
Gilthead Seabream Liver Integrative Proteomics and Metabolomics Analysis Reveals Regulation by Different Prosurvival Pathways in the Metabolic Adaptation to Stress

 ,
,  , ,
, , 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
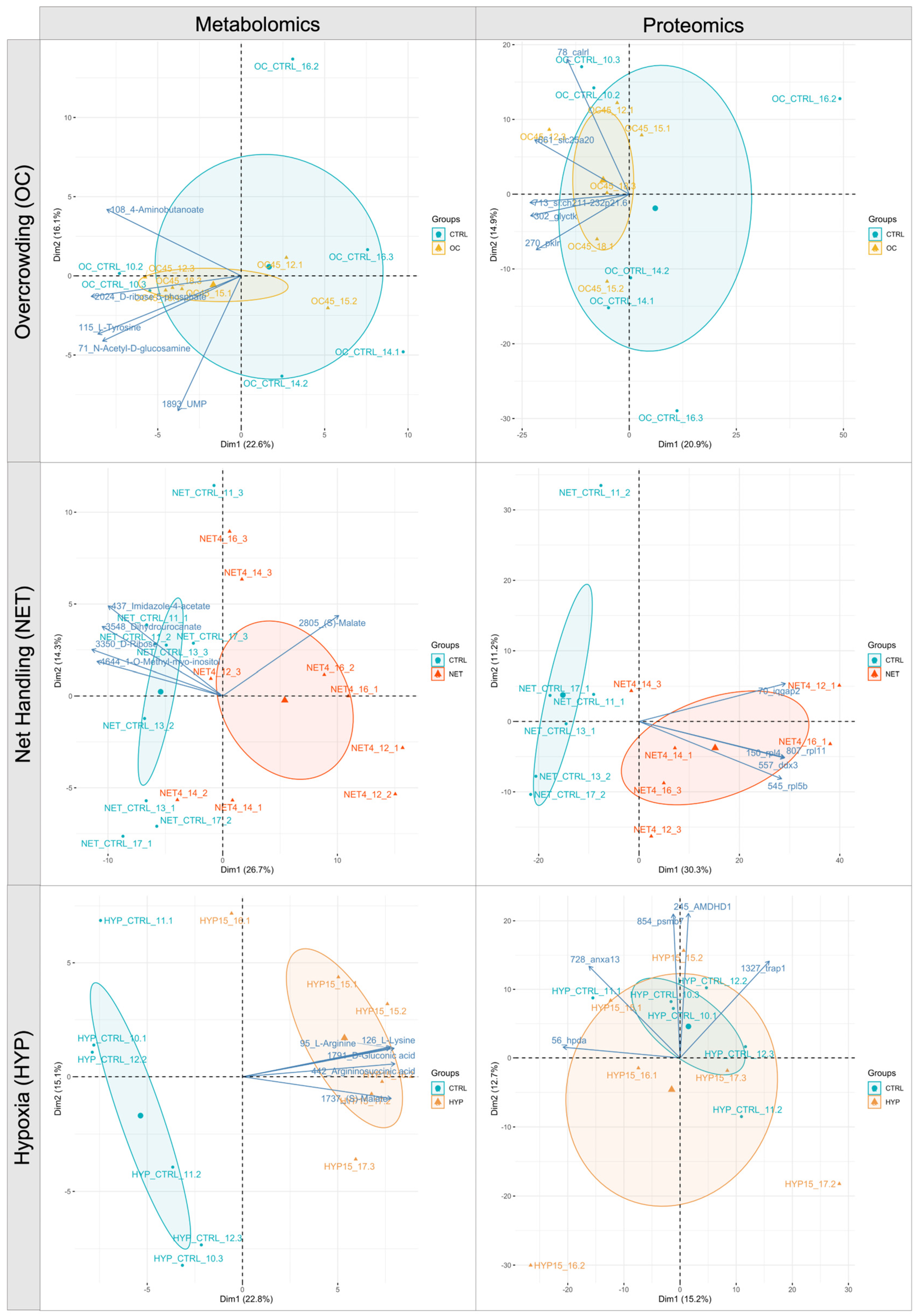
2.1. Proteomics and Metabolomics Data Overview
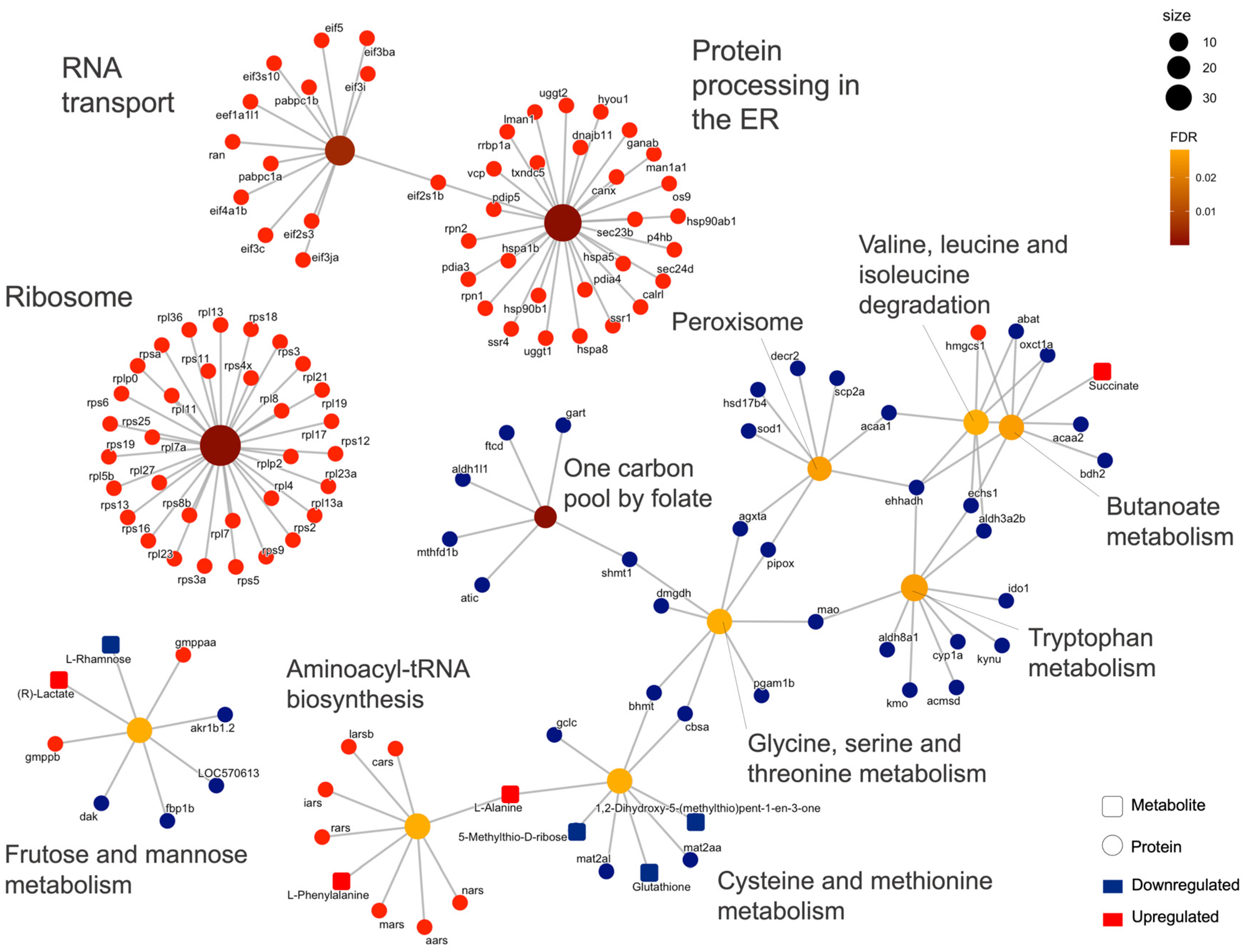
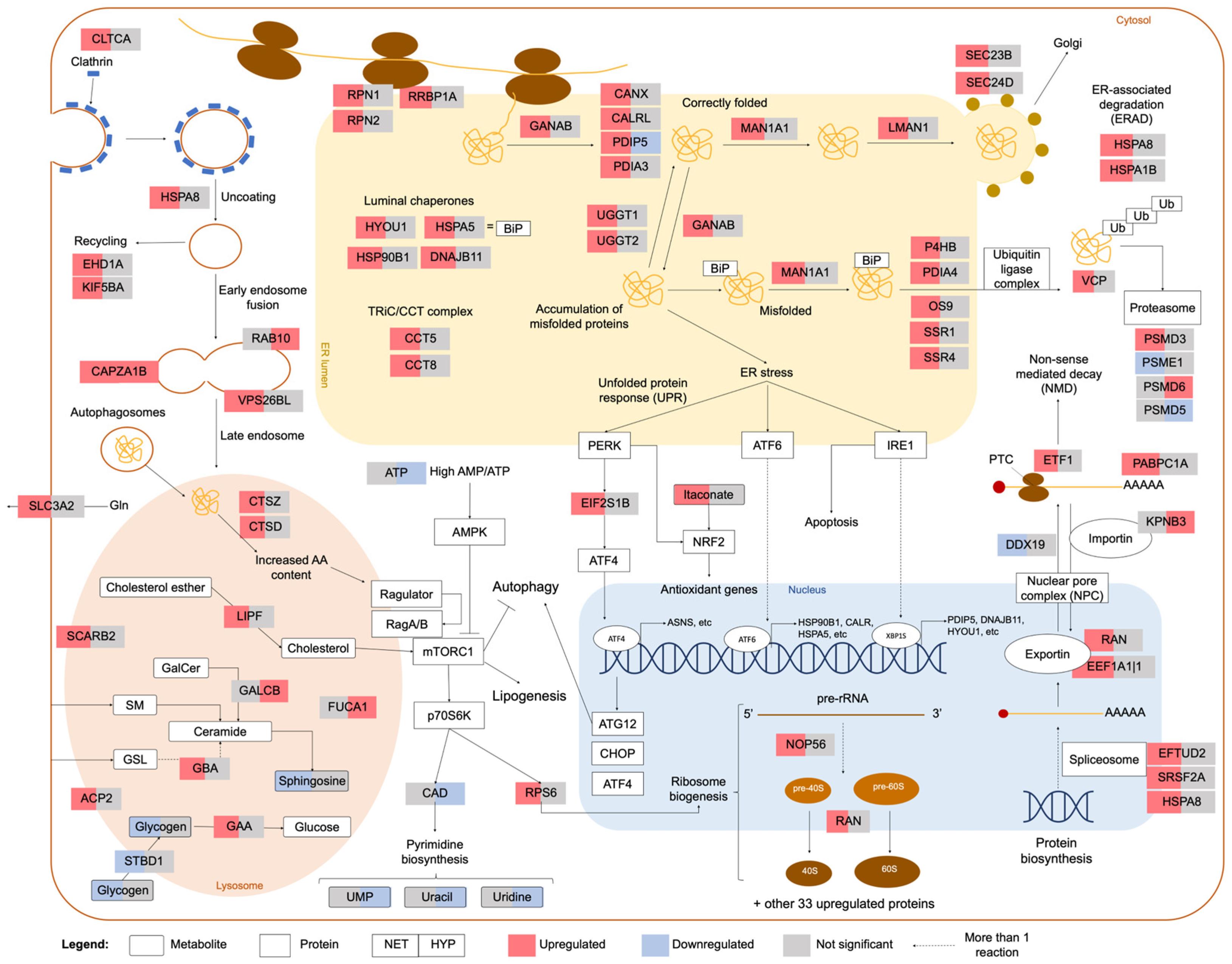
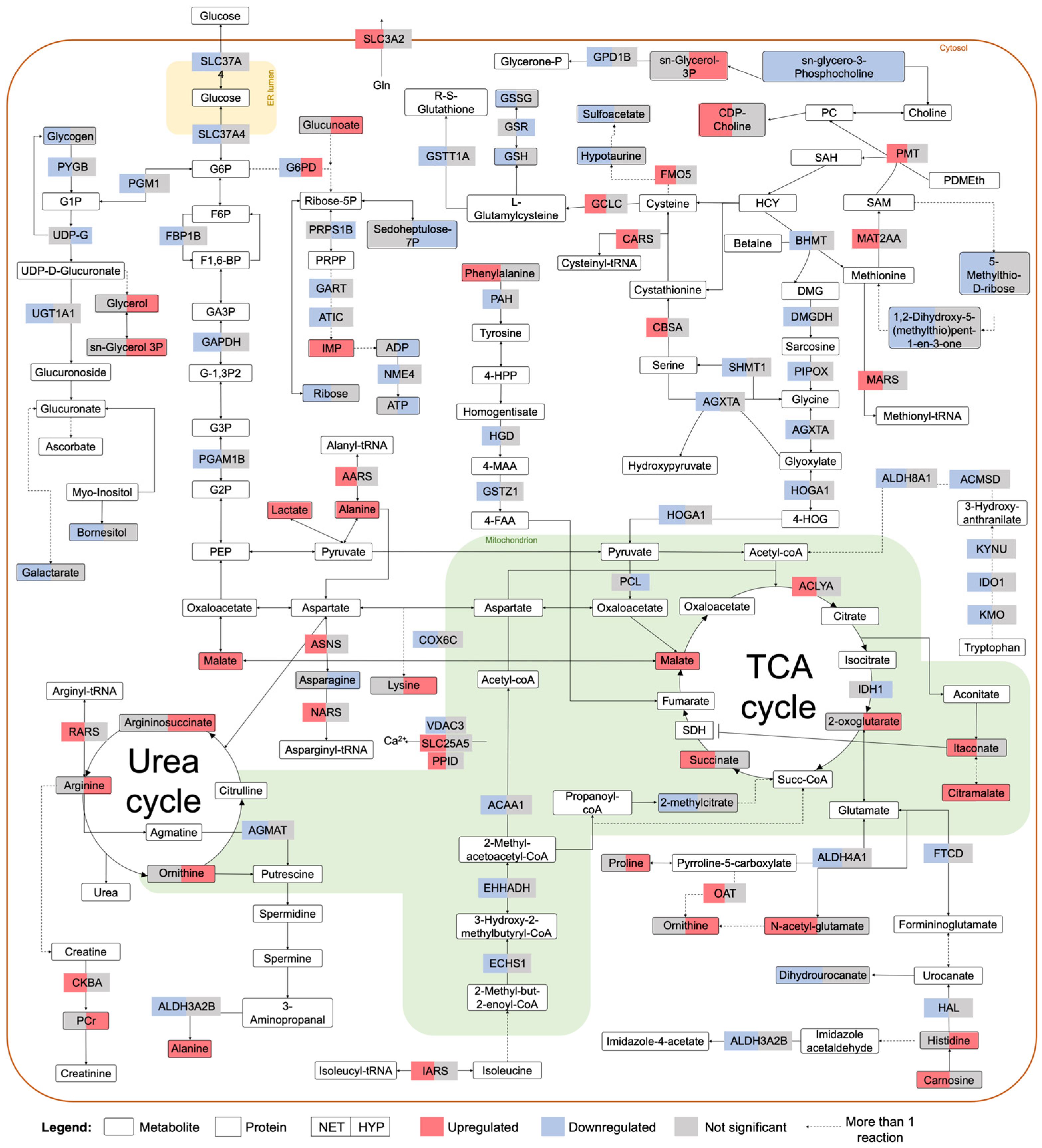
2.2. Integrated Stress Response Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Fish and Stocking Conditions
4.3. Experimental Design
4.4. Sampling
4.5. Sample Preparation
4.6. Label-Free Shotgun Proteomics Analysis
4.6.1. NanoLC-MS/MS Analysis
4.6.2. Protein Identification
4.7. Untargeted Metabolomics Analysis
4.8. Univariate and Multivariate Statistical Analyses
4.9. Functional Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huntingford, F.A.; Adams, C.; Braithwaite, V.A.; Kadri, S.; Pottinger, T.G.; Sandoe, P.; Turnbull, J.F. Current Issues in Fish Welfare. J. Fish Biol. 2006, 68, 332–372. [Google Scholar] [CrossRef]
- Iwama, G.K.; Afonso, L.O.B.; Vijayan, M.M. Stress in Fish. In Physiology of Fishes; Evans, D.H., Claiborne, J.B., Eds.; CRC, Taylor & Francis: Boca Ratón, FL, USA, 2006; pp. 320–342. ISBN 9780849320224. [Google Scholar]
- Mommsen, T.P.; Vijayan, M.M.; Moon, T.W. Cortisol in Teleosts: Dynamics, Mechanisms of Action, and Metabolic Regulation. Rev. Fish Biol. Fish. 1999, 9, 211–268. [Google Scholar] [CrossRef]
- Boonstra, R. Reality as the Leading Cause of Stress: Rethinking the Impact of Chronic Stress in Nature. Funct. Ecol. 2013, 27, 11–23. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.G.; Kültz, D. The Cellular Stress Response in Fish Exposed to Salinity Fluctuations. J. Exp. Zool. Part A Ecol. Integr. Physiol. 2020, 333, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Somero, G.N. The Cellular Stress Response and Temperature: Function, Regulation, and Evolution. J. Exp. Zool. Part A Ecol. Integr. Physiol. 2020, 333, 379–397. [Google Scholar] [CrossRef]
- Ghisaura, S.; Pagnozzi, D.; Melis, R.; Biosa, G.; Slawski, H.; Uzzau, S.; Anedda, R.; Addis, M.F. Liver Proteomics of Gilthead Sea Bream (Sparus aurata) Exposed to Cold Stress. J. Therm. Biol. 2019, 82, 234–241. [Google Scholar] [CrossRef]
- Causey, D.R.; N Pohl, M.A.; Stead, D.A.; M Martin, S.A.; Secombes, C.J.; Macqueen, D.J. High-Throughput Proteomic Profiling of the Fish Liver Following Bacterial Infection. BMC Genom. 2018, 19, 719. [Google Scholar] [CrossRef] [PubMed]
- Naderi, M.; Keyvanshokooh, S.; Ghaedi, A.; Salati, A.P. Effect of Acute Crowding Stress on Rainbow Trout (Oncorhynchus mykiss): A Proteomics Study. Aquaculture 2018, 495, 106–114. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, J.; Wen, X.; Zhao, C.; Zhang, H.; Li, X.; Yin, S. Comparative ITRAQ-Based Quantitative Proteomic Analysis of Pelteobagrus Vachelli Liver under Acute Hypoxia: Implications in Metabolic Responses. Proteomics 2017, 17, 1700140. [Google Scholar] [CrossRef]
- Gandar, A.; Laffaille, P.; Marty-Gasset, N.; Viala, D.; Molette, C.; Jean, S. Proteome Response of Fish under Multiple Stress Exposure: Effects of Pesticide Mixtures and Temperature Increase. Aquat. Toxicol. 2017, 184, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Pédron, N.; Artigaud, S.; Infante, J.-L.Z.; Le Bayon, N.; Charrier, G.; Pichereau, V.; Laroche, J. Proteomic Responses of European Flounder to Temperature and Hypoxia as Interacting Stressors: Differential Sensitivities of Populations. Sci. Total Environ. 2017, 586, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Raposo de Magalhães, C.; Schrama, D.; Nakharuthai, C.; Boonanuntanasarn, S.; Revets, D.; Planchon, S.; Kuehn, A.; Cerqueira, M.; Carrilho, R.; Farinha, A.P.; et al. Metabolic Plasticity of Gilthead Seabream Under Different Stressors: Analysis of the Stress Responsive Hepatic Proteome and Gene Expression. Front. Mar. Sci. 2021, 8, 676189. [Google Scholar] [CrossRef]
- Quan, J.; Kang, Y.; Li, L.; Zhao, G.; Sun, J.; Liu, Z. Proteome Analysis of Rainbow Trout (Oncorhynchus mykiss) Liver Responses to Chronic Heat Stress Using DIA/SWATH. J. Proteom. 2021, 233, 104079. [Google Scholar] [CrossRef]
- Ziarrusta, H.; Mijangos, L.; Picart-Armada, S.; Irazola, M.; Perera-Lluna, A.; Usobiaga, A.; Prieto, A.; Etxebarria, N.; Olivares, M.; Zuloaga, O. Non-Targeted Metabolomics Reveals Alterations in Liver and Plasma of Gilt-Head Bream Exposed to Oxybenzone. Chemosphere 2018, 211, 624–631. [Google Scholar] [CrossRef]
- Alfaro, A.C.; Nguyen, T.V.; Venter, L.; Ericson, J.A.; Sharma, S.; Ragg, N.L.C.; Mundy, C. The Effects of Live Transport on Metabolism and Stress Responses of Abalone (Haliotis iris). Metabolites 2021, 11, 748. [Google Scholar] [CrossRef]
- Jiao, S.; Nie, M.; Song, H.; Xu, D.; You, F. Physiological Responses to Cold and Starvation Stresses in the Liver of Yellow Drum (Nibea albiflora) Revealed by LC-MS Metabolomics. Sci. Total Environ. 2020, 715, 136940. [Google Scholar] [CrossRef]
- Raposo de Magalhães, C.; Cerqueira, M.; Schrama, D.; Moreira, M.; Boonanuntanasarn, S.; Rodrigues, P.M.L. A Proteomics and Other Omics Approach in the Context of Farmed Fish Welfare and Biomarker Discovery. Rev. Aquac. 2020, 12, 122–144. [Google Scholar] [CrossRef]
- Wei, Y.; Li, B.; Xu, H.; Liang, M. Liver Metabolome and Proteome Response of Turbot (Scophthalmus maximus) to Lysine and Leucine in Free and Dipeptide Forms. Front. Mar. Sci. 2021, 8, 743. [Google Scholar] [CrossRef]
- Zhu, Z.X.; Jiang, D.L.; Li, B.J.; Qin, H.; Meng, Z.N.; Lin, H.R.; Xia, J.H. Differential Transcriptomic and Metabolomic Responses in the Liver of Nile Tilapia (Oreochromis niloticus) Exposed to Acute Ammonia. Mar. Biotechnol. 2019, 21, 488–502. [Google Scholar] [CrossRef]
- Wen, X.; Hu, Y.; Zhang, X.; Wei, X.; Wang, T.; Yin, S. Integrated Application of Multi-Omics Provides Insights into Cold Stress Responses in Pufferfish Takifugu fasciatus. BMC Genom. 2019, 20, 563. [Google Scholar] [CrossRef] [PubMed]
- Colás-Ruiz, N.R.; Ramirez, G.; Courant, F.; Gomez, E.; Hampel, M.; Lara-Martín, P.A. Multi-Omic Approach to Evaluate the Response of Gilt-Head Sea Bream (Sparus aurata) Exposed to the UV Filter Sulisobenzone. Sci. Total Environ. 2022, 803, 150080. [Google Scholar] [CrossRef] [PubMed]
- Dale, K.; Yadetie, F.; Müller, M.B.; Pampanin, D.M.; Gilabert, A.; Zhang, X.; Tairova, Z.; Haarr, A.; Lille-Langøy, R.; Lyche, J.L.; et al. Proteomics and Lipidomics Analyses Reveal Modulation of Lipid Metabolism by Perfluoroalkyl Substances in Liver of Atlantic Cod (Gadus morhua). Aquat. Toxicol. 2020, 227, 105590. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Zhao, J.; Li, T.; Tafalla, C.; Zhang, Q.; Wang, X.; Gong, X.; Shen, Z.; Li, A. Transcriptomic and Proteomic Analyses of Splenic Immune Mechanisms of Rainbow Trout (Oncorhynchus Mykiss) Infected by Aeromonas salmonicida subsp. Salmonicida. J. Proteom. 2015, 122, 41–54. [Google Scholar] [CrossRef]
- Li, J.N.; Zhao, Y.T.; Cao, S.L.; Wang, H.; Zhang, J.J. Integrated Transcriptomic and Proteomic Analyses of Grass Carp Intestines after Vaccination with a Double-Targeted DNA Vaccine of Vibrio Mimicus. Fish Shellfish Immunol. 2020, 98, 641–652. [Google Scholar] [CrossRef]
- Ge, H.; Lin, K.; Zhou, C.; Lin, Q.; Zhang, Z.; Wu, J.; Zheng, L.; Yang, Q.; Wu, S.; Chen, W.; et al. A Multi-Omic Analysis of Orange-Spotted Grouper Larvae Infected with Nervous Necrosis Virus Identifies Increased Adhesion Molecules and Collagen Synthesis in the Persistent State. Fish Shellfish Immunol. 2020, 98, 595–604. [Google Scholar] [CrossRef]
- FAO. The State of World Fisheries and Aquaculture (SOFIA) 2022; Towards Blue Transformation; FAO: Rome, Italy, 2022. [Google Scholar]
- Wendelaar Bonga, S.E. The Stress Response in Fish. Physiol. Rev. 1997, 77, 591–625. [Google Scholar] [CrossRef]
- Vijayan, M.M.; Aluru, N.; Leatherland, J.F. Stress Response and the Role of Cortisol. In Fish Diseases and Disorders; Leatherland, J.F., Woo, P., Eds.; Vol 2: Non-infectious disorders; CAB International: Oxfordshire, UK, 2010; pp. 182–201. [Google Scholar]
- Petitjean, Q.; Jean, S.; Gandar, A.; Côte, J.; Laffaille, P.; Jacquin, L. Stress Responses in Fish: From Molecular to Evolutionary Processes. Sci. Total Environ. 2019, 684, 371–380. [Google Scholar] [CrossRef]
- Raposo de Magalhães, C.; Schrama, D.; Farinha, A.P.; Revets, D.; Kuehn, A.; Planchon, S.; Rodrigues, P.M.; Cerqueira, M. Protein Changes as Robust Signatures of Fish Chronic Stress: A Proteomics Approach to Fish Welfare Research. BMC Genom. 2020, 21, 309. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular Stress Responses: Cell Survival and Cell Death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef]
- Zhao, X.; Li, L.; Li, C.; Liu, E.; Zhu, H.; Ling, Q. Heat Stress-Induced Endoplasmic Reticulum Stress Promotes Liver Apoptosis in Largemouth Bass (Micropterus salmoides). Aquaculture 2022, 546, 737401. [Google Scholar] [CrossRef]
- Iwama, G.K.; Afonso, L.O.B.; Todgham, A.; Ackerman, P.; Nakano, K. Are Hsps Suitable for Indicating Stressed States in Fish? J. Exp. Biol. 2004, 207, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Morro, B.; Broughton, R.; Balseiro, P.; Handeland, S.O.; Mackenzie, S.; Doherty, M.K.; Whitfield, P.D.; Shimizu, M.; Gorissen, M.; Sveier, H.; et al. Endoplasmic Reticulum Stress as a Key Mechanism in Stunted Growth of Seawater Rainbow Trout (Oncorhynchus mykiss). BMC Genom. 2021, 22, 824. [Google Scholar] [CrossRef]
- Jia, R.; Du, J.; Cao, L.; Feng, W.; He, Q.; Xu, P.; Yin, G. Chronic Exposure of Hydrogen Peroxide Alters Redox State, Apoptosis and Endoplasmic Reticulum Stress in Common Carp (Cyprinus carpio). Aquat. Toxicol. 2020, 229, 105657. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, C.A.; Ponce, C.; Zuloaga, R.; González, P.; Avendaño-Herrera, R.; Valdés, J.A.; Molina, A. Effects of Crowding on the Three Main Proteolytic Mechanisms of Skeletal Muscle in Rainbow Trout (Oncorhynchus mykiss). BMC Vet. Res. 2020, 16, 294. [Google Scholar] [CrossRef] [PubMed]
- Mininni, A.N.; Milan, M.; Ferraresso, S.; Petochi, T.; Di Marco, P.; Marino, G.; Livi, S.; Romualdi, C.; Bargelloni, L.; Patarnello, T. Liver Transcriptome Analysis in Gilthead Sea Bream upon Exposure to Low Temperature. BMC Genom. 2014, 15, 765. [Google Scholar] [CrossRef]
- Tepedelen, B.E.; Kirmizibayrak, P.B. Endoplasmic Reticulum-Associated Degradation (ERAD). In Endoplasmic Reticulum; Catala, A., Brzozowski, T., Eds.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Rajan, B.; Lokesh, J.; Kiron, V.; Brinchmann, M.F. Differentially Expressed Proteins in the Skin Mucus of Atlantic Cod (Gadus morhua) upon Natural Infection with Vibrio Anguillarum. BMC Vet. Res. 2013, 9, 103. [Google Scholar] [CrossRef]
- Nuez-Ortín, W.G.; Carter, C.G.; Nichols, P.D.; Cooke, I.R.; Wilson, R. Liver Proteome Response of Pre-Harvest Atlantic Salmon Following Exposure to Elevated Temperature. BMC Genom. 2018, 19, 133. [Google Scholar] [CrossRef]
- Lescat, L.; Véeron, V.; Mourot, B.; Péron, S.; Chenais, N.; Dias, K.; Riera-Heredia, N.; Beaumatin, F.; Pinel, K.; Priault, M.; et al. Chaperone-Mediated Autophagy in the Light of Evolution: Insight from Fish. Mol. Biol. Evol. 2020, 37, 2887–2899. [Google Scholar] [CrossRef]
- Lescat, L.; Herpin, A.; Mourot, B.; Véron, V.; Guiguen, Y.; Bobe, J.; Seiliez, I. CMA Restricted to Mammals and Birds: Myth or Reality? Autophagy 2018, 14, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, O.D.; Silva, T.S.; Alves, R.N.; Costas, B.; Wulff, T.; Richard, N.; de Vareilles, M.; Conceição, L.E.C.; Rodrigues, P.M. Changes in Liver Proteome Expression of Senegalese Sole (Solea senegalensis) in Response to Repeated Handling Stress. Mar. Biotechnol. 2012, 14, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Matos, E.; Silva, T.S.; Wulff, T.; Valente, L.M.P.; Sousa, V.; Sampaio, E.; Gonçalves, A.; Silva, J.M.G.; Guedes de Pinho, P.; Dinis, M.T.; et al. Influence of Supplemental Maslinic Acid (Olive-Derived Triterpene) on the Post-Mortem Muscle Properties and Quality Traits of Gilthead Seabream. Aquaculture 2013, 396–399, 146–155. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy: Many Paths to the Same End. Mol. Cell. Biochem. 2004, 263, 55–72. [Google Scholar] [CrossRef] [PubMed]
- López-Hernández, T.; Haucke, V.; Maritzen, T. Endocytosis in the Adaptation to Cellular Stress. Cell Stress 2020, 4, 230. [Google Scholar] [CrossRef]
- Wiseman, S.; Osachoff, H.; Bassett, E.; Malhotra, J.; Bruno, J.; VanAggelen, G.; Mommsen, T.P.; Vijayan, M.M. Gene Expression Pattern in the Liver during Recovery from an Acute Stressor in Rainbow Trout. Comp. Biochem. Physiol.-Part D Genom. Proteom. 2007, 2, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Salmerón, C.; Navarro, I.; Johnston, I.A.; Gutiérrez, J.; Capilla, E. Characterisation and Expression Analysis of Cathepsins and Ubiquitin-Proteasome Genes in Gilthead Sea Bream (Sparus aurata) Skeletal Muscle. BMC Res. Notes 2015, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Faught, E. Mechanisms of Cortisol Action in Fish Hepatocytes. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2016, 199, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.; Bánhegyi, G. The ER–Glycogen Particle–Phagophore Triangle: A Hub Connecting Glycogenolysis and Glycophagy? Pathol. Oncol. Res. 2018, 24, 821–826. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Zoncu, R. The Lysosome as a Cellular Centre for Signalling, Metabolism and Quality Control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Saftig, P.; Puertollano, R. How Lysosomes Sense, Integrate, and Cope with Stress. Trends Biochem. Sci. 2021, 46, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Lakpa, K.L.; Khan, N.; Afghah, Z.; Chen, X.; Geiger, J.D. Lysosomal Stress Response (LSR): Physiological Importance and Pathological Relevance. J. Neuroimmune Pharmacol. 2021, 16, 219. [Google Scholar] [CrossRef]
- Köhler, A.; Wahl, E.; Söffker, K. Functional and Morphological Changes of Lysosomes as Prognostic Biomarkers of Toxic Liver Injury in a Marine Flatfish (Platichthys flesus (L.)). Environ. Toxicol. Chem. 2002, 21, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Heberle, A.M.; Prentzell, M.T.; van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular Mechanisms of MTOR Regulation by Stress. Mol. Cell. Oncol. 2015, 2, e970489. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional Transport of Amino Acids Regulates MTOR and Autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-TRNA Synthetase Is an Intracellular Leucine Sensor for the MTORC1-Signaling Pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef]
- Johnston, I.A.; Bower, N.I.; Macqueen, D.J. Growth and the Regulation of Myotomal Muscle Mass in Teleost Fish. J. Exp. Biol. 2011, 214, 1617–1628. [Google Scholar] [CrossRef]
- Vélez, E.J.; Lutfi, E.; Jiménez-Amilburu, V.; Riera-Codina, M.; Capilla, E.; Navarro, I.; Gutiérrez, J. IGF-I and Amino Acids Effects through TOR Signaling on Proliferation and Differentiation of Gilthead Sea Bream Cultured Myocytes. Gen. Comp. Endocrinol. 2014, 205, 296–304. [Google Scholar] [CrossRef]
- Farinha, A.P.; Schrama, D.; Silva, T.; Conceição, L.E.C.; Colen, R.; Engrola, S.; Rodrigues, P.; Cerqueira, M. Evaluating the Impact of Methionine-Enriched Diets in the Liver of European Seabass through Label-Free Shotgun Proteomics. J. Proteom. 2021, 232, 104047. [Google Scholar] [CrossRef]
- Han, S.L.; Wang, J.; Li, L.Y.; Lu, D.L.; Chen, L.Q.; Zhang, M.L.; Du, Z.Y. The Regulation of Rapamycin on Nutrient Metabolism in Nile Tilapia Fed with High-Energy Diet. Aquaculture 2020, 520, 734975. [Google Scholar] [CrossRef]
- Sáez-Arteaga, A.; Wu, Y.; Silva-Marrero, J.I.; Rashidpour, A.; Almajano, M.P.; Fernández, F.; Baanante, I.V.; Metón, I. Gene Markers of Dietary Macronutrient Composition and Growth in the Skeletal Muscle of Gilthead Sea Bream (Sparus aurata). Aquaculture 2022, 555, 738221. [Google Scholar] [CrossRef]
- Martos-Sitcha, J.A.; Mancera, J.M.; Calduch-Giner, J.A.; Yúfera, M.; Martínez-Rodríguez, G.; Pérez-Sánchez, J. Unraveling the Tissue-Specific Gene Signatures of Gilthead Sea Bream (Sparus aurata L.) after Hyper- and Hypo-Osmotic Challenges. PLoS ONE 2016, 11, e0148113. [Google Scholar] [CrossRef]
- Yoo, H.-C.; Han, J.-M.; Yoo, H.-C.; Han, J.-M. Amino Acid Metabolism in Cancer Drug Resistance. Cells 2022, 11, 140. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. AMPK–MTOR Signaling and Cellular Adaptations in Hypoxia. Int. J. Mol. Sci. 2021, 22, 9765. [Google Scholar] [CrossRef] [PubMed]
- Jibb, L.A.; Richards, J.G. AMP-Activated Protein Kinase Activity during Metabolic Rate Depression in the Hypoxic Goldfish, Carassius Auratus. J. Exp. Biol. 2008, 211, 3111–3122. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Cassidy, A.A.; Verhille, C.E.; Lamarre, S.G.; MacCormack, T.J. Diel Cycling Hypoxia Enhances Hypoxia Tolerance in Rainbow Trout (Oncorhynchus mykiss): Evidence of Physiological and Metabolic Plasticity. J. Exp. Biol. 2019, 222, jeb206045. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.A.; Lamarre, S.G. Activation of Oxygen-Responsive Pathways Is Associated with Altered Protein Metabolism in Arctic Char Exposed to Hypoxia. J. Exp. Biol. 2019, 222, jeb203901. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274. [Google Scholar] [CrossRef] [PubMed]
- Kierans, S.J.; Taylor, C.T. Regulation of Glycolysis by the Hypoxia-Inducible Factor (HIF): Implications for Cellular Physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Richards, J.G. Chapter 10 Metabolic and Molecular Responses of Fish to Hypoxia, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2009; Volume 27. [Google Scholar]
- Lardon, I.; Eyckmans, M.; Vu, T.N.; Laukens, K.; De Boeck, G.; Dommisse, R. 1H-NMR Study of the Metabolome of a Moderately Hypoxia-Tolerant Fish, the Common Carp (Cyprinus carpio). Metabolomics 2013, 9, 1216–1227. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an Intermediate in Metabolism, Signal Transduction, ROS, Hypoxia, and Tumorigenesis. Biochim. Biophys. Acta-Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, P.; Wang, Y.; Tao, K. Itaconate: A Metabolite Regulates Inflammation Response and Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5404780. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Deng, M.; Scott, M.J.; Fu, G.; Loughran, P.A.; Lei, Z.; Li, S.; Sun, P.; Yang, C.; Li, W.; et al. Immune-Responsive Gene 1/Itaconate Activates Nuclear Factor Erythroid 2–Related Factor 2 in Hepatocytes to Protect Against Liver Ischemia–Reperfusion Injury. Hepatology 2020, 72, 1394–1411. [Google Scholar] [CrossRef] [PubMed]
- Scarl, R.T.; Lawrence, C.M.; Gordon, H.M.; Nunemaker, C.S. STEAP4: Its Emerging Role in Metabolism and Homeostasis of Cellular and Copper. J. Endocrinol. 2017, 234, R123. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-Pot, Solid-Phase-Enhanced Sample Preparation for Proteomics Experiments. Nat. Protoc. 2018, 14, 68–85. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical Statistical Model to Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange Consortium in 2020: Enabling ‘Big Data’ Approaches in Proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Gloaguen, Y.; Morton, F.; Daly, R.; Gurden, R.; Rogers, S.; Wandy, J.; Wilson, D.; Barrett, M.; Burgess, K. PiMP My Metabolome: An Integrated, Web-Based Tool for LC-MS Metabolomics Data. Bioinformatics 2017, 33, 4007–4009. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Scheltema, R.A.; Jankevics, A.; Jansen, R.C.; Swertz, M.A.; Breitling, R. PeakML/MzMatch: A File Format, Java Library, R Library, and Tool-Chain for Mass Spectrometry Data Analysis. Anal. Chem. 2011, 83, 2786–2793. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A Resource Evolving in Response to the Needs of Its Scientific Community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; ISBN 3-900051-07-0. [Google Scholar]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package. Version 1.0.7. 2020. Available online: https://cran.r-project.org/web/packages/factoextra/readme/README.html (accessed on 29 August 2022).
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. MixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. R Packag. Version 1.16.0. 2022. Available online: https://www.bioconductor.org/packages/devel/bioc/manuals/enrichplot/man/enrichplot.pdf (accessed on 29 August 2022).
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction Networks of Chemicals and Proteins. Nucleic Acids Res. 2008, 36, D684. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome Pathway Analysis: A High-Performance in-Memory Approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raposo de Magalhães, C.; Farinha, A.P.; Blackburn, G.; Whitfield, P.D.; Carrilho, R.; Schrama, D.; Cerqueira, M.; Rodrigues, P.M. Gilthead Seabream Liver Integrative Proteomics and Metabolomics Analysis Reveals Regulation by Different Prosurvival Pathways in the Metabolic Adaptation to Stress. Int. J. Mol. Sci. 2022, 23, 15395. https://doi.org/10.3390/ijms232315395
Raposo de Magalhães C, Farinha AP, Blackburn G, Whitfield PD, Carrilho R, Schrama D, Cerqueira M, Rodrigues PM. Gilthead Seabream Liver Integrative Proteomics and Metabolomics Analysis Reveals Regulation by Different Prosurvival Pathways in the Metabolic Adaptation to Stress. International Journal of Molecular Sciences. 2022; 23(23):15395. https://doi.org/10.3390/ijms232315395
Chicago/Turabian StyleRaposo de Magalhães, Cláudia, Ana Paula Farinha, Gavin Blackburn, Phillip D. Whitfield, Raquel Carrilho, Denise Schrama, Marco Cerqueira, and Pedro M. Rodrigues. 2022. "Gilthead Seabream Liver Integrative Proteomics and Metabolomics Analysis Reveals Regulation by Different Prosurvival Pathways in the Metabolic Adaptation to Stress" International Journal of Molecular Sciences 23, no. 23: 15395. https://doi.org/10.3390/ijms232315395
APA StyleRaposo de Magalhães, C., Farinha, A. P., Blackburn, G., Whitfield, P. D., Carrilho, R., Schrama, D., Cerqueira, M., & Rodrigues, P. M. (2022). Gilthead Seabream Liver Integrative Proteomics and Metabolomics Analysis Reveals Regulation by Different Prosurvival Pathways in the Metabolic Adaptation to Stress. International Journal of Molecular Sciences, 23(23), 15395. https://doi.org/10.3390/ijms232315395