
Placental Transcriptome Profiling in Subtypes of Diabetic Pregnancies Is Strongly Confounded by Fetal Sex

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics of the Study Cohort
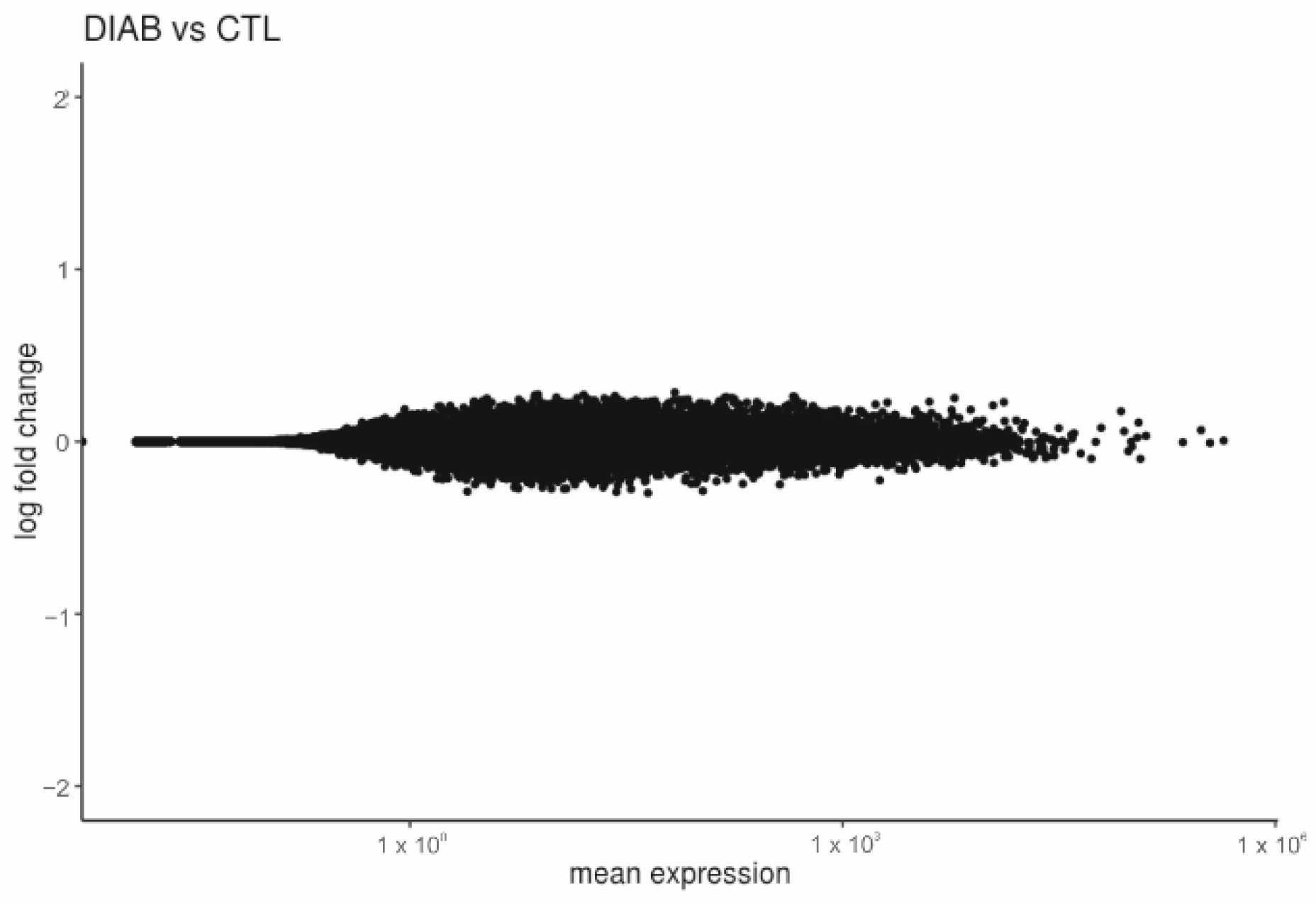
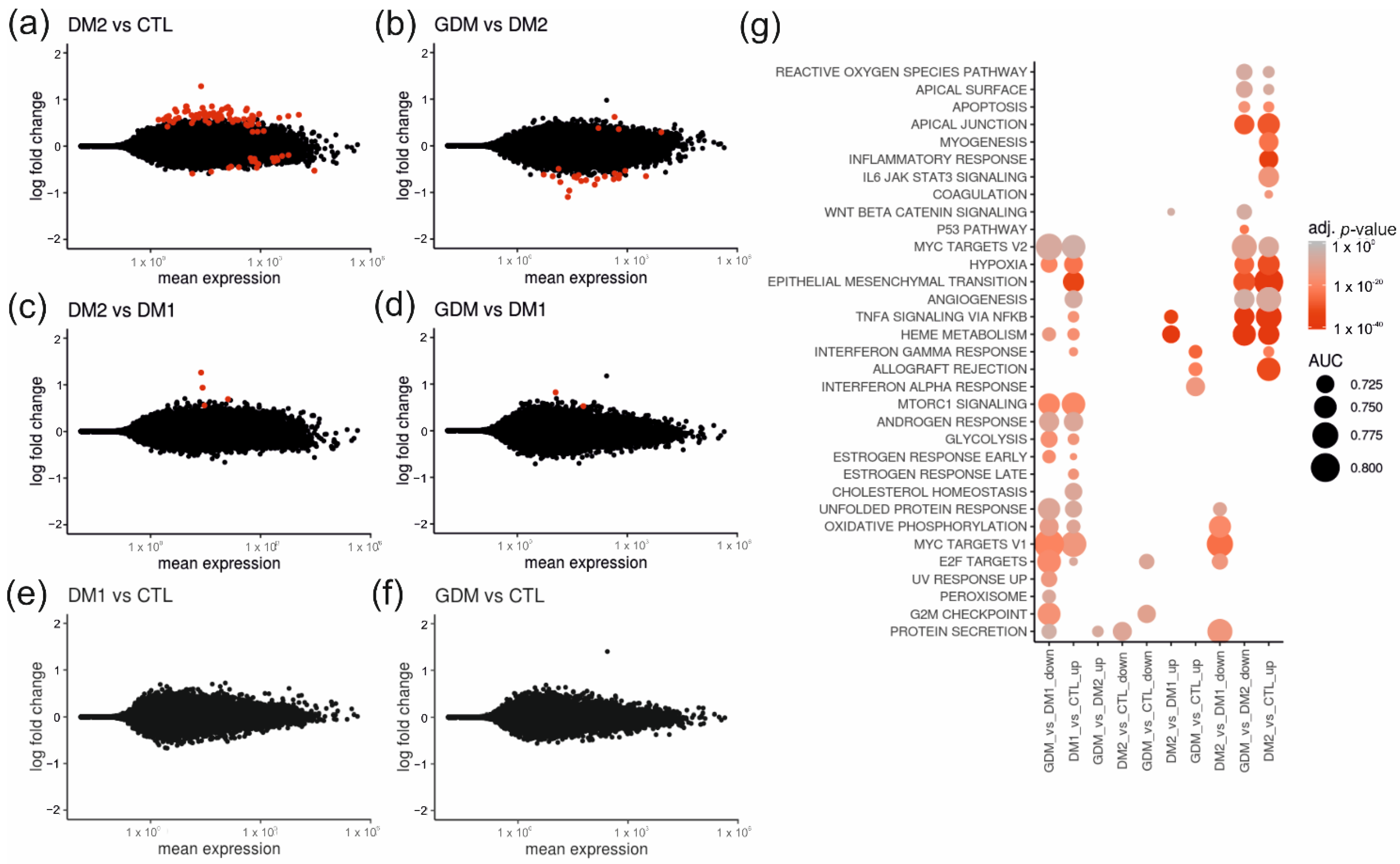
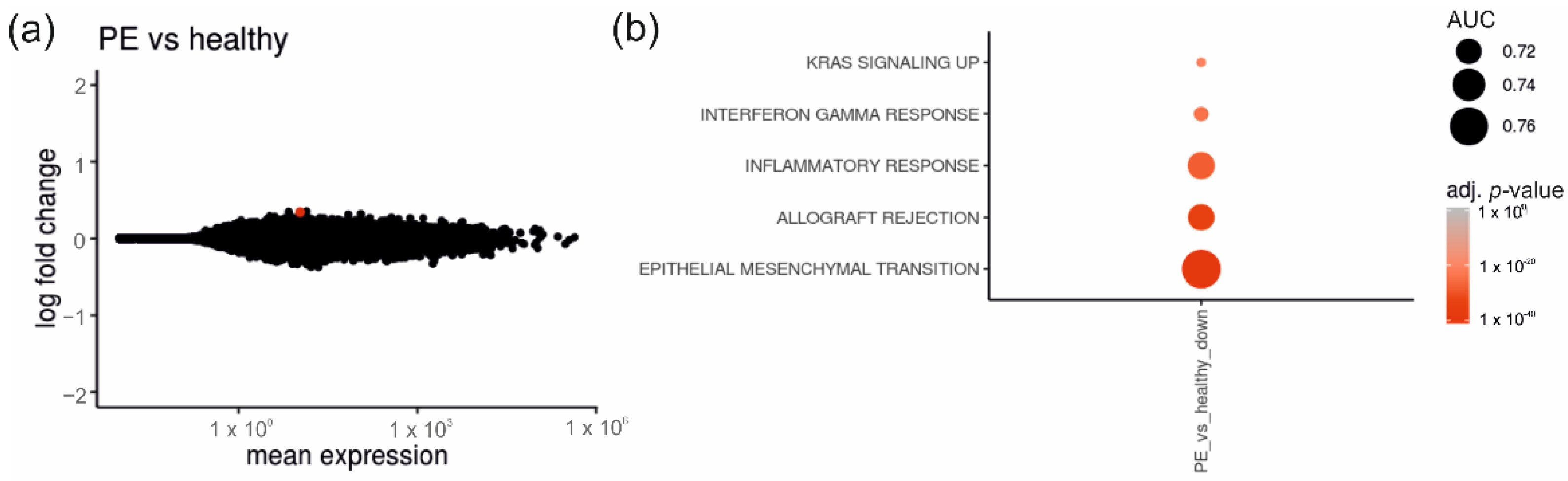
2.2. Placentas of Patients with Diabetes Reveal Differentially Expressed Genes
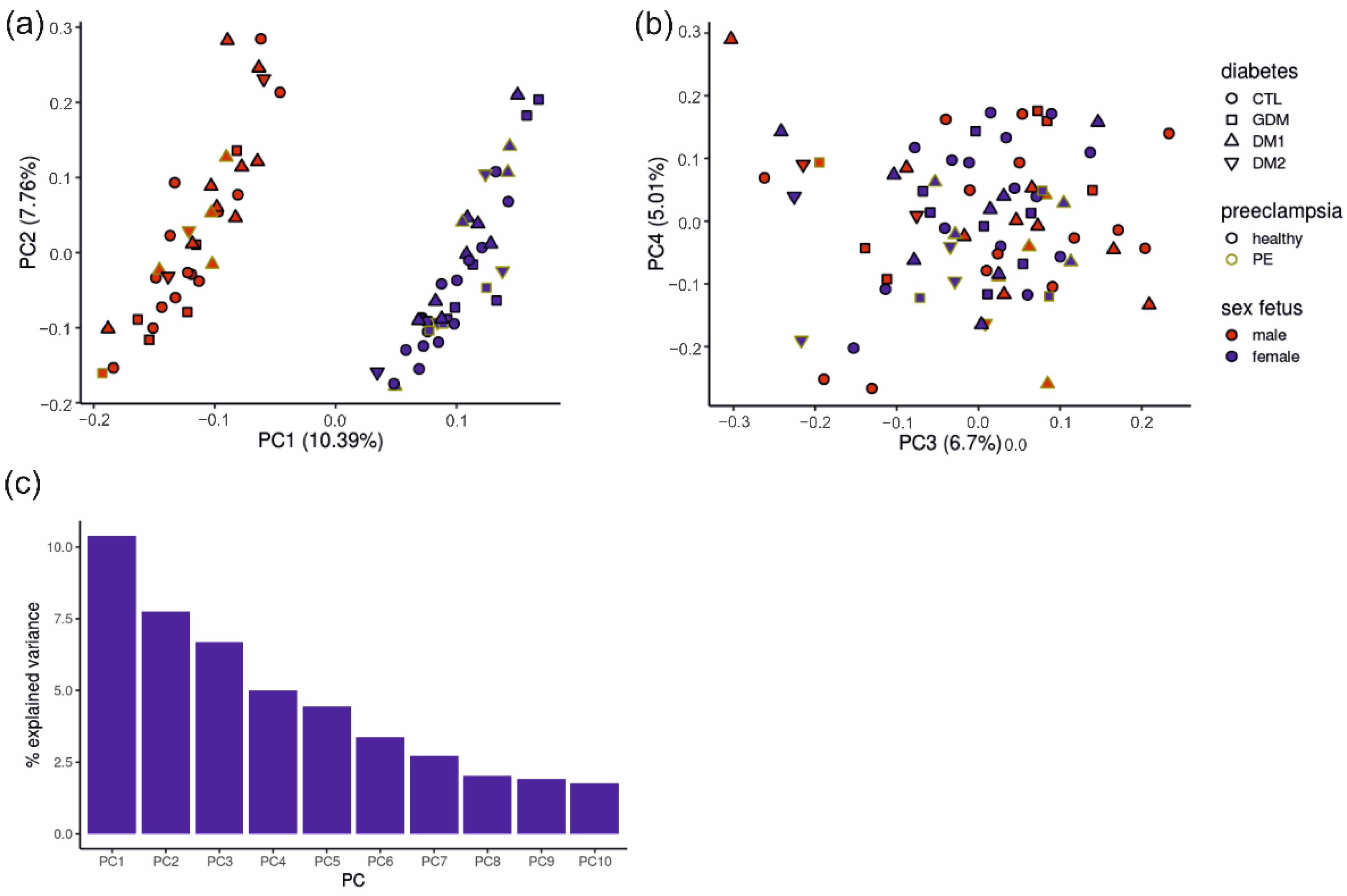
2.3. Placental RNA Sequencing Samples Group Mainly According to Fetal Sex
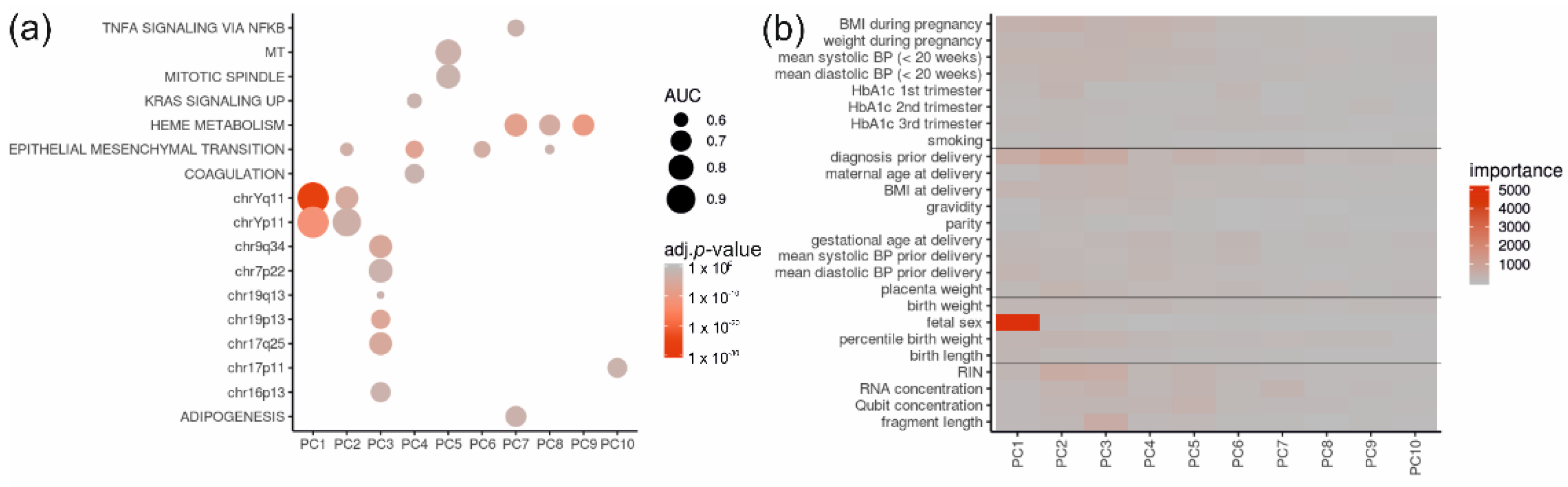
2.4. Of All Gene Sets and Clinical Data, the Fetal Sex and Diagnosis Contribute Most to Principal Components
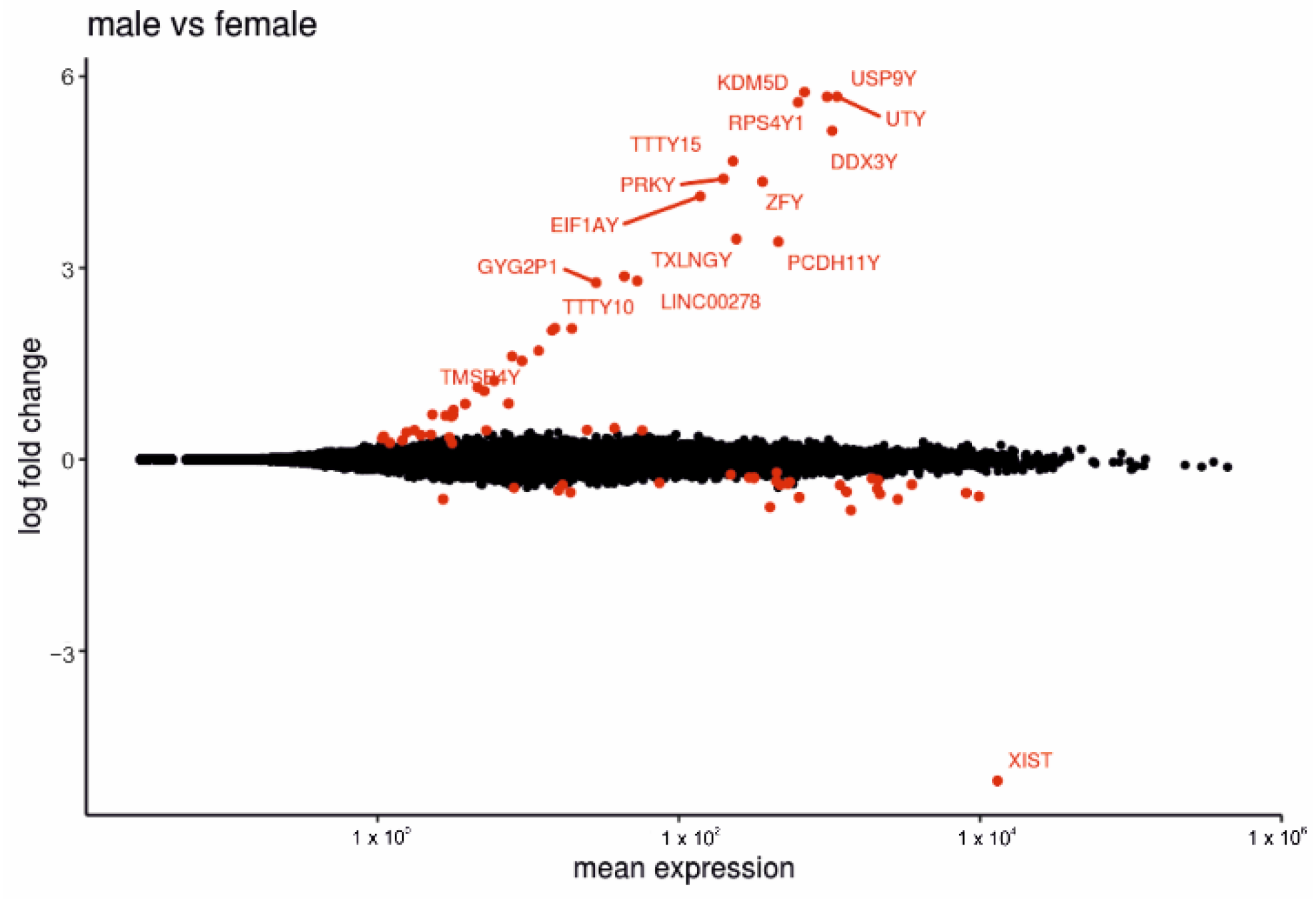
2.5. Comparison between Placentas of Male or Female Fetus Display Several DEGs
3. Discussion
4. Materials and Methods
4.1. Study Population and Sample Collection
4.2. RNA Isolation
4.3. RNA Sequencing
4.4. Sequencing Data Processing
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aplin, J.D.; Myers, J.E.; Timms, K.; Westwood, M. Tracking placental development in health and disease. Nat. Rev. Endocrinol. 2020, 16, 479–494. [Google Scholar] [CrossRef]
- Desoye, G.; Hauguel-De Mouzon, S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care 2007, 30 (Suppl. S2), S120–S126. [Google Scholar] [CrossRef]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. Hypertensive Disorders of Pregnancy: ISSHP Classification, Diagnosis, and Management Recommendations for International Practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Benirschke, K.; Burton, G.J.; Baergen, R.N. (Eds.) Pathology of the Human Placenta, 6th ed.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- American Diabetes Association. 14. Management of Diabetes in Pregnancy: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43 (Suppl. S1), S183–S192. [Google Scholar] [CrossRef] [PubMed]
- Desoye, G.; Cervar-Zivkovic, M. Diabetes Mellitus, Obesity, and the Placenta. Obstet. Gynecol. Clin. N. Am. 2020, 47, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Dawson, D.; Roberts, D.; Bentley-Lewis, R. A systematic review of placental pathology in maternal diabetes mellitus. Placenta 2015, 36, 101–114. [Google Scholar] [CrossRef]
- Ferrara, A. Increasing prevalence of gestational diabetes mellitus: A public health perspective. Diabetes Care 2007, 30 (Suppl. S2), S141–S146. [Google Scholar] [CrossRef]
- Yessoufou, A.; Moutairou, K. Maternal diabetes in pregnancy: Early and long-term outcomes on the offspring and the concept of “metabolic memory”. Exp. Diabetes Res. 2011, 2011, 218598. [Google Scholar] [CrossRef]
- Nelson, D.M. How the placenta affects your life, from womb to tomb. Am. J. Obstet. Gynecol. 2015, 213 (Suppl. S4), S12–S13. [Google Scholar] [CrossRef] [PubMed]
- Duckitt, K.; Harrington, D. Risk factors for pre-eclampsia at antenatal booking: Systematic review of controlled studies. BMJ 2005, 330, 565. [Google Scholar] [CrossRef]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.T. IFPA Award in Placentology Lecture: Complicated interactions between genes and the environment in placentation, pregnancy outcome and long term health. Placenta 2010, 31, S47–S53. [Google Scholar] [CrossRef] [PubMed]
- Gauster, M.; Desoye, G.; Tötsch, M.; Hiden, U. The placenta and gestational diabetes mellitus. Curr. Diab. Rep. 2012, 12, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Micoogullari, M.; Hoang, N.A.; Bergheim, I.; Klotz, L.-O.; Sies, H. Selenium-binding protein 1 (SELENBP1) is a marker of mature adipocytes. Redox Biol. 2019, 20, 489–495. [Google Scholar] [CrossRef]
- Huang, H.-H.; Condello, G.; Chen, C.-Y. Towards a link between magnesium, exercise, and risk of type 2 diabetes mellitus. J Chin. Med. Assoc. 2019, 82, 527–528. [Google Scholar] [CrossRef]
- Zampieri, T.T.; Ramos-Lobo, A.M.; Furigo, I.C.; Pedroso, J.A.B.; Buonfiglio, D.C.; Donato, J. SOCS3 deficiency in leptin receptor-expressing cells mitigates the development of pregnancy-induced metabolic changes. Mol. Metab. 2015, 4, 237–245. [Google Scholar] [CrossRef]
- Duttaroy, A.K. Transport of fatty acids across the human placenta: A review. Prog. Lipid Res. 2009, 48, 52–61. [Google Scholar] [CrossRef]
- Chalise, J.P.; Hashimoto, S.; Parajuli, G.; Kang, S.; Singh, S.K.; Gemechu, Y.; Metwally, H.; Nyati, K.K.; Dubey, P.K.; Zaman, M.M.-U.; et al. Feedback regulation of Arid5a and Ppar-γ2 maintains adipose tissue homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 15128–15133. [Google Scholar] [CrossRef]
- Liu, G.-M.; Zeng, H.-D.; Zhang, C.-Y.; Xu, J.-W. Key genes associated with diabetes mellitus and hepatocellular carcinoma. Pathol. Res. Pract. 2019, 215, 152510. [Google Scholar] [CrossRef]
- Yeo, S.-H.; Noh, J.-R.; Kim, Y.-H.; Gang, G.-T.; Kim, S.-W.; Kim, K.-S.; Hwang, J.H.; Shong, M.; Lee, C.-H. Increased vulnerability to β-cell destruction and diabetes in mice lacking NAD(P)H:quinone oxidoreductase 1. Toxicol. Lett. 2013, 219, 35–41. [Google Scholar] [CrossRef]
- Lash, G.E.; Otun, H.A.; Innes, B.A.; Bulmer, J.N.; Searle, R.F.; Robson, S.C. Low oxygen concentrations inhibit trophoblast cell invasion from early gestation placental explants via alterations in levels of the urokinase plasminogen activator system. Biol. Reprod. 2006, 74, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, D.; Cao, B.; Li, Y.-X.; Herva, R.; Piao, Y.-S.; Wang, Y.-L. Expression and localization of SWAP-70 in human fetomaternal interface and placenta during tubal pregnancy and normal placentation. J. Histochem. Cytochem. 2007, 55, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, T.L.; Sun, T.; Koeppel, A.F.; Lee, B.; Wang, E.T.; Farber, C.R.; Rich, S.S.; Sundheimer, L.W.; Buttle, R.A.; Chen, Y.-D.I.; et al. Sex differences in the late first trimester human placenta transcriptome. Biol. Sex Differ. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Yong, H.E.J.; Chan, S.-Y. Current approaches and developments in transcript profiling of the human placenta. Hum. Reprod. Update 2020, 26, 799–840. [Google Scholar] [CrossRef]
- Herse, F.; Dechend, R.; Harsem, N.K.; Wallukat, G.; Janke, J.; Qadri, F.; Hering, L.; Muller, D.N.; Luft, F.C.; Staff, A.C. Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension 2007, 49, 604–611. [Google Scholar] [CrossRef]
- Gong, S.; Gaccioli, F.; Dopierala, J.; Sovio, U.; Cook, E.; Volders, P.-J.; Martens, L.; Kirk, P.D.W.; Richardson, S.; Smith, G.C.S.; et al. The RNA landscape of the human placenta in health and disease. Nat. Commun. 2021, 12, 2639. [Google Scholar] [CrossRef]
- Sõber, S.; Reiman, M.; Kikas, T.; Rull, K.; Inno, R.; Vaas, P.; Teesalu, P.; Marti, J.M.L.; Mattila, P.; Laan, M. Extensive shift in placental transcriptome profile in preeclampsia and placental origin of adverse pregnancy outcomes. Sci. Rep. 2015, 5, 13336. [Google Scholar] [CrossRef]
- Lekva, T.; Lyle, R.; Roland, M.C.P.; Friis, C.; Bianchi, D.W.; Jaffe, I.Z.; Norwitz, E.R.; Bollerslev, J.; Henriksen, T.; Ueland, T. Gene expression in term placentas is regulated more by spinal or epidural anesthesia than by late-onset preeclampsia or gestational diabetes mellitus. Sci. Rep. 2016, 6, 29715. [Google Scholar] [CrossRef]
- Aye, I.L.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol. Reprod. 2014, 90, 129. [Google Scholar] [CrossRef] [PubMed]
- Magee, T.R.; Ross, M.G.; Wedekind, L.; Desai, M.; Kjos, S.; Belkacemi, L. Gestational diabetes mellitus alters apoptotic and inflammatory gene expression of trophobasts from human term placenta. J. Diabetes Complicat. 2014, 28, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Enquobahrie, D.A.; Williams, M.A.; Qiu, C.; Meller, M.; Sorensen, T.K. Global placental gene expression in gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2009, 200, 206.e1–206.e13. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Guo, F.; Zhang, Y.; Liu, X.-M.; Xiang, Y.-Q.; Zhang, C.; Liu, Z.-W.; Sheng, J.-Z.; Huang, H.-F.; Zhang, J.; et al. Integrated Transcriptome Sequencing Analysis Reveals Role of miR-138-5p/ TBL1X in Placenta from Gestational Diabetes Mellitus. Cell Physiol. Biochem. 2018, 51, 630–646. [Google Scholar] [CrossRef]
- Binder, A.M.; LaRocca, J.; Lesseur, C.; Marsit, C.J.; Michels, K.B. Epigenome-wide and transcriptome-wide analyses reveal gestational diabetes is associated with alterations in the human leukocyte antigen complex. Clin. Epigenetics 2015, 7, 79. [Google Scholar] [CrossRef]
- Tang, L.; Li, P.; Li, L. Whole transcriptome expression profiles in placenta samples from women with gestational diabetes mellitus. J. Diabetes Investig. 2020, 11, 1307–1317. [Google Scholar] [CrossRef]
- Radaelli, T.; Varastehpour, A.; Catalano, P.; Hauguel-De Mouzon, S. Gestational diabetes induces placental genes for chronic stress and inflammatory pathways. Diabetes 2003, 52, 2951–2958. [Google Scholar] [CrossRef]
- Alexander, J.; Teague, A.M.; Chen, J.; Aston, C.E.; Leung, Y.-K.; Chernausek, S.; Simmons, R.; Pinney, S.E. Offspring sex impacts DNA methylation and gene expression in placentae from women with diabetes during pregnancy. PLoS ONE 2018, 13, e0190698. [Google Scholar] [CrossRef]
- Radaelli, T.; Lepercq, J.; Varastehpour, A.; Basu, S.; Catalano, P.M.; Hauguel-De Mouzon, S. Differential regulation of genes for fetoplacental lipid pathways in pregnancy with gestational and type 1 diabetes mellitus. Am. J. Obstet. Gynecol. 2009, 201, 209.e1–209.e10. [Google Scholar] [CrossRef]
- Coton, S.J.; Nazareth, I.; Petersen, I. A cohort study of trends in the prevalence of pregestational diabetes in pregnancy recorded in UK general practice between 1995 and 2012. BMJ Open 2016, 6, e009494. [Google Scholar] [CrossRef] [PubMed]
- Lindegaard, M.L.S.; Damm, P.; Mathiesen, E.R.; Nielsen, L.B. Placental triglyceride accumulation in maternal type 1 diabetes is associated with increased lipase gene expression. J. Lipid Res. 2006, 47, 2581–2588. [Google Scholar] [CrossRef] [PubMed]
- Iciek, R.; Wender-Ozegowska, E.; Zawiejska, A.; Mikolajczak, P.; Mrozikiewicz, P.M.; Pietryga, M.; Brazert, J. Placental leptin and its receptor genes expression in pregnancies complicated by type 1 diabetes. J. Physiol. Pharmacol. 2013, 64, 579–585. [Google Scholar] [PubMed]
- Kaartokallio, T.; Cervera, A.; Kyllönen, A.; Laivuori, K.; Kere, J.; Laivuori, H. Gene expression profiling of pre-eclamptic placentae by RNA sequencing. Sci. Rep. 2015, 5, 14107. [Google Scholar] [CrossRef] [PubMed]
- Buckberry, S.; Bianco-Miotto, T.; Bent, S.J.; Clifton, V.; Shoubridge, C.; Shankar, K.; Roberts, C.T. Placental transcriptome co-expression analysis reveals conserved regulatory programs across gestation. BMC Genom. 2017, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Benton, S.J.; Leavey, K.; Grynspan, D.; Cox, B.J.; Bainbridge, S.A. The clinical heterogeneity of preeclampsia is related to both placental gene expression and placental histopathology. Am. J. Obstet. Gynecol. 2018, 219, 604.e1–604.e25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-P.; Chen, X.; Li, M.-Q. Gestational diabetes induces chronic hypoxia stress and excessive inflammatory response in murine placenta. Int. J. Clin. Exp. Pathol. 2013, 6, 650–659. [Google Scholar]
- Gauster, M.; Majali-Martinez, A.; Maninger, S.; Gutschi, E.; Greimel, P.H.; Ivanisevic, M.; Djelmis, J.; Desoye, G.; Hiden, U. Maternal Type 1 diabetes activates stress response in early placenta. Placenta 2017, 50, 110–116. [Google Scholar] [CrossRef]
- Kang, Q.; Li, W.; Xiao, J.; Yu, N.; Fan, L.; Sha, M.; Ma, S.; Wu, J.; Chen, S. Integrated analysis of multiple microarray studies to identify novel gene signatures in preeclampsia. Placenta 2021, 105, 104–118. [Google Scholar] [CrossRef]
- Sood, R.; Zehnder, J.L.; Druzin, M.L.; Brown, P.O. Gene expression patterns in human placenta. Proc. Natl. Acad. Sci. USA 2006, 103, 5478–5483. [Google Scholar] [CrossRef]
- Gong, S.; Sovio, U.; Aye, I.L.; Gaccioli, F.; Dopierala, J.; Johnson, M.D.; Wood, A.M.; Cook, E.; Jenkins, B.J.; Koulman, A.; et al. Placental polyamine metabolism differs by fetal sex, fetal growth restriction, and preeclampsia. JCI Insight 2018, 3, e120723. [Google Scholar] [CrossRef]
- Clifton, V.L. Review: Sex and the human placenta: Mediating differential strategies of fetal growth and survival. Placenta 2010, 31, S33–S39. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.; Zimmet, P.Z. for the WHO Consultation. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Sugulle, M.; Dechend, R.; Herse, F.; Weedon-Fekjaer, M.S.; Johnsen, G.M.; Brosnihan, K.B.; Anton, L.; Luft, F.; Wollert, K.C.; Kempf, T.; et al. Circulating and placental growth-differentiation factor 15 in preeclampsia and in pregnancy complicated by diabetes mellitus. Hypertension 2009, 54, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Lindheimer, M.D.; de Swiet, M.; Van Assche, A.; Moutquin, J.M. The classification and diagnosis of the hypertensive disorders of pregnancy: Statement from the International Society for the Study of Hypertension in Pregnancy (ISSHP). Hypertens Pregnancy 2001, 20, IX–XIV. [Google Scholar] [CrossRef]
- Harsem, N.K.; Staff, A.C.; He, L.; Roald, B. The decidual suction method: A new way of collecting decidual tissue for functional and morphological studies. Acta Obstet. Gynecol. Scand. 2004, 83, 724–730. [Google Scholar] [CrossRef]
- Staff, A.C.; Ranheim, T.; Khoury, J.; Henriksen, T. Increased contents of phospholipids, cholesterol, and lipid peroxides in decidua basalis in women with preeclampsia. Am. J. Obstet. Gynecol. 1999, 180, 587–592. [Google Scholar] [CrossRef]
- Weedon-Fekjær, M.; Sheng, Y.; Sugulle, M.; Johnsen, G.; Herse, F.; Redman, C.; Lyle, R.; Dechend, R.; Staff, A. Placental miR-1301 is dysregulated in early-onset preeclampsia and inversely correlated with maternal circulating leptin. Placenta 2014, 35, 709–717. [Google Scholar] [CrossRef]
- Moe, K.; Alnaes-Katjavivi, P.; Størvold, G.L.; Sugulle, M.; Johnsen, G.M.; Redman, C.W.; Dechend, R.; Staff, A.C. Classical Cardiovascular Risk Markers in Pregnancy and Associations to Uteroplacental Acute Atherosis. Hypertension 2018, 72, 695–702. [Google Scholar] [CrossRef]
- Rieber-Mohn, A.B.; Sugulle, M.; Wallukat, G.; Alnæs-Katjavivi, P.; Størvold, G.L.; Bolstad, N.; Redman, C.W.; Dechend, R.; Staff, A.C. Auto-antibodies against the angiotensin II type I receptor in women with uteroplacental acute atherosis and preeclampsia at delivery and several years postpartum. J. Reprod. Immunol. 2018, 128, 23–29. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Zyla, J.; Marczyk, M.; Domaszewska, T.; Kaufmann, S.H.E.; Polanska, J.; Weiner, J. Gene set enrichment for reproducible science: Comparison of CERNO and eight other algorithms. Bioinformatics 2019, 35, 5146–5154. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | CTL (n = 29) | GDM (n = 12) | p-Value CTL vs. GDM | DM1 (n = 17) | p-Value CTL vs. DM1 | DM2 (n = 3) | p-Value CTL vs. DM2 |
|---|---|---|---|---|---|---|---|
| maternal characteristics <20 weeks gestation | |||||||
| BMI | 23.68 ± 4.06 | 28.03 ± 5.65 | 0.02 | 25.22 ± 3.95 | 0.81 | 34.58 ± 1.20 | 0.00 |
| SBP, mmHg | 111.03 ± 9.41 | 113.08 ± 9.27 | 0.99 | 113.81 ± 8.42 (16) | 0.98 | 127.67 ± 19.14 | 0.15 |
| DBP, mmHg | 66.21 ± 7.04 | 70.42 ± 7.35 | 0.48 | 70.06 ± 5.92 (16) | 0.48 | 75 ± 1.73 | 0.29 |
| HbA1c 2. Trimester, % | NA | 5.2 ± 0.62 (4) | NA | 6.17 ± 0.62 (15) | NA | 5.87 ± 0.09 | NA |
| smoking, % (n) | 13.79 (4) | 16.67 (2) | NA | 5.88 (1) | NA | 33.3 (1) | NA |
| height, m | 1.70 ± 0.06 | 1.67 ± 0.06 | 0.87 | 1.67 ± 0.05 | 0.75 | 1.68 ± 0.05 | 0.99 |
| weight, kg | 68.17 ± 11.96 | 78.34 ± 14.77 | 0.13 | 70.53 ± 11.48 | 0.99 | 98 ± 5.29 | 0.00 |
| maternal characteristics at delivery | |||||||
| BMI at delivery | 28.79 ± 3.98 | 33.2 ± 6.77 (11) | 0.05 | 29.7 ± 4.03 | 0.99 | 38.57 ± 2.89 | 0.00 |
| pre-operative SBP, mmHg | 124.42 ± 12.22 (26) | 132 ± 10.89 (9) | 0.72 | 138.93 ± 14.35 (15) | 0.02 | 144.5 ± 0.70 (2) | 0.35 |
| pre-operative DBP, mmHg | 72.96 ± 10.78 (26) | 80.44 ± 15.48 (9) | 0.42 | 79.13 ± 9.95 (15) | 0.44 | 73.0 ± 2.83 (2) | >0.99 |
| proteinuria, % (n) | 4 (1) | 10 (1) | NA | 13,32 (1) | NA | 0 (0) | NA |
| HbA1c 3. Trimester, % | NA | 5.79 ± 0.55 (11) | NA | 6.27 ± 0.57 (15) | NA | 6.2 ± 0.22 | NA |
| medication, % (n) [insulin, metformin] | 0 (0) | 41.67 (5) | NA | 88.24 (15) | NA | 100 (3) | NA |
| gestational age, days | 271.93 ± 7.94 | 267.67 ± 13.03 | 0.94 | 260.65 ± 17.19 | 0.06 | 269.67 ± 4.16 | >0.99 |
| age, years | 32.66± 4.50 | 34.58 ± 3.61 | 0.77 | 33.59 ± 3.76 | 0.98 | 31.0 ± 6.56 | 0.99 |
| parity, count | 0.93 ± 0.88 | 1.08 ± 0.90 | 0.99 | 0.71 ± 0.59 | 0.93 | 0.33 ± 0.58 | 0.78 |
| gravidity, count | 2.72 ± 1.25 | 3.25 ± 1.42 | 0.77 | 2.29 ± 1.16 | 0.83 | 2.00 ± 1.00 | 0.91 |
| blood sugar, mmol/l | 3.98 ± 0.48 (9) | 4.78 ± 1.05 (9) | 0.72 | 6.57 ± 1.56 (13) | 0.00 | 6.53 ± 1.45 | 0.03 |
| birth outcome | |||||||
| fetal sex, female/male, count | 15/14 | 7/5 | NA | 8/9 | NA | 1/2 | NA |
| birth weight, g | 3342 ± 450 | 3697 ± 844 | 0.72 | 3611 ± 864 (16) | 0.85 | 4515 ± 788 | 0.09 |
| birth length, cm | 50.05 ± 1.49 (21) | 50.55 ± 3.55 (10) | 0.99 | 49.57 ± 4.09 (14) | 0.99 | 53.67 ± 2.89 | 0.34 |
| percentile birthweight | 51.04 ± 27.68 | 73.18 ± 33.06 | 0.22 | 78.87 ± 30.69 (16) | 0.03 | 97.19 ± 2.99 | 0.09 |
| placental + umbilical cord weight, g | 584.23 ± 107.9 (26) | 680.11 ± 114.4 (9) | 0.51 | 653.53 ± 198.7 (15) | 0.67 | 906.33 ± 164.5 | 0.00 |
| Characteristics | CTL (n = 29) | GDM + PE (n = 4) | p-Value CTL vs. GDM + PE | DM1 + PE (n = 8) | p-Value CTL vs. DM1 + PE | DM2 + PE (n = 4) | p-Value CTL vs. DM2 + PE |
|---|---|---|---|---|---|---|---|
| maternal characteristics <20 weeks gestation | |||||||
| BMI | 23.68 ± 4.06 | 31.91 ± 4.47 | 0.00 | 24.68 ± 2.85 | 0.99 | 28.07 ± 5.69 | 0.30 |
| SBP, mmHg | 111.03 ± 9.41 | 118.5 ± 11.21 | 0.82 | 125.88 ± 16.3 | 0.02 | 124 ± 29.02 | 0.25 |
| DBP, mmHg | 66.21 ± 7.04 | 76.25 ± 8.96 | 0.08 | 72.25 ± 6.5 | 0.25 | 69.25 ± 16.4 | 0.97 |
| HbA1c 2. Trimester, % | NA | 5.5 ± 0.1 (2) | NA | 6.63 ± 0.66 | NA | 5.67 ± 0.12 (2) | NA |
| smoking, % (n) | 13.79 (4) | 25 (1) | NA | 25 (2) | NA | 0 (0) | NA |
| height, m | 1.70 ± 0.06 | 1.61 ± 0.11 | 0.06 | 1.69 ± 0.04 | >0.99 | 1.57 ± 0.14 | 0.00 |
| weight, kg | 68.17 ± 11.96 | 83.5 ± 20.76 | 0.16 | 68.75 ± 7.15 | >0.99 | 70.25 ± 20.76 | 0.99 |
| maternal characteristics at delivery | |||||||
| BMI at delivery | 28.79 ± 3.98 | 38.05 ± 3.55 | 0.00 | 32.07 ± 3.65 | 0.39 | 31.54 ± 7.39 | 0.85 |
| pre-operative SBP, mmHg | 124.42 ± 12.22 (26) | 153 ± 16.92 (3) | 0.03 | 169.38 ± 25.05 | 0.00 | 152.67 ± 13.65 (3) | 0.02 |
| pre-operative DBP, mmHg | 72.96 ± 10.78 (26) | 90.33 ± 8.51 (3) | 0.08 | 99.13 ± 10.91 | 0.00 | 88.0 ± 8.66 (3) | 0.17 |
| proteinuria, % (n) | 4 (1) | 100 (4) | NA | 100 (8) | NA | 100 (4) | NA |
| HbA1c 3. Trimester, % | NA | 6.25 ± 0.05 (2) | NA | 6.6 ± 0.56 | NA | 6.65 ± 0.25 (2) | NA |
| medication, % (n) [insulin, metformin] | 0 (0) | 25 (1) | NA | 100 (8) | NA | 100 (4) | NA |
| gestational age, days | 271.93 ± 7.94 | 247.25 ± 18.77 | 0.00 | 248.38 ± 13.75 | 0.00 | 249.25 ± 31.10 | 0.02 |
| age, years | 32.66± 4.50 | 29.0 ± 4.08 | 0.57 | 32.13 ± 6.11 | 0.98 | 34.25 ± 5.38 | 0.98 |
| parity, count | 0.93 ± 0.88 | 0.75 ± 0.50 | 0.99 | 0.5 ± 0.76 | 0.70 | 0.5 ± 1.00 | 0.90 |
| gravidity, count | 2.72 ± 1.25 | 1.75 ± 0.50 | 0.60 | 2.0 ± 1.07 | 0.60 | 1.75 ± 1.50 | 0.60 |
| blood sugar, mmol/l | 3.98 ± 0.48 (9) | 4.28 ± 0.50 | 0.99 | 6.32 ± 1.8 (7) | 0.00 | 3.9 ± 0.30 (2) | >0.99 |
| birth outcome | |||||||
| fetal sex, female/male, count | 15/14 | 3/1 | NA | 4/4 | NA | 3/1 | NA |
| birth weight, g | 3342 ± 450 | 2422 ± 671 | 0.17 | 3305 ± 830 | >0.99 | 2989 ± 1845 | 0.95 |
| birth length, cm | 50.05 ± 1.49 (21) | 46.75 ± 2.06 | 0.33 | 48.8 ± 0.84 (5) | 0.96 | 38 ± 11.30 (2) | 0.00 |
| percentile birthweight | 51.04 ± 27.68 | 28.29 ± 26.60 | 0.68 | 76.97 ± 31.06 | 0.21 | 51.6 ± 55.17 | >0.99 |
| placental + umbilical cord weight, g | 584.23 ± 107.9 (26) | 494.25 ± 111.7 | 0.86 | 615.75 ± 201.3 | 0.99 | 577.75 ± 218.0 | >0.99 |
|
CTL (n = 29) |
Diabetes (n = 32) | p-Value CTL vs. Diabetes |
Diabetes + PE (n = 16) | p-Value CTL vs. Diabetes + PE | |
|---|---|---|---|---|---|
| maternal characteristics <20 weeks gestation | |||||
| BMI | 23.68 ± 4.06 | 27.15 ± 5.21 | 0.01 | 27.33 ± 4.87 | 0.03 |
| SBP, mmHg (n) | 111.03 ± 9.41 | 114.87 ± 10.47 (31) | 0.40 | 123.56 ± 18.09 | 0.00 |
| DBP, mmHg (n) | 66.21 ± 7.04 | 70.68 ± 6.28 (31) | 0.04 | 72.50 ± 9.81 | 0.02 |
| HbA1c 2. Trimester, % (n) | NA | 5.95 ± 0.69 (22) | NA | 6.23 ± 0.73 (13) | NA |
| smoking, % (n) | 13.79 (4) | 12.50 (4) | NA | 18.75 (3) | NA |
| height, m | 1.70 ± 0.06 | 1.76 ± 0.05 | 0.33 | 1.64 ± 0.10 | 0.01 |
| weight, kg | 68.17 ± 11.96 | 76.03 ± 14.58 | 0.06 | 72.81 ± 15.40 | 0.49 |
| maternal characteristics at delivery | |||||
| BMI at delivery | 28.79 ± 3.98 | 31.80 ± 5.69 (31) | 0.04 | 33.43 ± 5.22 | 0.00 |
| pre-operative SBP, mmHg | 124.42 ± 12.22 (26) | 136.96 ± 13.02 (26) | 0.00 | 161.93 ± 22.15 (14) | 0.00 |
| pre-operative DBP, mmHg | 72.96 ± 10.78 (26) | 79.12 ± 11.66 (26) | 0.09 | 94.86 ± 10.65 (14) | 0.00 |
| Proteinuria, % (n) | 4 (1) | 6.25 (2) | NA | 100 (16) | NA |
| HbA1c 3. Trimester, % | NA | 6.08 ± 0.58 (29) | NA | 6.55 ± 0.49 (12) | NA |
| medication metformin, % (n) | 0 (0) | 9.38 (3) | NA | 18.75 (3) | NA |
| insulin, % (n) | 0 (0) | 68.75 (22) | NA | 68.75 (11) | NA |
| early-onset PE % (n) | 0 (0) | 0 (0) | NA | 37.5 (6) | NA |
| age, years | 32.66± 4.50 | 33.72 ± 3.96 | 0.59 | 31.88 ± 5.50 | 0.82 |
| parity, count | 0.93 ± 0.88 | 0.81 ± 0.74 | 0.81 | 0.56 ± 0.72 | 0.26 |
| gravidity, count | 2.72 ± 1.25 | 2.63 ± 1.31 | 0.94 | 1.88 ± 1.03 | 0.06 |
| blood sugar, mmol/l | 3.98 ± 0.48 (9) | 5.92 ± 1.59 (25) | 0.00 | 5.32 ± 1.74 (13) | 0.08 |
| birth outcome | |||||
| fetal sex, female/male, count | 15/14 | 16/16 | NA | 10/6 | NA |
| birth weight, g | 3342 ± 450 | 3732 ± 863 (31) | 0.12 | 3005 ± 1110 | 0.32 |
| birth length, cm | 50.05 ± 1.49 (21) | 50.39 ± 3.87 (27) | 0.94 | 46.09 ± 5.59 (11) | 0.01 |
| percentile birthweight | 51.04 ± 27.68 | 78.44 ± 30.31 (31) | 0.00 | 58.46 ± 40.48 | 0.70 |
| placental + umbilical cord weight, g | 584.23 ± 107.9 (26) | 690.48 ± 183.2 (27) | 0.03 | 575.88 ± 183.1 | 0.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kedziora, S.M.; Obermayer, B.; Sugulle, M.; Herse, F.; Kräker, K.; Haase, N.; Langmia, I.M.; Müller, D.N.; Staff, A.C.; Beule, D.; et al. Placental Transcriptome Profiling in Subtypes of Diabetic Pregnancies Is Strongly Confounded by Fetal Sex. Int. J. Mol. Sci. 2022, 23, 15388. https://doi.org/10.3390/ijms232315388
Kedziora SM, Obermayer B, Sugulle M, Herse F, Kräker K, Haase N, Langmia IM, Müller DN, Staff AC, Beule D, et al. Placental Transcriptome Profiling in Subtypes of Diabetic Pregnancies Is Strongly Confounded by Fetal Sex. International Journal of Molecular Sciences. 2022; 23(23):15388. https://doi.org/10.3390/ijms232315388
Chicago/Turabian StyleKedziora, Sarah M., Benedikt Obermayer, Meryam Sugulle, Florian Herse, Kristin Kräker, Nadine Haase, Immaculate M. Langmia, Dominik N. Müller, Anne Cathrine Staff, Dieter Beule, and et al. 2022. "Placental Transcriptome Profiling in Subtypes of Diabetic Pregnancies Is Strongly Confounded by Fetal Sex" International Journal of Molecular Sciences 23, no. 23: 15388. https://doi.org/10.3390/ijms232315388
APA StyleKedziora, S. M., Obermayer, B., Sugulle, M., Herse, F., Kräker, K., Haase, N., Langmia, I. M., Müller, D. N., Staff, A. C., Beule, D., & Dechend, R. (2022). Placental Transcriptome Profiling in Subtypes of Diabetic Pregnancies Is Strongly Confounded by Fetal Sex. International Journal of Molecular Sciences, 23(23), 15388. https://doi.org/10.3390/ijms232315388