105. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, H.; Nakahara, K.; Kaneko, Y.; Akiyama, S.; Okuda, K.; Iwawaki, T.; Fujimura, M.; Kumagai, Y.; Takasugi, N.; Uehara, T. Modulation of unfolded protein response by methylmercury. Biol. Pharm. Bull. 2017, 40, 1595–1598. [Google Scholar] [CrossRef] [PubMed]
- Nakato, R.; Ohkubo, Y.; Konishi, A.; Shibata, M.; Kaneko, Y.; Iwawaki, T.; Nakamura, T.; Lipton, S.A.; Uehara, T. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep. 2015, 5, 14812. [Google Scholar] [CrossRef] [PubMed]
- Remondelli, P.; Renna, M. The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front Mol. Neurosci. 2017, 10, 187. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Kohno, K.; Miura, M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 2004, 10, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Yorifuji, T.; Tsuda, T.; Sekikawa, T.; Nakadaira, H.; Saito, H. Methyl mercury exposure at Niigata, Japan: Results of neurological examinations of 103 adults. J. Biomed. Biotechnol. 2012, 2012, 635075. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Minamata Disease (NIMD). Ministry of the Environment NIMD Annual Report; Ministy of the Environment: Tokyo, Japan, 2006. (In Japanese)
- Hiraoka, H.; Nomura, R.; Takasugi, N.; Akai, R.; Iwawaki, T.; Kumagai, Y.; Fujimura, M.; Uehara, T. Spatiotemporal analysis of the UPR transition induced by methylmercury in the mouse brain. Arch. Toxicol. 2021, 95, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F. In situ different antioxidative systems contribute to the site-specific methylmercury neurotoxicity in mice. Toxicology 2017, 392, 55–63. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response to decrease the load in the ER; these processes are collectively termed the unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef]
- Sakanyan, V. Reactive Chemicals and Electrophilic Stress in Cancer: A Minireview. High-Throughput 2018, 7, 12. [Google Scholar] [CrossRef]
- Zimniak, P. Free Radical Biology & Medicine Relationship of electrophilic stress to aging. Free Radic. Biol. Med. 2011, 51, 1087–1105. [Google Scholar] [CrossRef]
- Groeger, A.L.; Freeman, B.A. Signaling Actions of Electrophiles: Anti-inflammatory Therapeutic Candidates. Mol. Interv. 2010, 10, 39–50. [Google Scholar] [CrossRef]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.I.; Lipton, S.A. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. The binary switch between life and death of endoplasmic reticulum-stressed β cells. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 107–112. [Google Scholar] [CrossRef]
- Chang, S.H.; Lee, H.J.; Kang, B.; Yu, K.N.; Arash, M.T.; Lee, S.; Kim, S.U.; Cho, M.H. Methylmercury induces caspase-dependent apoptosis and autophagy in human neural stem cells. J. Toxicol. Sci. 2013, 38, 823–831. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nomura, R.; Takasugi, N.; Hiraoka, H.; Iijima, Y.; Iwawaki, T.; Kumagai, Y.; Fujimura, M.; Uehara, T. Alterations in UPR Signaling by Methylmercury Trigger Neuronal Cell Death in the Mouse Brain. Int. J. Mol. Sci. 2022, 23, 15412. https://doi.org/10.3390/ijms232315412
Nomura R, Takasugi N, Hiraoka H, Iijima Y, Iwawaki T, Kumagai Y, Fujimura M, Uehara T. Alterations in UPR Signaling by Methylmercury Trigger Neuronal Cell Death in the Mouse Brain. International Journal of Molecular Sciences. 2022; 23(23):15412. https://doi.org/10.3390/ijms232315412
Chicago/Turabian StyleNomura, Ryosuke, Nobumasa Takasugi, Hideki Hiraoka, Yuta Iijima, Takao Iwawaki, Yoshito Kumagai, Masatake Fujimura, and Takashi Uehara. 2022. "Alterations in UPR Signaling by Methylmercury Trigger Neuronal Cell Death in the Mouse Brain" International Journal of Molecular Sciences 23, no. 23: 15412. https://doi.org/10.3390/ijms232315412
APA StyleNomura, R., Takasugi, N., Hiraoka, H., Iijima, Y., Iwawaki, T., Kumagai, Y., Fujimura, M., & Uehara, T. (2022). Alterations in UPR Signaling by Methylmercury Trigger Neuronal Cell Death in the Mouse Brain. International Journal of Molecular Sciences, 23(23), 15412. https://doi.org/10.3390/ijms232315412