

Branched Poly(ε-caprolactone)-Based Copolyesters of Different Architectures and Their Use in the Preparation of Anticancer Drug-Loaded Nanoparticles

 ,
,  ,
,  and
and 
Abstract
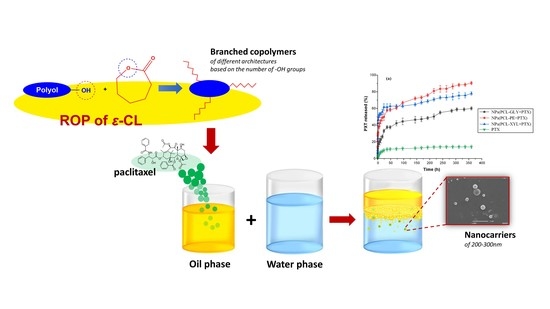
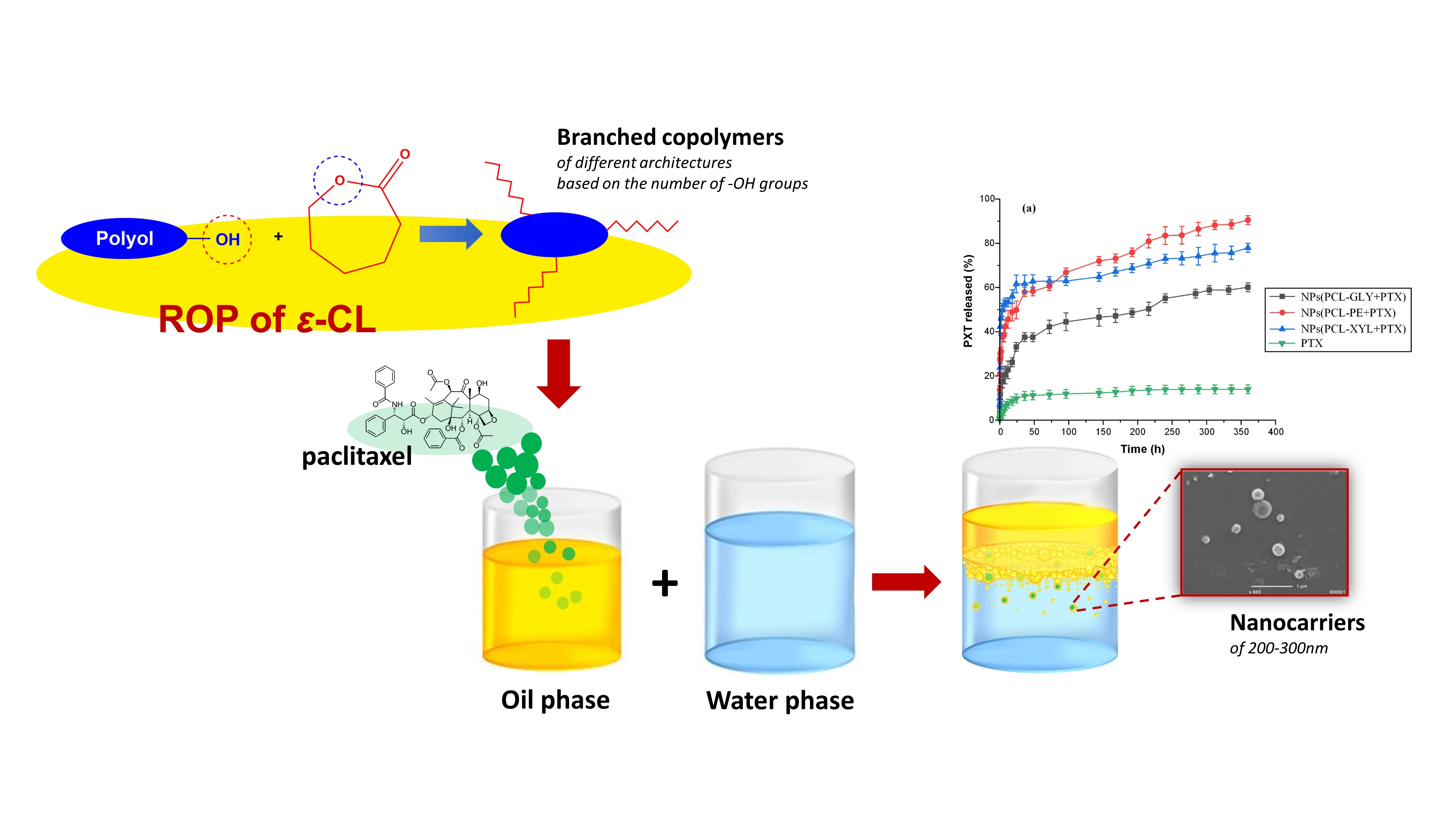
1. Introduction
2. Results and Discussion
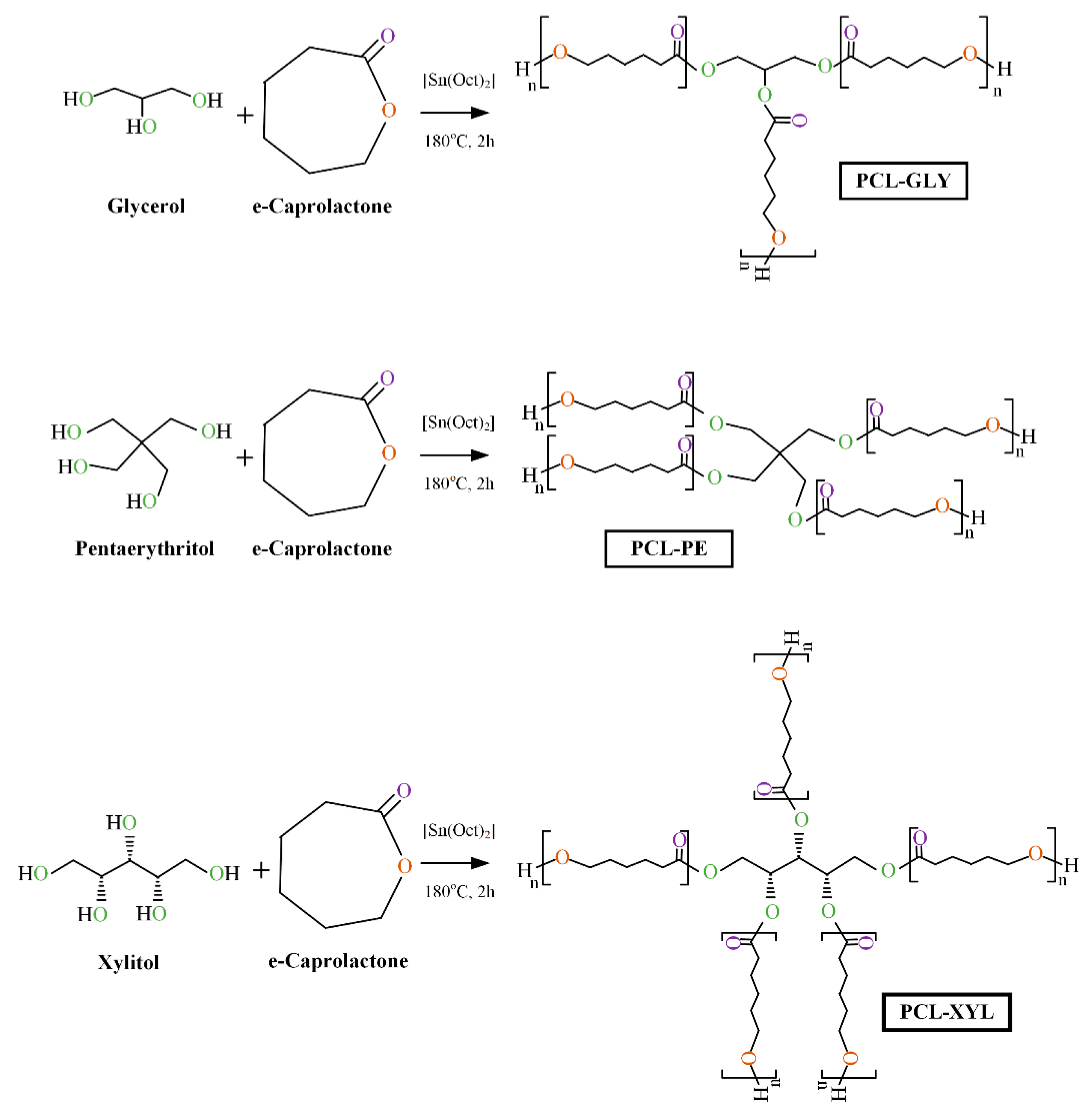
2.1. Branched PCL/Polyol Copolymers Synthesis
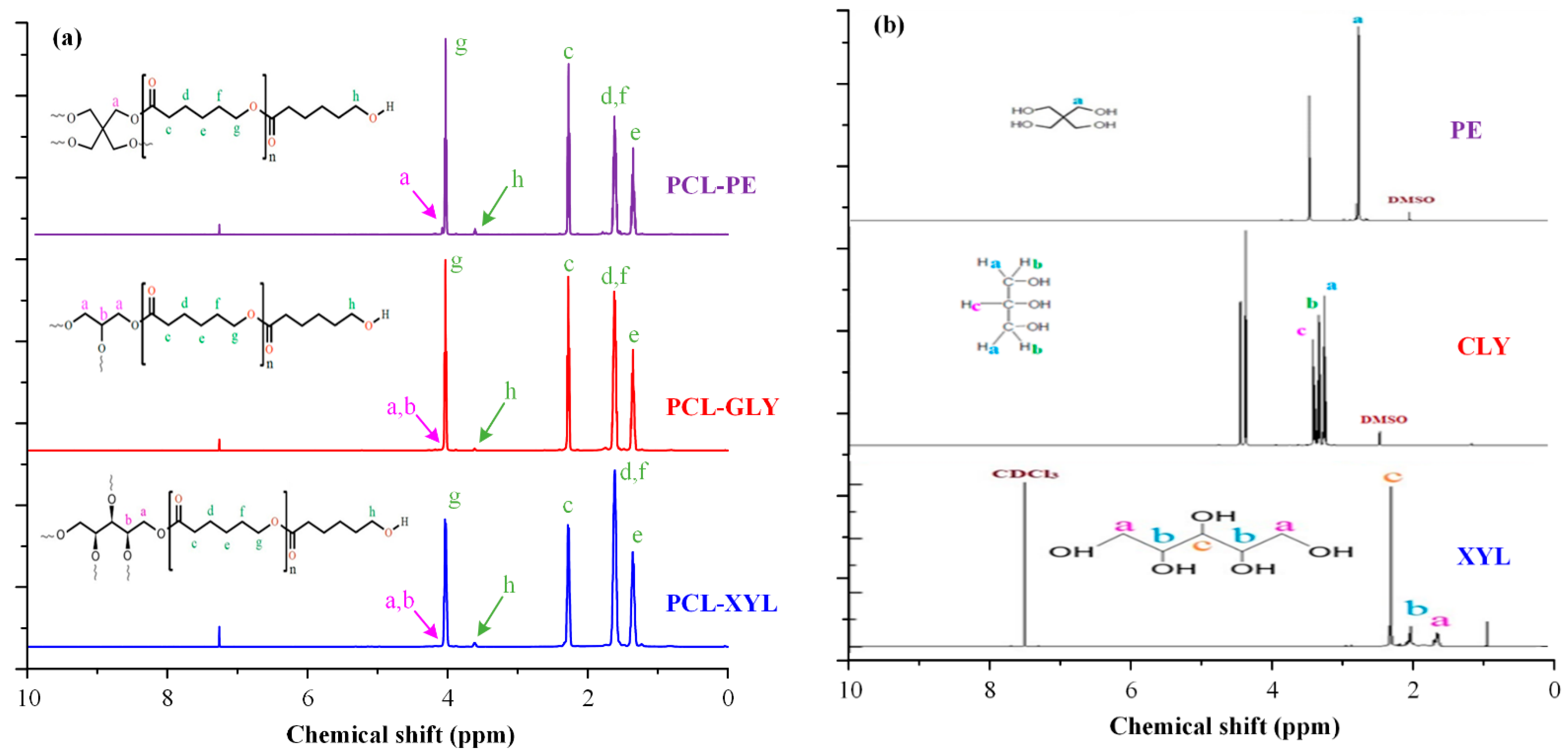
2.1.1. H NMR Analysis
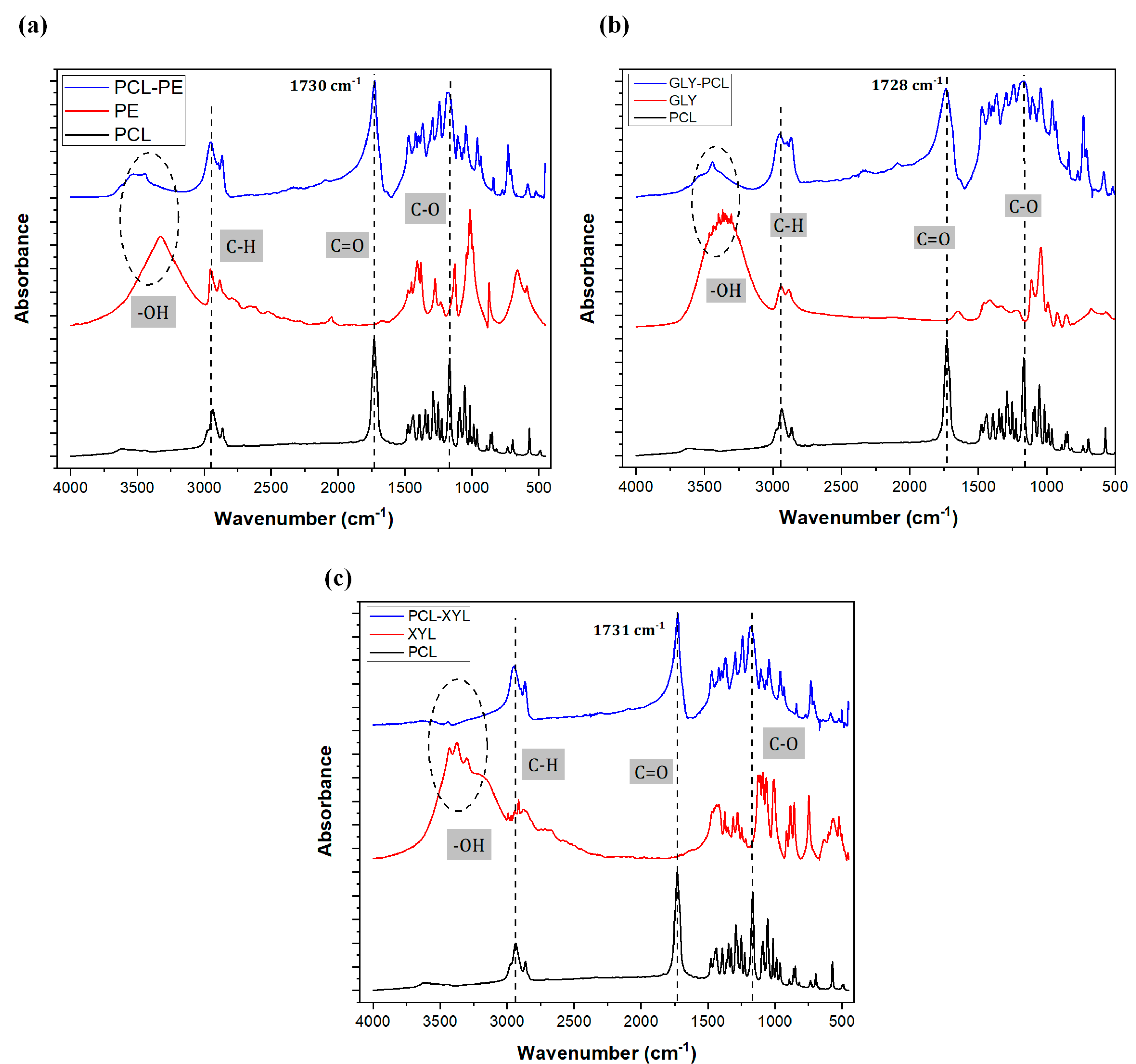
2.1.2. FTIR Analysis
2.2. Copolymer Characterization
2.2.1. Molecular Weight and Intrinsic Viscosity
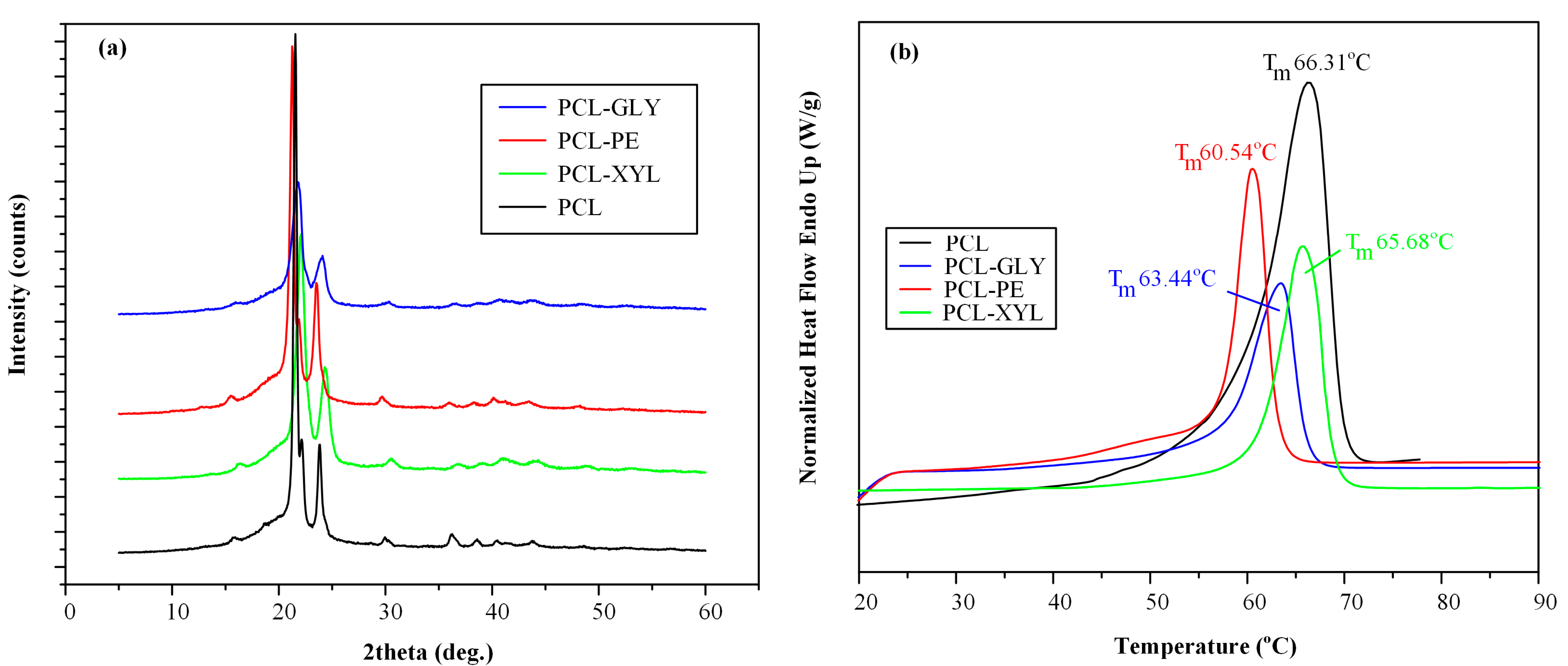
2.2.2. Physical State and Thermal Properties Evaluation
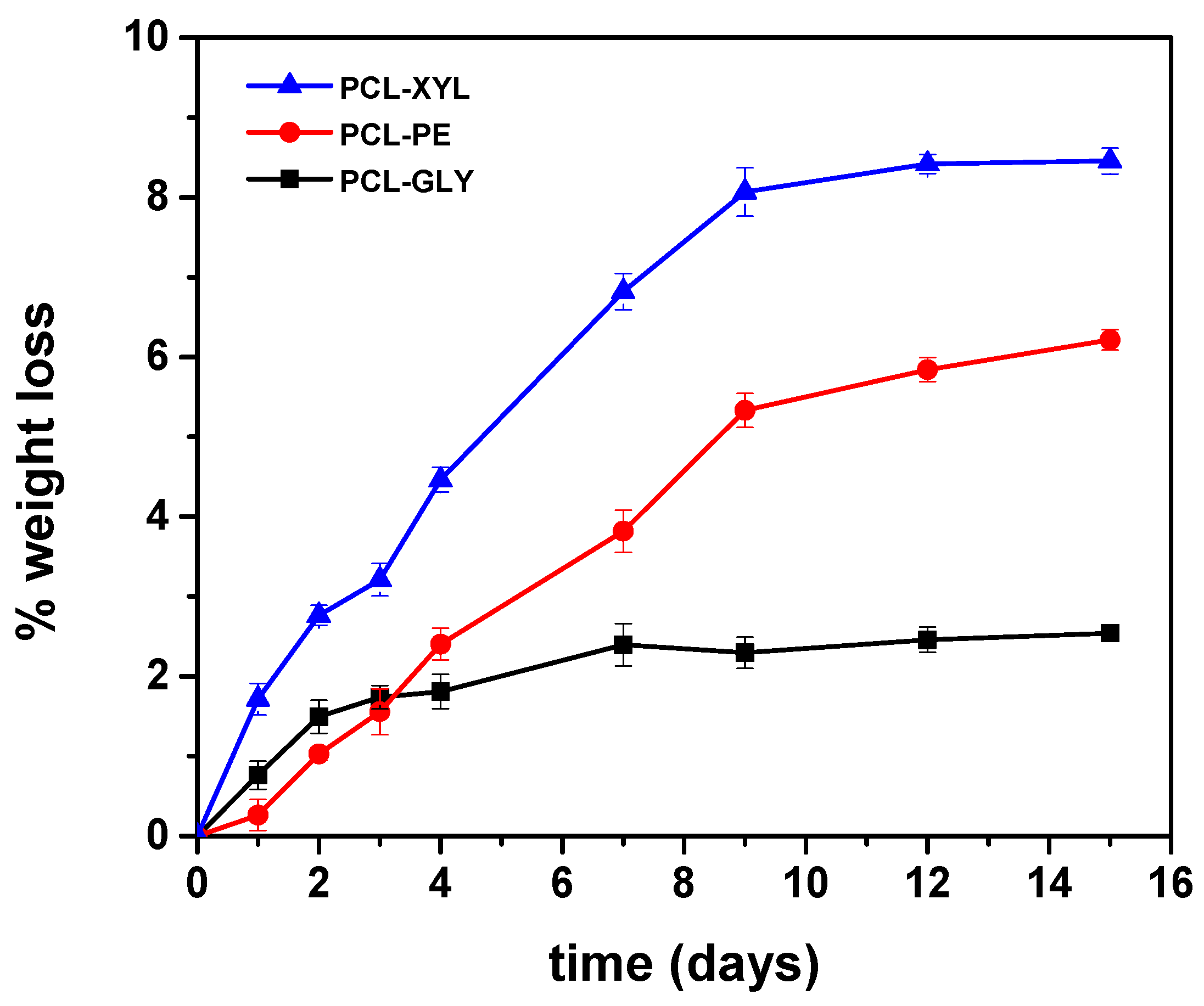
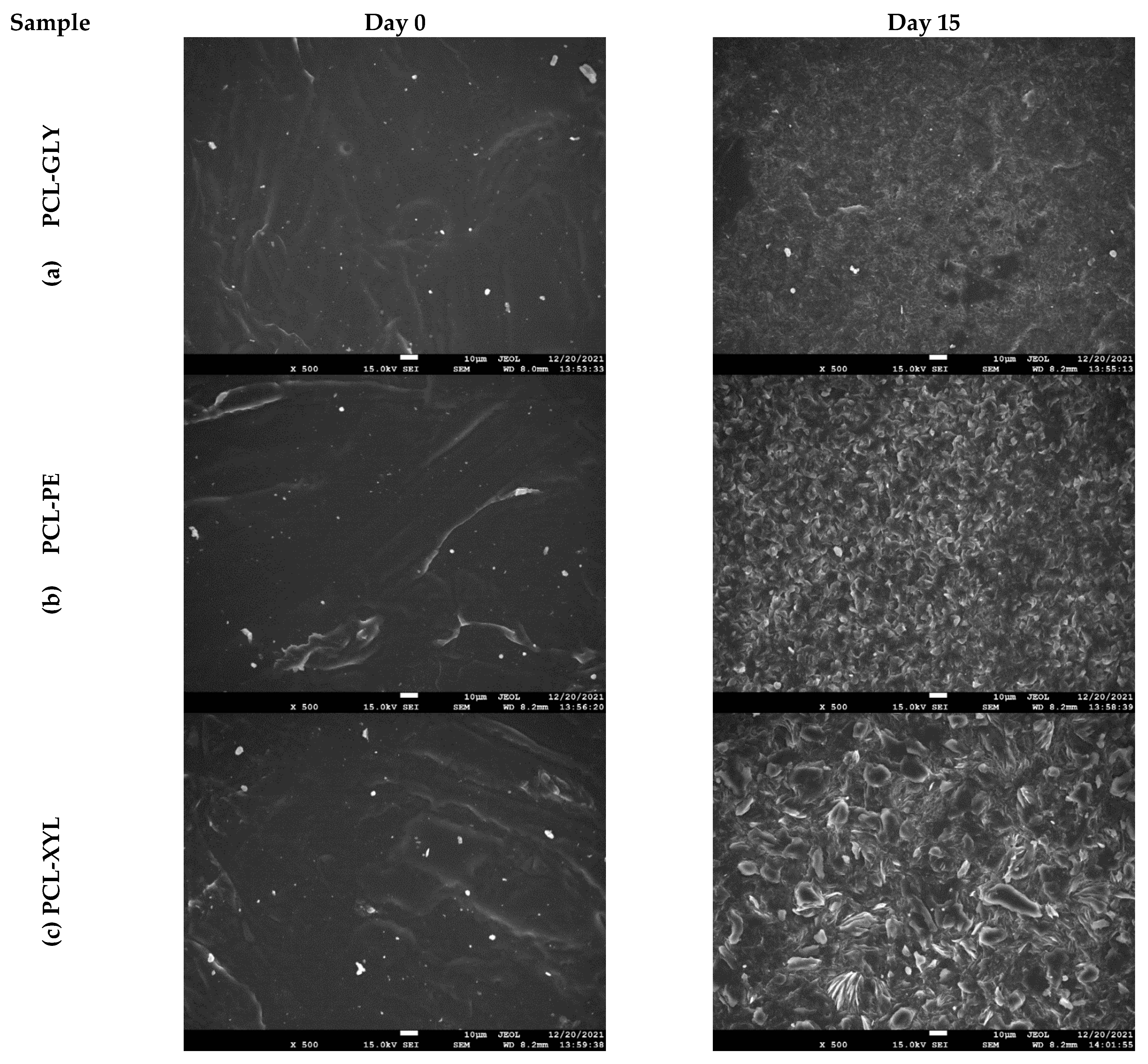
2.2.3. Enzymatic Hydrolysis
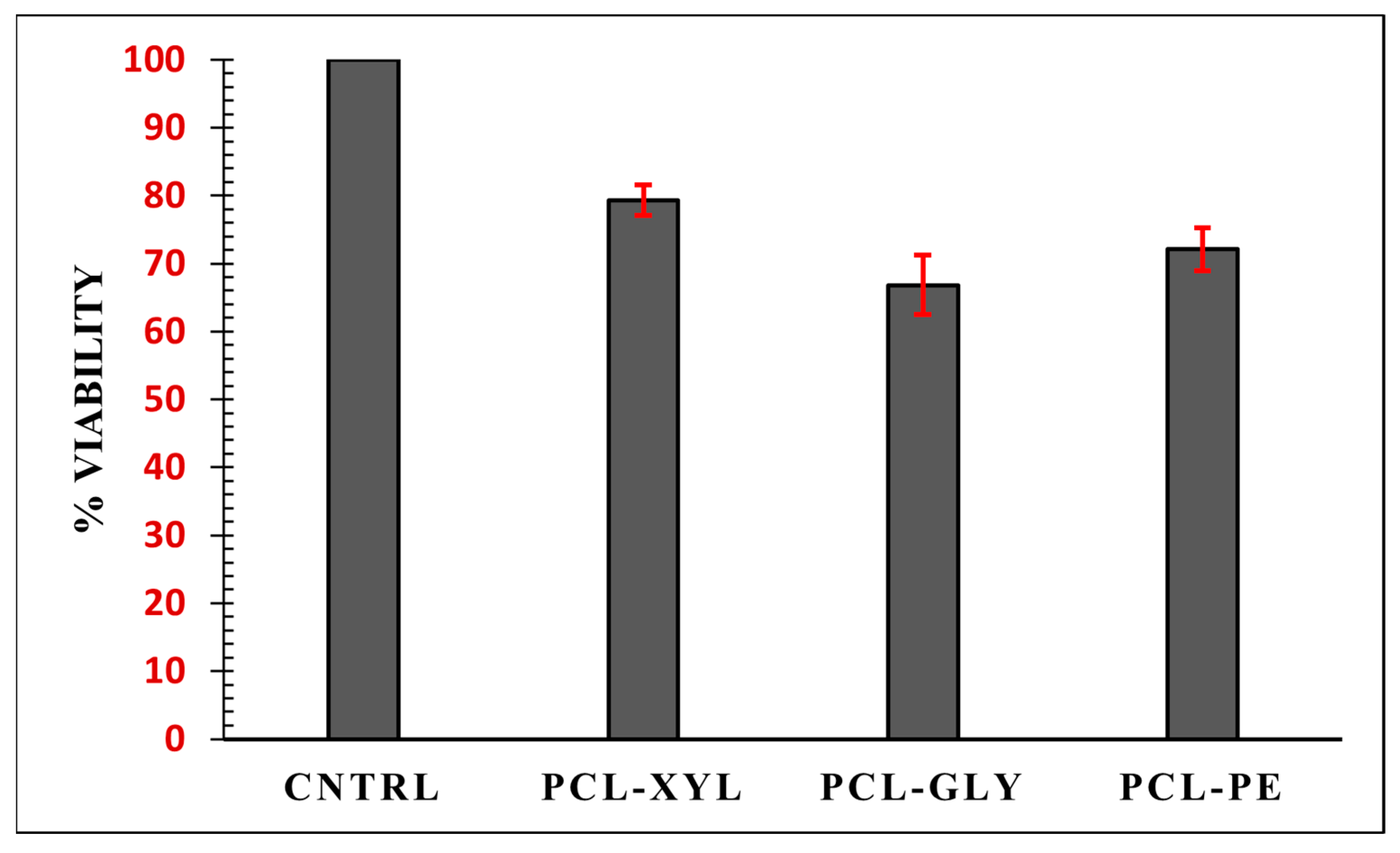
2.2.4. Cytotoxicity Evaluation
2.3. PTX-Loaded NPs Characterization
2.3.1. NPs’ Morphology and Size
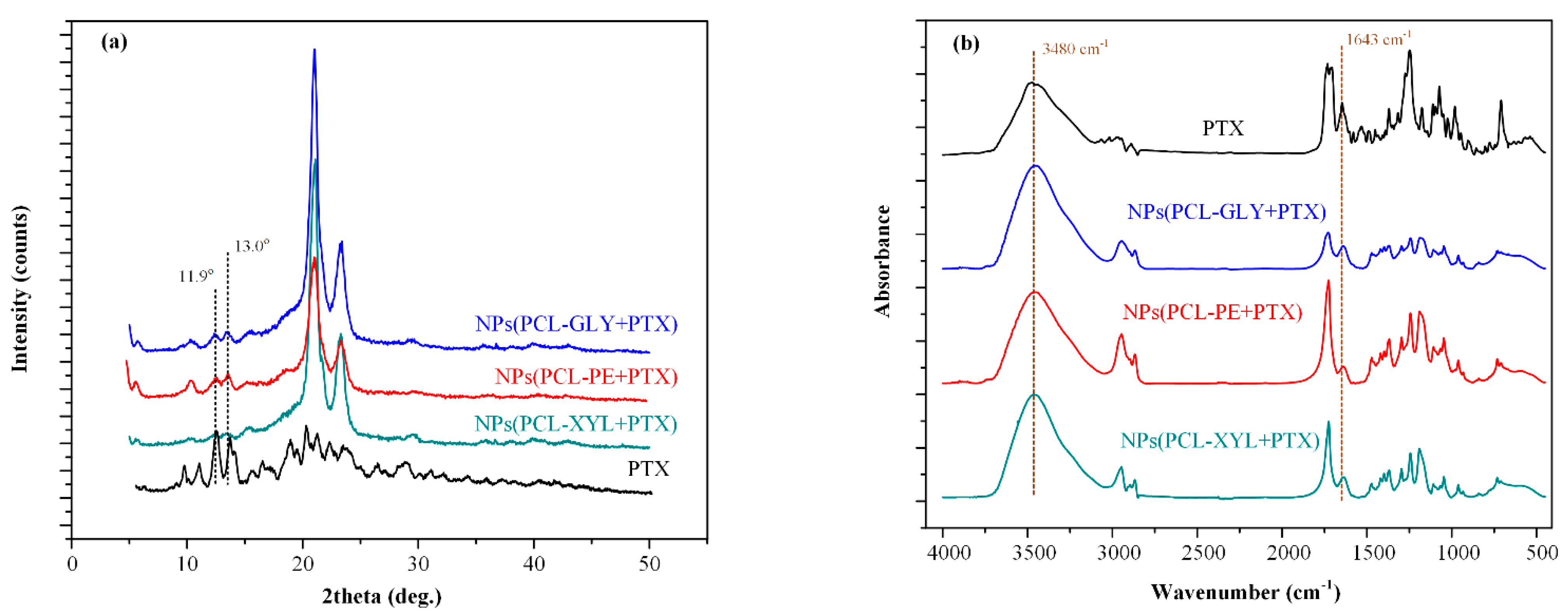
2.3.2. PTX Physical State Evaluation
2.3.3. PTX-Copolymer Molecular Interactions
2.3.4. Drug Loading, Encapsulation Efficiency (EE), and Yield
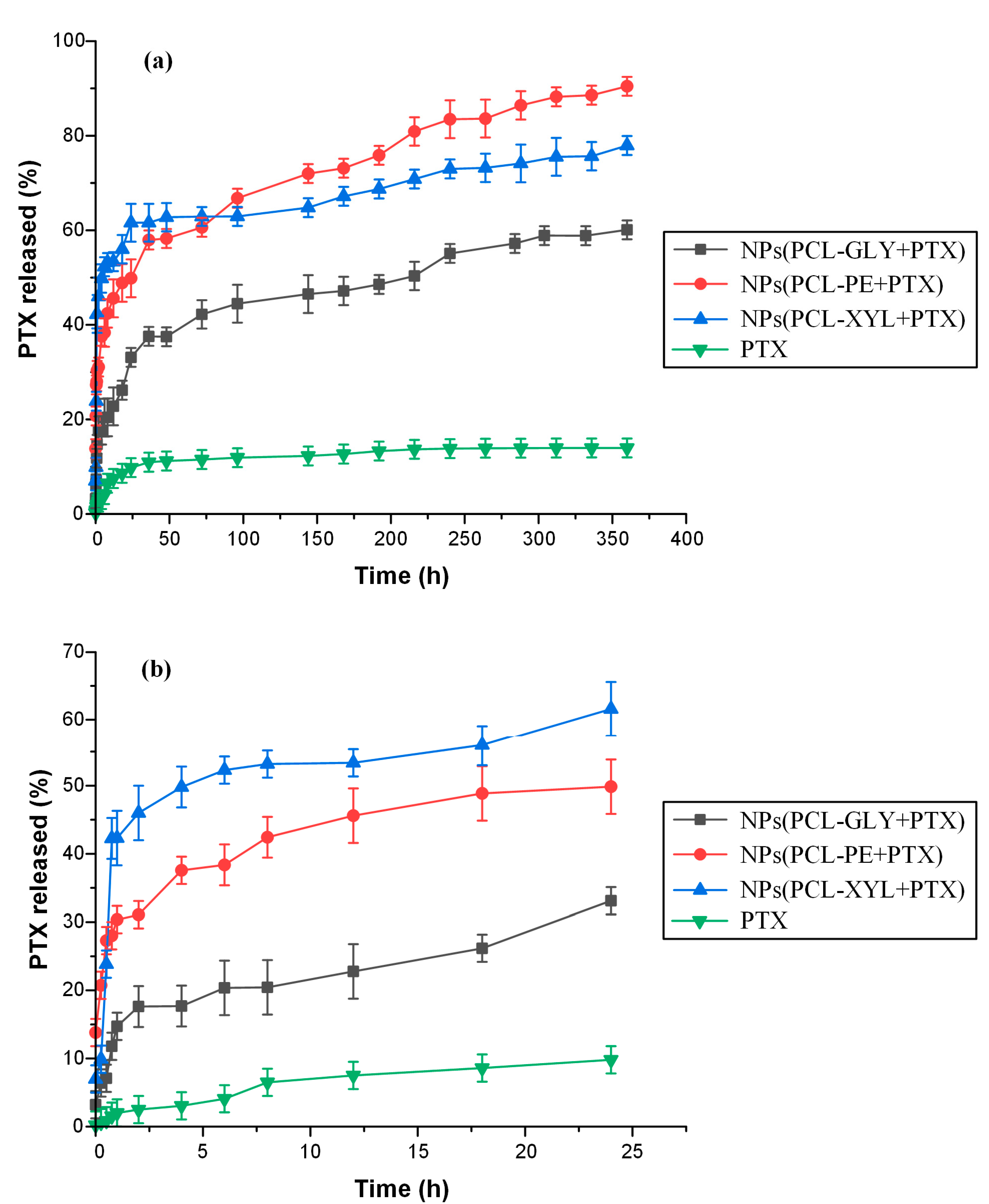
2.3.5. In Vitro Drug Dissolution Studies
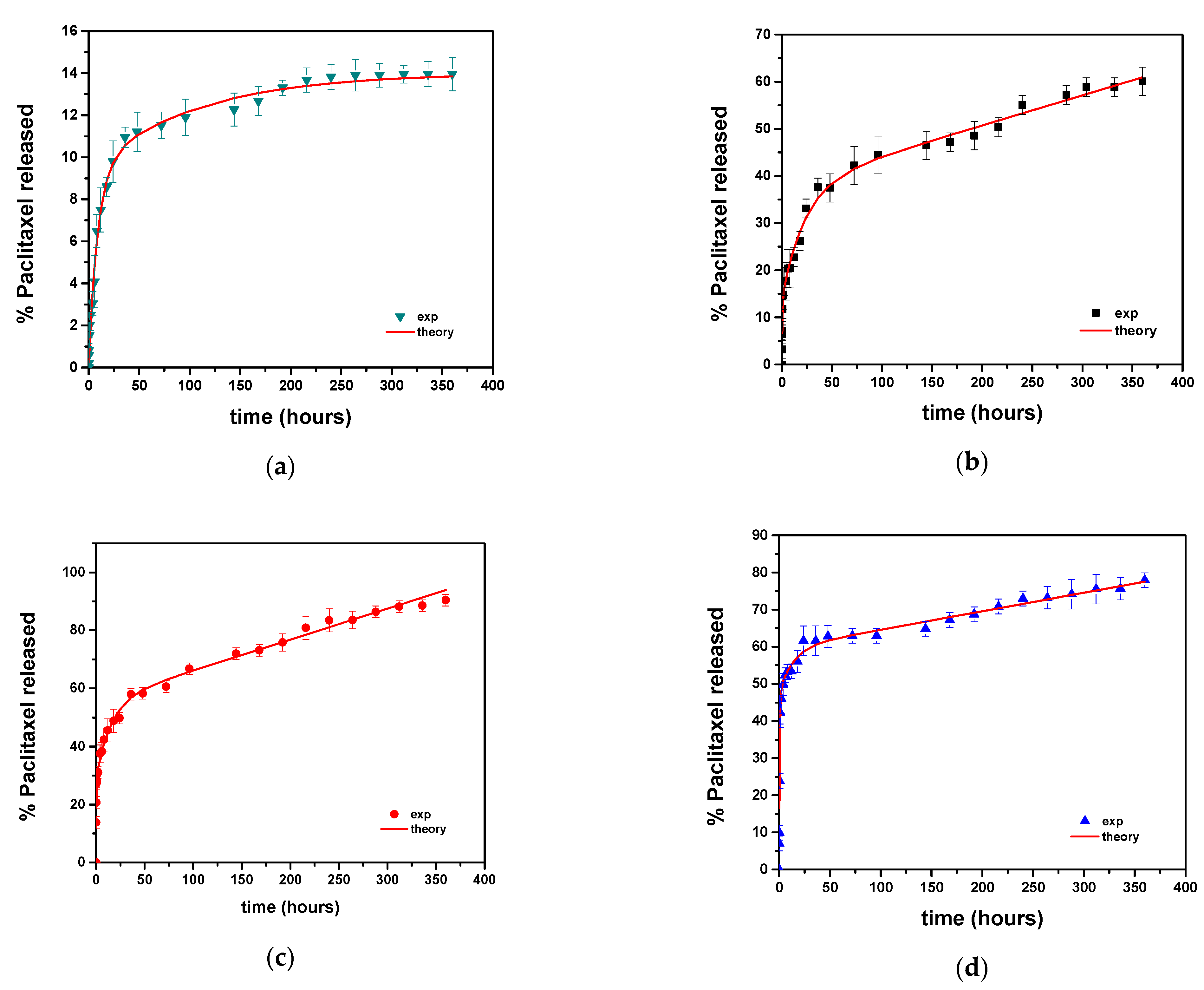
2.3.6. Modeling of Release Kinetics
3. Materials and Methods
3.1. Materials
3.2. Preparation of the Synthesized Branched PCL Copolymers
3.3. Characterization of the Synthesized Branched PCL Copolymers
3.3.1. Nuclear Magnetic Resonance Spectroscopy
3.3.2. Intrinsic Viscosity
3.3.3. Powder X-Ray (pXRD) Diffractometry
3.3.4. Fourier-Transform Infrared (FTIR) Spectroscopy
3.3.5. Differential Scanning Calorimetry (DSC)
3.3.6. Size Exclusion Chromatography (SEC)
3.3.7. Enzymatic Hydrolysis
3.3.8. Cytotoxicity Studies
3.4. Preparation of NPs
3.5. Characterization of NPs
3.5.1. Physical State Evaluation
3.5.2. Evaluation of Molecular Interactions
3.5.3. Evaluation of Particle Size Distribution (PSD) and NPs’ Morphology
3.5.4. Drug Loading, Encapsulation Efficiency (EE), and Yield
3.5.5. In Vitro Dissolution Studies
3.5.6. HPLC Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grossen, P.; Witzigmann, D.; Sieber, S.; Huwyler, J. Peg-pcl-based nanomedicines: A biodegradable drug delivery system and its application. J. Control. Release 2017, 260, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, E.A.J.; de Jong, W.H.; Geertsma, R.E.; Groenewold, M.; Heugens, E.H.W.; Koers-Jacquemijns, M.; van de Meent, D.; Popma, J.R.; Rietveld, A.G.; Wijnhoven, S.W.P.; et al. Considerations on the eu definition of a nanomaterial: Science to support policy making. Regul. Toxicol. Pharmacol. 2013, 65, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. The first targeted delivery of sirna in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef]
- Huang, H.-C.; Barua, S.; Sharma, G.; Dey, S.K.; Rege, K. Inorganic nanoparticles for cancer imaging and therapy. J. Control. Release 2011, 155, 344–357. [Google Scholar] [CrossRef]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for her2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Fang, J.-Y.; Al-Suwayeh, S.A. Nanoparticles as delivery carriers for anticancer prodrugs. Expert Opin. Drug Deliv. 2012, 9, 657–669. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase iii trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]
- Mauri, E.; Papa, S.; Masi, M.; Veglianese, P.; Rossi, F. Novel functionalization strategies to improve drug delivery from polymers. Expert Opin. Drug Deliv. 2017, 14, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Masotti, A. Multifunctional nanoparticles, nanocages and degradable polymers as a potential novel generation of non-invasive molecular and cellular imaging systems. Recent Pat. Nanotechnol. 2011, 5, 163–177. [Google Scholar] [CrossRef]
- Shaw, Z.; Patel, A.; Butcher, T.; Banerjee, T.; Bean, R.; Santra, S. Pseudo-branched polyester copolymer: An efficient drug delivery system to treat cancer. Biomater. Sci. 2020, 8, 1592–1603. [Google Scholar] [CrossRef]
- Guzmán, E.; Cavallo, J.A.; Chuliá-Jordán, R.; Gómez, C.; Strumia, M.C.; Ortega, F.; Rubio, R.G. Ph-induced changes in the fabrication of multilayers of poly(acrylic acid) and chitosan: Fabrication, properties, and tests as a drug storage and delivery system. Langmuir 2011, 27, 6836–6845. [Google Scholar] [CrossRef]
- Chen, H.; He, S. Pla–peg coated multifunctional imaging probe for targeted drug delivery. Mol. Pharm. 2015, 12, 1885–1892. [Google Scholar] [CrossRef]
- Laquintana, V.; Denora, N.; Lopalco, A.; Lopedota, A.; Cutrignelli, A.; Lasorsa, F.M.; Agostino, G.; Franco, M. Translocator protein ligand–plga conjugated nanoparticles for 5-fluorouracil delivery to glioma cancer cells. Mol. Pharm. 2014, 11, 859–871. [Google Scholar] [CrossRef]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized plga–peg nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef]
- Joshi, G.; Patel, M.; Chaudhary, D.; Sawant, K. Preparation and surface modification of polymeric nanoparticles for drug delivery: State of the art. Recent Pat. Drug Deliv. Formul. 2020, 14, 201–213. [Google Scholar] [CrossRef]
- Mahmoudi Saber, M. Strategies for surface modification of gelatin-based nanoparticles. Colloids Surf. B Biointerfaces 2019, 183, 110407. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sheng, Y.; Shi, J.; Yu, B.; Yu, Z.; Liao, G. Long circulating polymeric nanoparticles for gene/drug delivery. Curr. Drug Metab. 2018, 19, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Severino, P.; da Silva, C.F.; Andrade, L.N.; de Lima Oliveira, D.; Campos, J.; Souto, E.B. Alginate nanoparticles for drug delivery and targeting. Curr. Pharm. Des. 2019, 25, 1312–1334. [Google Scholar] [PubMed]
- Xiang, F.; Stuart, M.; Vorenkamp, J.; Roest, S.; Timmer-Bosscha, H.; Stuart, M.C.; Fokkink, R.; Loontjens, T. One-pot synthesis for biocompatible amphiphilic hyperbranched polyurea micelles. Macromolecules 2013, 46, 4418–4425. [Google Scholar] [CrossRef]
- Kainthan, R.K.; Brooks, D.E. Unimolecular micelles based on hydrophobically derivatized hyperbranched polyglycerols: Biodistribution studies. Bioconjug. Chem. 2008, 19, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, H.; Liu, J.; Deng, L.; Liu, J.; Dong, A.; Zhang, J. Comb-like amphiphilic copolymers bearing acetal-functionalized backbones with the ability of acid-triggered hydrophobic-to-hydrophilic transition as effective nanocarriers for intracellular release of curcumin. Biomacromolecules 2013, 14, 3973–3984. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.E.; Wei, H.; Tan, N.; Boydston, A.J.; Pun, S.H. Sunflower polymers for folate-mediated drug delivery. Biomacromolecules 2016, 17, 69–75. [Google Scholar] [CrossRef]
- Hong, S.; Leroueil, P.R.; Janus, E.K.; Peters, J.L.; Kober, M.-M.; Islam, M.T.; Orr, B.G.; Baker, J.R.; Banaszak Holl, M.M. Interaction of polycationic polymers with supported lipid bilayers and cells: Nanoscale hole formation and enhanced membrane permeability. Bioconjug. Chem. 2006, 17, 728–734. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Y.; Wei, L.; Teng, Y.G.; Honda, T.; Ojima, I. Design, synthesis, and biological evaluations of asymmetric bow-tie pamam dendrimer-based conjugates for tumor-targeted drug delivery. ACS Omega 2018, 3, 3717–3736. [Google Scholar] [CrossRef]
- Li, S.; Chen, B.; Qu, Y.; Yan, X.; Wang, W.; Ma, X.; Wang, B.; Liu, S.; Yu, X. Ros-response-induced zwitterionic dendrimer for gene delivery. Langmuir 2019, 35, 1613–1620. [Google Scholar] [CrossRef]
- Gao, C.; Yan, D.; Frey, H. Promising dendritic materials: An introduction to hyperbranched polymers. In Hyperbranched Polymers; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 1–26. [Google Scholar]
- Espinoza, S.M.; Patil, H.I.; San Martin Martinez, E.; Casañas Pimentel, R.; Ige, P.P. Poly-ε-caprolactone (pcl), a promising polymer for pharmaceutical and biomedical applications: Focus on nanomedicine in cancer. Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 85–126. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Sinha, V.R.; Bansal, K.; Kaushik, R.; Kumria, R.; Trehan, A. Poly-ϵ-caprolactone microspheres and nanospheres: An overview. Int. J. Pharm. 2004, 278, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Croisier, F.; Atanasova, G.; Poumay, Y.; Jérôme, C. Polysaccharide-coated pcl nanofibers for wound dressing applications. Adv. Healthc. Mater. 2014, 3, 2032–2039. [Google Scholar] [CrossRef]
- Mouro, C.; Simões, M.; Gouveia, I.C. Emulsion electrospun fiber mats of pcl/pva/chitosan and eugenol for wound dressing applications. Adv. Polym. Technol. 2019, 2019, 9859506. [Google Scholar] [CrossRef]
- Eskandarinia, A.; Kefayat, A.; Agheb, M.; Rafienia, M.; Amini Baghbadorani, M.; Navid, S.; Ebrahimpour, K.; Khodabakhshi, D.; Ghahremani, F. A novel bilayer wound dressing composed of a dense polyurethane/propolis membrane and a biodegradable polycaprolactone/gelatin nanofibrous scaffold. Sci. Rep. 2020, 10, 3063. [Google Scholar] [CrossRef]
- Corden, T.J.; Jones, I.A.; Rudd, C.D.; Christian, P.; Downes, S. Initial development into a novel technique for manufacturing a long fibre thermoplastic bioabsorbable composite: In-situ polymerisation of poly-ϵ-caprolactone. Compos. Part A Appl. Sci. Manuf. 1999, 30, 737–746. [Google Scholar] [CrossRef]
- Barbosa, J.A.P.; Franco, E.S.; Silva, C.V.N.S.; Bezerra, T.O.; Santana, M.A.N.; Júnior, C.H.R.C.; Silva, T.G.; Santos, N.P.S.; Maia, M.B.S. Poly-ε-caprolactone microsphere polymers containing usnic acid: Acute toxicity and anti-inflammatory activity. Evid.-Based Complement. Altern. Med. 2017, 2017, 7392891. [Google Scholar] [CrossRef]
- Cabeza, L.; Ortiz, R.; Prados, J.; Delgado, Á.V.; Martín-Villena, M.J.; Clares, B.; Perazzoli, G.; Entrena, J.M.; Melguizo, C.; Arias, J.L. Improved antitumor activity and reduced toxicity of doxorubicin encapsulated in poly(ε-caprolactone) nanoparticles in lung and breast cancer treatment: An in vitro and in vivo study. Eur. J. Pharm. Sci. 2017, 102, 24–34. [Google Scholar] [CrossRef]
- Forrest, M.L.; Yáñez, J.A.; Remsberg, C.M.; Ohgami, Y.; Kwon, G.S.; Davies, N.M. Paclitaxel prodrugs with sustained release and high solubility in poly(ethylene glycol)-b-poly(ε-caprolactone) micelle nanocarriers: Pharmacokinetic disposition, tolerability, and cytotoxicity. Pharm. Res. 2008, 25, 194–206. [Google Scholar] [CrossRef]
- Karanam, V.; Marslin, G.; Krishnamoorthy, B.; Chellan, V.; Siram, K.; Natarajan, T.; Bhaskar, B.; Franklin, G. Poly (ɛ-caprolactone) nanoparticles of carboplatin: Preparation, characterization and in vitro cytotoxicity evaluation in u-87 mg cell lines. Colloids Surf. B Biointerfaces 2015, 130, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, G.; Gupta, B.S.; Tonelli, A.E. Estimation of the poly (ε-caprolactone) [pcl] and α-cyclodextrin [α-cd] stoichiometric ratios in their inclusion complexes [ics], and evaluation of porosity and fiber alignment in pcl nanofibers containing these ics. Data Brief 2015, 5, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Bronich, T.K.; Kabanov, A.V.; Rauh, R.D.; Roovers, J. Synthesis and characterization of star poly(ε-caprolactone)-b-poly(ethylene glycol) and poly(l-lactide)-b-poly(ethylene glycol) copolymers: Evaluation as drug delivery carriers. Bioconjug. Chem. 2008, 19, 1423–1429. [Google Scholar] [CrossRef]
- Elzein, T.; Nasser-Eddine, M.; Delaite, C.; Bistac, S.; Dumas, P. Ftir study of polycaprolactone chain organization at interfaces. J. Colloid Interface Sci. 2004, 273, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Sasmal, A.; Nanda, R.; Phani, A.R.; Nayak, P.L. Synthesis of chitosan–polycaprolactone blend for control delivery of ofloxacin drug. Carbohydr. Polym. 2010, 79, 106–113. [Google Scholar] [CrossRef]
- Kredatusová, J.; Svobodová, J.; Brožek, J. Preparation of poly(ε-caprolactone) and its nanocomposites in ε-caprolactam. e-Polymers 2008, 8, 144. [Google Scholar] [CrossRef]
- Bikiaris, N.D.; Ainali, N.M.; Christodoulou, E.; Kostoglou, M.; Kehagias, T.; Papasouli, E.; Koukaras, E.N.; Nanaki, S.G. Dissolution enhancement and controlled release of paclitaxel drug via a hybrid nanocarrier based on mpeg-pcl amphiphilic copolymer and fe-btc porous metal-organic framework. Nanomaterials 2020, 10, 2490. [Google Scholar] [CrossRef]
- Christodoulou, E.; Nerantzaki, M.; Nanaki, S.; Barmpalexis, P.; Giannousi, K.; Dendrinou-Samara, C.; Angelakeris, M.; Gounari, E.; Anastasiou, A.D.; Bikiaris, D.N. Paclitaxel magnetic core–shell nanoparticles based on poly(lactic acid) semitelechelic novel block copolymers for combined hyperthermia and chemotherapy treatment of cancer. Pharmaceutics 2019, 11, 213. [Google Scholar] [CrossRef]
- Dordunoo, S.K.; Burt, H.M. Solubility and stability of taxol: Effects of buffers and cyclodextrins. Int. J. Pharm. 1996, 133, 191–201. [Google Scholar] [CrossRef]
- Koletti, A.E.; Tsarouchi, E.; Kapourani, A.; Kontogiannopoulos, K.N.; Assimopoulou, A.N.; Barmpalexis, P. Gelatin nanoparticles for nsaid systemic administration: Quality by design and artificial neural networks implementation. Int. J. Pharm. 2020, 578, 119118. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in ther own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Pappa, C.; Nanaki, S.; Giliopoulos, D.; Triantafyllidis, K.; Kostoglou, M.; Avgeropoulos, A.; Bikiaris, D. Nanostructured composites of sodium montmorillonite clay and peo used in dissolution improvement of aprepitant drug by melt mixing. Appl. Sci. 2018, 8, 786. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Crank, J.; Crank, E.P.J. The Mathematics of Diffusion; Clarendon Press: Oxford, UK, 1979. [Google Scholar]
- Brenner, H. Adsorption Calculations and Modelling; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Christodoulou, E.; Klonos, P.A.; Tsachouridis, K.; Zamboulis, A.; Kyritsis, A.; Bikiaris, D.N. Synthesis, crystallization, and molecular mobility in poly(ε-caprolactone) copolyesters of different architectures for biomedical applications studied by calorimetry and dielectric spectroscopy. Soft Matter 2020, 16, 8187–8201. [Google Scholar] [CrossRef] [PubMed]
- Solomon, O.F.; Ciutǎ, I.Z. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962, 6, 683–686. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn (g/mol) | Mw (g/mol) | PDI |
|---|---|---|---|
| PCL-GLY | 43,300 | 60,400 | 1.39 |
| PCL-PE | 29,000 | 32,900 | 1.13 |
| PCL-XYL | 21,116 | 26,438 | 1.25 |
| Sample | Yield (%) | Drug Loading (%) | EE (%) |
|---|---|---|---|
| NPs(PCL-GLY + PTΧ) | 96 ± 1.38 | 5.97 ± 1.39 | 60.09 ± 2.02 |
| NPs(PCL-PE + PTΧ) | 90 ± 2.56 | 6.58 ± 2.02 | 66.26 ± 2.85 |
| NPs(PCL-XYL + PTΧ) | 86 ± 1.84 | 7.42 ± 1.98 | 72.01 ± 2.70 |
| Sample | φ1 | φ2 | K1 (h−1) | K2 (h−1) | K3 (h−1) |
|---|---|---|---|---|---|
| PCL-GLY | 14.1 | 23.9 | 1.95 | 0.043 | 0.064 |
| PCL-PE | 29 | 26.4 | 4.65 | 0.066 | 0.106 |
| PCL-XYL | 47.7 | 11.9 | 1.68 | 0.072 | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christodoulou, E.; Notopoulou, M.; Nakiou, E.; Kostoglou, M.; Barmpalexis, P.; Bikiaris, D.N. Branched Poly(ε-caprolactone)-Based Copolyesters of Different Architectures and Their Use in the Preparation of Anticancer Drug-Loaded Nanoparticles. Int. J. Mol. Sci. 2022, 23, 15393. https://doi.org/10.3390/ijms232315393
Christodoulou E, Notopoulou M, Nakiou E, Kostoglou M, Barmpalexis P, Bikiaris DN. Branched Poly(ε-caprolactone)-Based Copolyesters of Different Architectures and Their Use in the Preparation of Anticancer Drug-Loaded Nanoparticles. International Journal of Molecular Sciences. 2022; 23(23):15393. https://doi.org/10.3390/ijms232315393
Chicago/Turabian StyleChristodoulou, Evi, Maria Notopoulou, Eirini Nakiou, Margaritis Kostoglou, Panagiotis Barmpalexis, and Dimitrios N. Bikiaris. 2022. "Branched Poly(ε-caprolactone)-Based Copolyesters of Different Architectures and Their Use in the Preparation of Anticancer Drug-Loaded Nanoparticles" International Journal of Molecular Sciences 23, no. 23: 15393. https://doi.org/10.3390/ijms232315393
APA StyleChristodoulou, E., Notopoulou, M., Nakiou, E., Kostoglou, M., Barmpalexis, P., & Bikiaris, D. N. (2022). Branched Poly(ε-caprolactone)-Based Copolyesters of Different Architectures and Their Use in the Preparation of Anticancer Drug-Loaded Nanoparticles. International Journal of Molecular Sciences, 23(23), 15393. https://doi.org/10.3390/ijms232315393