
Increased Expression of Galectin-3 in Skin Fibrosis: Evidence from In Vitro and In Vivo Studies
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
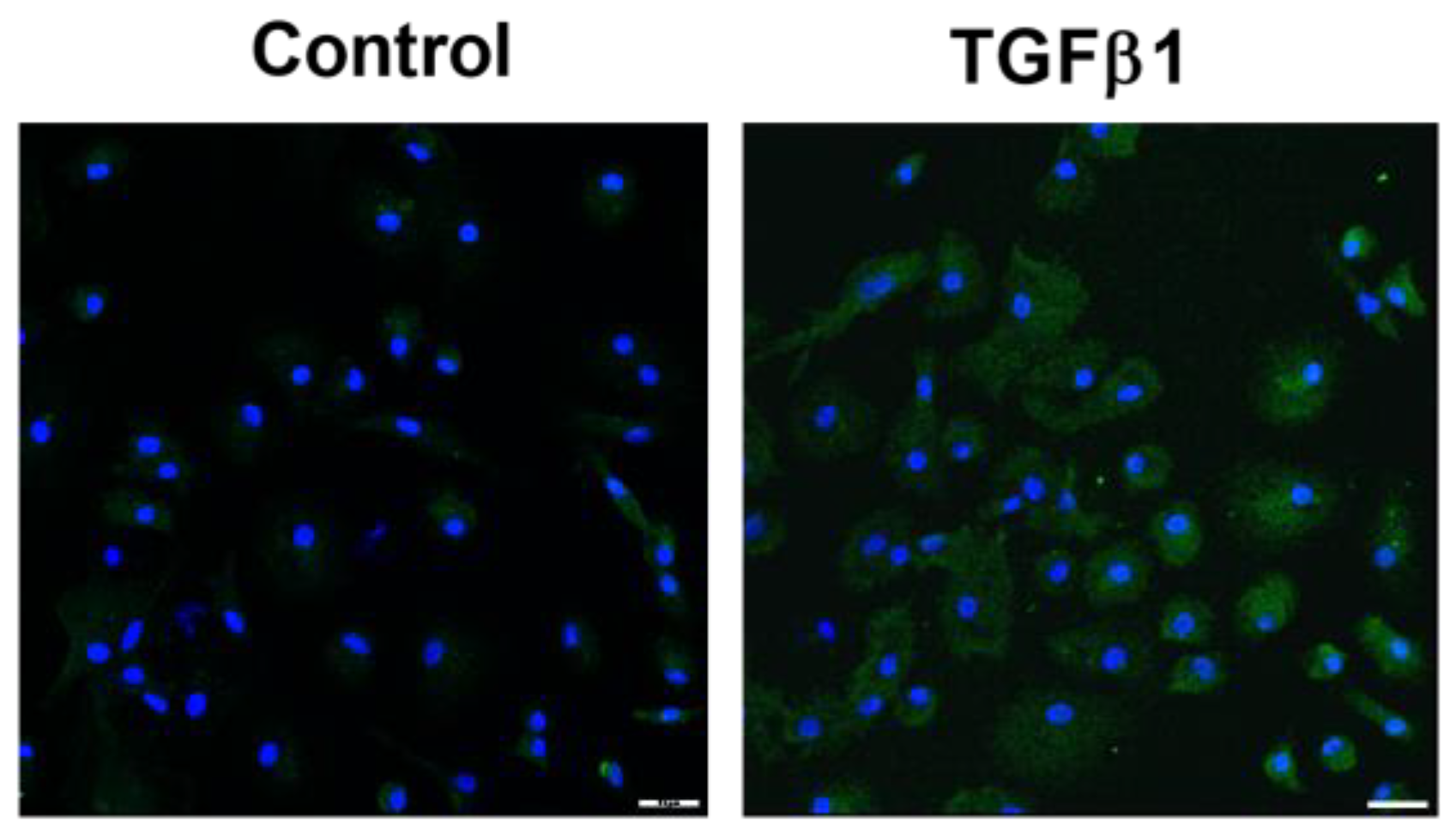
2.1. GAL-3 Expression in Skin Fibrosis In Vitro Models with TGFB1 Stimulation
2.1.1. Fibroblast to Myofibroblast Transition Model
2.1.2. Human Fibrocytes Model
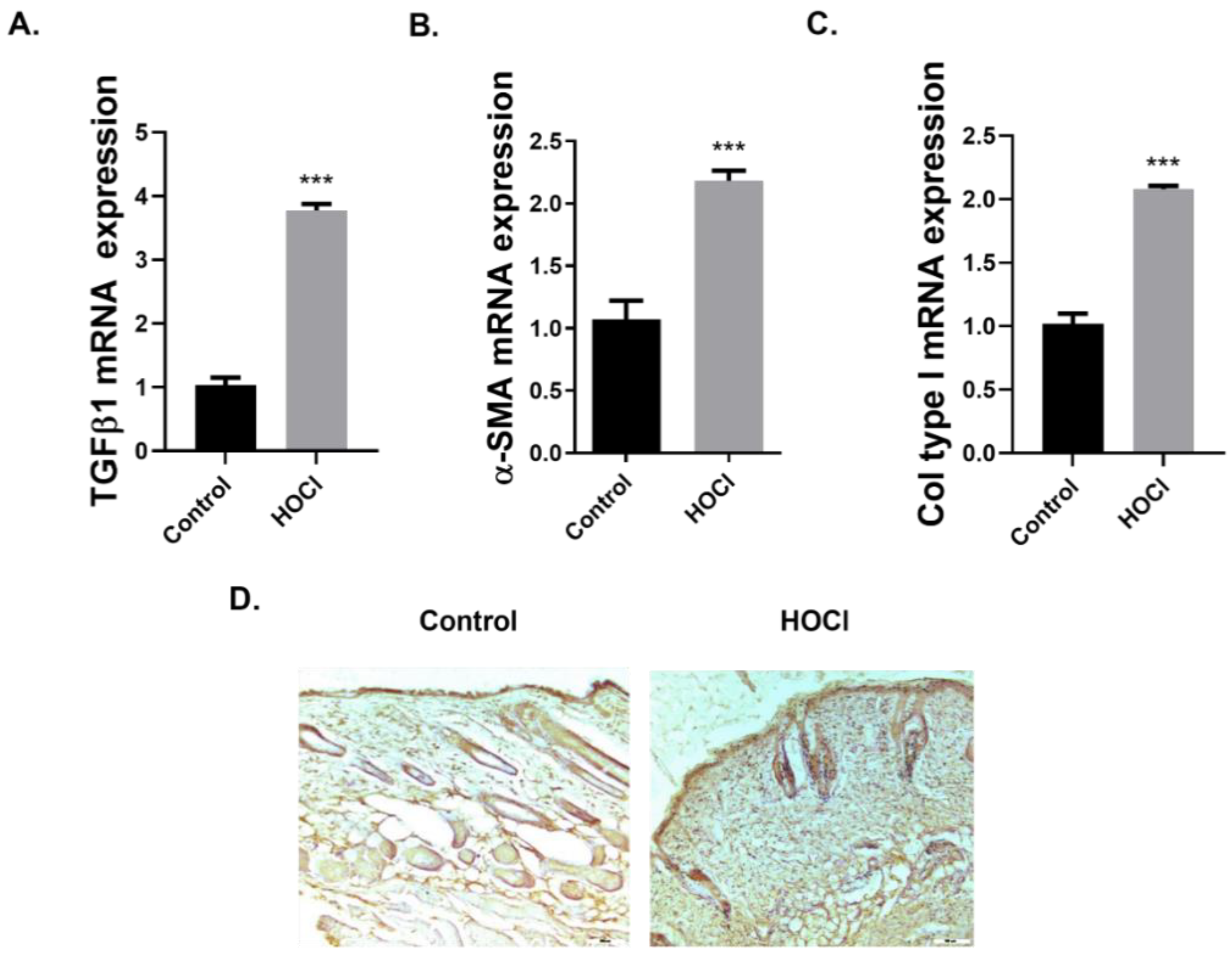
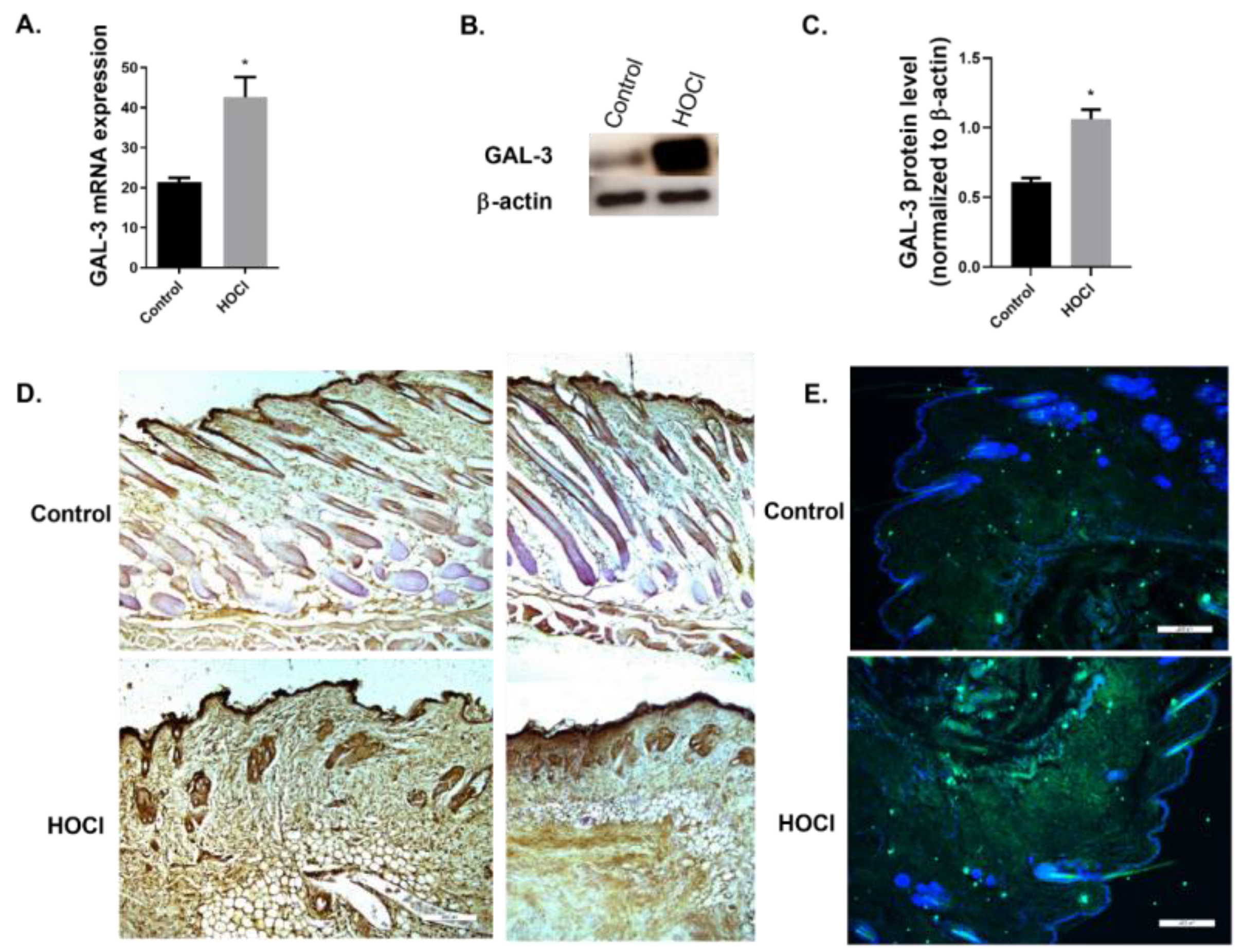
2.2. GAL-3 Expression in In Vivo Models
GAL-3 Expression Is Increased in a Skin Fibrosis Murine Model
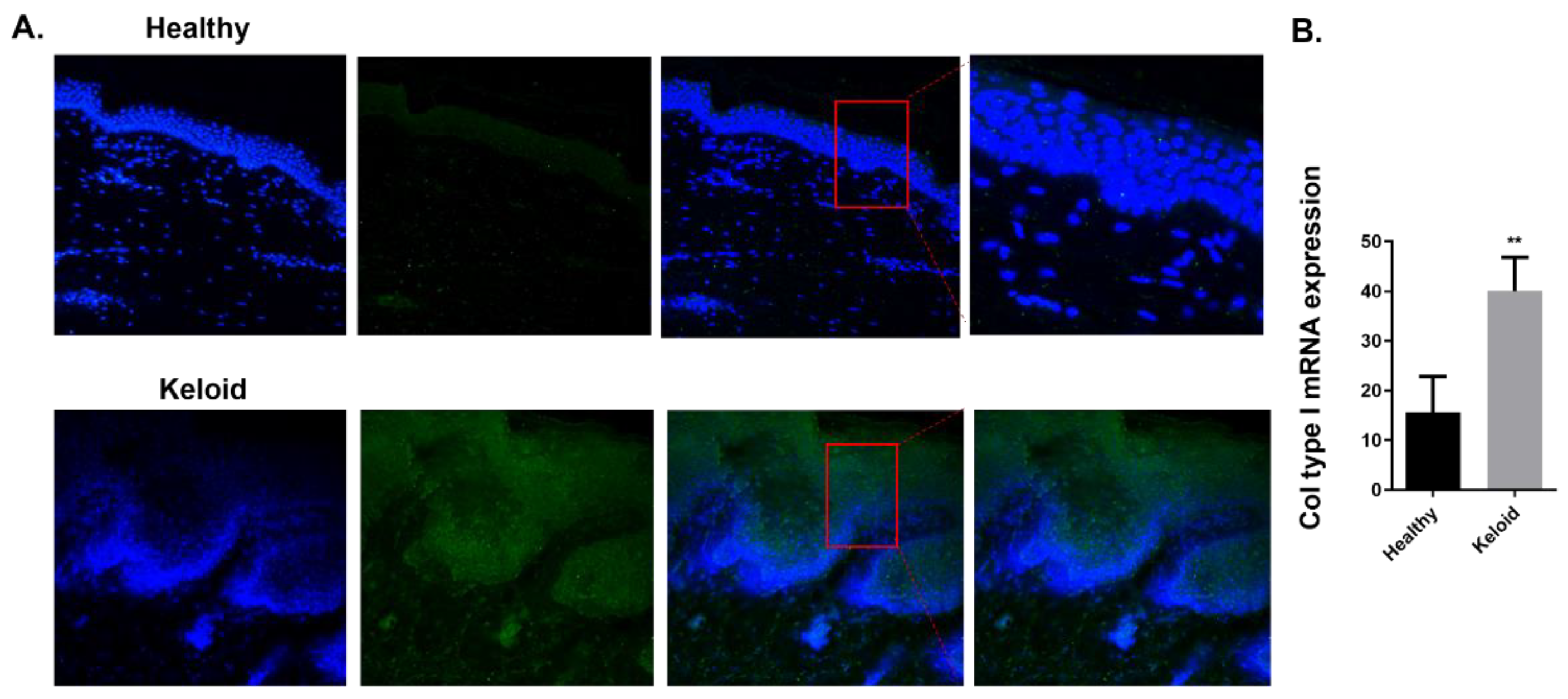
2.3. GAL-3 Expression in Skin Biopsies of Human Keloids
2.3.1. Expression of Fibrotic Markers Is Increased in Human Keloids Compared to Human Healthy Skin Tissue
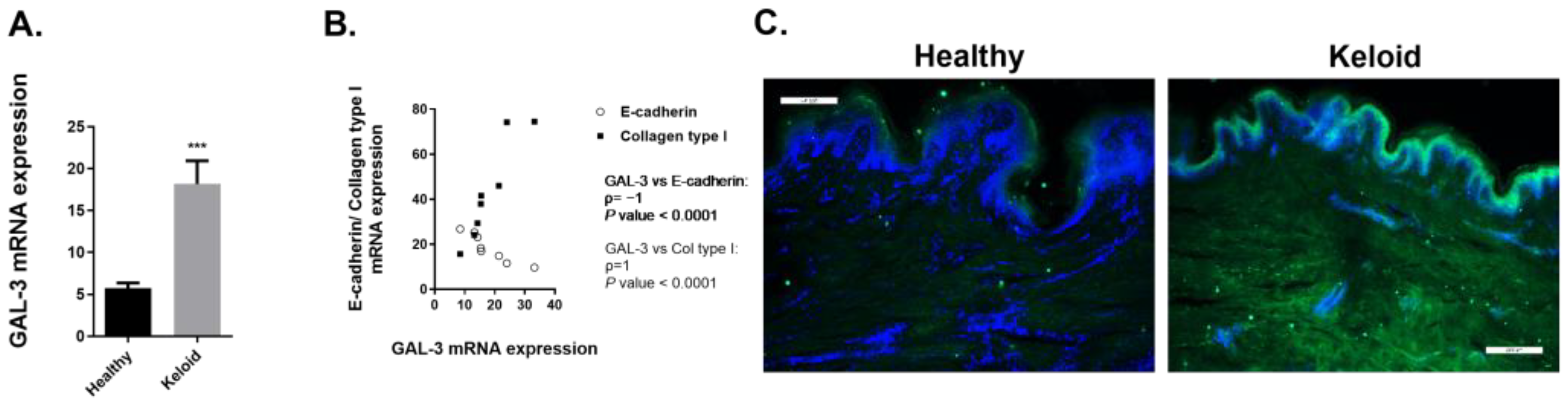
2.3.2. Expression of GAL-3 Is Increased in Human Keloids Compared to Human Healthy Skin Tissue
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Skin Biopsies: Healthy vs. Keloid Human Tissue
4.3. Skin Fibrosis Animal Model
4.4. Real-Time RT-PCR
4.5. Histological, Immunohistochemical and Immunofluorescence Studies
4.6. Western Blotting Analysis
4.7. ELISA
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Do, N.N.; Eming, S.A. Skin fibrosis: Models and mechanisms. Curr. Res. Transl. Med. 2016, 64, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.P.; Marttala, J.; Macarak, E.; Rosenbloom, J.; Uitto, J. Keloids: The paradigm of skin fibrosis—Pathomechanisms and treatment. Matrix Biol. 2016, 51, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Shroff, A.; Mamalis, A.; Jagdeo, J. Oxidative Stress and Skin Fibrosis. Curr. Pathobiol. Rep. 2014, 2, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Hao, C.; Xie, Y.; Peng, M.; Ma, L.; Zhou, Y.; Zhang, Y.; Kang, W.; Wang, J.; Bai, X.; Wang, P.; et al. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation in vitro and prevents liver fibrosis in vivo. Clin. Exp. Med. 2014, 14, 141–150. [Google Scholar] [CrossRef]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell. Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef]
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004, 15, 255–273. [Google Scholar] [CrossRef]
- Speca, S.; Giusti, I.; Rieder, F.; Latella, G. Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. 2012, 18, 3635–3661. [Google Scholar] [CrossRef]
- Pasmatzi, E.; Papadionysiou, C.; Monastirli, A.; Badavanis, G.; Tsambaos, D. Galectin 3: An extraordinary multifunctional protein in dermatology. Current knowledge and perspectives. An. Bras. Dermatol. 2019, 94, 348–354. [Google Scholar] [CrossRef]
- Saegusa, J.; Hsu, D.K.; Chen, H.-Y.; Yu, L.; Fermin, A.; Fung, M.A.; Liu, F.-T. Galectin-3 Is Critical for the Development of the Allergic Inflammatory Response in a Mouse Model of Atopic Dermatitis. Am. J. Pathol. 2009, 174, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Alvarez, N.; Gonçalves, B.; Andrews, A.R.; Wang, Z.; Wang, Z.; Harrington, E.; Shah, J.; Sachs, D.H.; Eliaz, I.; Huang, C.A. The effects of galectin-3 depletion apheresis on induced skin inflammation in a porcine model. J. Clin. Apher. 2018, 33, 486–493. [Google Scholar] [CrossRef]
- Saravanan, C.; Cao, Z.; Head, S.R.; Panjwani, N. Detection of Differentially Expressed Wound-Healing–Related Glycogenes in Galectin-3–Deficient Mice. Investig. Opthalmology Vis. Sci. 2009, 50, 5690–5696. [Google Scholar] [CrossRef]
- Panjwani, N. Role of galectins in re-epithelialization of wounds. Ann. Transl. Med. 2014, 2, 89. [Google Scholar] [CrossRef]
- Liu, W.; Hsu, D.K.; Chen, H.Y.; Yang, R.Y.; Carraway, K.L., 3rd; Isseroff, R.R.; Liu, F.T. Galectin-3 regulates intracellular trafficking of EGFR through Alix and promotes keratinocyte migration. J Invest Dermatol. 2012, 132, 2828–2837. [Google Scholar] [CrossRef]
- McLeod, K.; Walker, J.T.; Hamilton, D.W. Galectin-3 regulation of wound healing and fibrotic processes: Insights for chronic skin wound therapeutics. J. Cell Commun. Signal. 2018, 12, 281–287. [Google Scholar] [CrossRef]
- Pepe, D.; Elliott, C.G.; Forbes, T.L.; Hamilton, D.W. Detection of galectin-3 and localization of advanced glycation end products (AGE) in human chronic skin wounds. Histol. Histopathol. 2014, 29, 251–258. [Google Scholar] [CrossRef]
- Shi, Z.R.; Tan, G.Z.; Cao, C.X.; Han, Y.F.; Meng, Z.; Man, X.Y.; Jiang, Z.X.; Zhang, Y.P.; Dang, N.N.; Wei, K.H.; et al. Decrease of galectin-3 in keratinocytes: A potential diagnostic marker and a critical contributor to the pathogenesis of psoriasis. J. Autoimmun. 2018, 89, 30–40. [Google Scholar] [CrossRef]
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One Molecule for an Alphabet of Diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379. [Google Scholar] [CrossRef]
- Ritchie, S.; Neal, D.; Shlevin, H.; Allgood, A.; Traber, P. A phase 2a, open-label pilot study of the galectin-3 inhibitor GR-MD-02 for the treatment of moderate-to-severe plaque psoriasis. J. Am. Acad. Dermatol. 2017, 77, 753–755. [Google Scholar] [CrossRef]
- Koca, S.S.; Akbas, F.; Ozgen, M.; Yolbas, S.; Ilhan, N.; Gundogdu, B.; Isik, A. Serum galectin-3 level in systemic sclerosis. Clin. Rheumatol. 2014, 33, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Asano, Y.; Akamata, K.; Noda, S.; Masui, Y.; Yamada, D.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Tamaki, Z.; et al. Serum Levels of Galectin-3: Possible Association with Fibrosis, Aberrant Angiogenesis, and Immune Activation in Patients with Systemic Sclerosis. J. Rheumatol. 2012, 39, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Faludi, R.; Nagy, G.; Tőkés-Füzesi, M.; Kovács, K.; Czirják, L.; Komócsi, A. Galectin-3 is an independent predictor of survival in systemic sclerosis. Int. J. Cardiol. 2017, 233, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Sundblad, V.; Gomez, R.A.; Stupirski, J.C.; Hockl, P.F.; Pino, M.S.; Laborde, H.; Rabinovich, G.A. Circulating Galectin-1 and Galectin-3 in Sera From Patients With Systemic Sclerosis: Associations With Clinical Features and Treatment. Front. Pharmacol. 2021, 12, 650605. [Google Scholar] [CrossRef]
- Cunha, E.G.C.; de Lima, C.A.D.; Vilar, K.D.M.; de Nóbrega, M.F.; de Almeida, A.R.; Pereira, M.C.; Dantas, A.T.; Gonçalves, R.S.G.; Rêgo, M.J.B.D.M.; Duarte, A.L.B.P.; et al. Genetic variants in LGALS3 are related to lower galectin-3 serum levels and clinical outcomes in systemic sclerosis patients: A case-control study. Autoimmunity 2021, 54, 187–194. [Google Scholar] [CrossRef]
- Mora, G.F.; Zubieta, M.R. Galectin-1 and Galectin-3 Expression in Lesional Skin of Patients With Systemic Sclerosis—Association With Disease Severity. JCR: J. Clin. Rheumatol. 2020, 27, 317–323. [Google Scholar] [CrossRef]
- Arciniegas, E.; Carrillo, L.M.; Rojas, H.; Ramírez, R.; Chopite, M. Galectin-1 and Galectin-3 and Their Potential Binding Partners in the Dermal Thickening of Keloid Tissues. Am. J. Dermatopathol. 2019, 41, 193–204. [Google Scholar] [CrossRef]
- Servettaz, A.; Goulvestre, C.; Kavian, N.; Nicco, C.; Guilpain, P.; Chéreau, C.; Vuiblet, V.; Guillevin, L.; Mouthon, L.; Weill, B.; et al. Selective Oxidation of DNA Topoisomerase 1 Induces Systemic Sclerosis in the Mouse. J. Immunol. 2009, 182, 5855–5864. [Google Scholar] [CrossRef]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Korman, B. Evolving insights into the cellular and molecular pathogenesis of fibrosis in systemic sclerosis. Transl. Res. 2019, 209, 77–89. [Google Scholar] [CrossRef]
- Michalik, M.; Wójcik-Pszczoła, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pękala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. 2018, 75, 3943–3961. [Google Scholar] [CrossRef]
- Inohara, H.; Akahani, S.; Raz, A. Galectin-3 Stimulates Cell Proliferation. Exp. Cell Res. 1998, 245, 294–302. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhang, J.; Guan, X.; Chen, M.; Li, Y.; Zhang, L. Galectin-3 contributes to vascular fibrosis in monocrotaline-induced pulmonary arterial hypertension rat model. J. Biochem. Mol. Toxicol. 2017, 31, e21879. [Google Scholar] [CrossRef]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef]
- MacKinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef]
- Jiang, J.X.; Chen, X.; Hsu, D.K.; Baghy, K.; Serizawa, N.; Scott, F.; Takada, Y.; Takada, Y.; Fukada, H.; Chen, J.; et al. Galectin-3 modulates phagocytosis-induced stellate cell activation and liver fibrosis in vivo. Am. J. Physiol. Liver Physiol. 2012, 302, G439–G446. [Google Scholar] [CrossRef]
- Walker, J.T.; Elliott, C.G.; Forbes, T.L.; Hamilton, D.W. Genetic Deletion of Galectin-3 Does Not Impair Full-Thickness Excisional Skin Healing. J. Investig. Dermatol. 2016, 136, 1042–1050. [Google Scholar] [CrossRef]
- Quan, T.E.; Cowper, S.E.; Bucala, R. The role of circulating fibrocytes in fibrosis. Curr. Rheumatol. Rep. 2006, 8, 145–150. [Google Scholar] [CrossRef]
- Schmidt, M.; Sun, G.; Stacey, M.A.; Mori, L.; Mattoli, S. Identification of Circulating Fibrocytes as Precursors of Bronchial Myofibroblasts in Asthma. J. Immunol. 2003, 171, 380–389. [Google Scholar] [CrossRef]
- Abe, R.; Donnelly, S.; Peng, T.; Bucala, R.; Metz, C.N. Peripheral Blood Fibrocytes: Differentiation Pathway and Migration to Wound Sites. J. Immunol. 2001, 166, 7556–7562. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, G.; Bitto, A.; Pizzino, G.; Irrera, N.; Sangari, D.; Cinquegrani, M.; Roberts, W.N.; Cerinic, M.M.; Squadrito, F.; Altavilla, D.; et al. Simvastatin attenuates the development of pulmonary and cutaneous fibrosis in a murine model of systemic sclerosis. Rheumatology 2013, 52, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Maria, A.T.J.; Toupet, K.; Maumus, M.; Rozier, P.; Vozenin, M.-C.; Le Quellec, A.; Jorgensen, C.; Noël, D.; Guilpain, P. Fibrosis Development in HOCl-Induced Systemic Sclerosis: A Multistage Process Hampered by Mesenchymal Stem Cells. Front. Immunol. 2018, 9, 2571. [Google Scholar] [CrossRef] [PubMed]
- Maria, A.T.J.; Toupet, K.; Bony, C.; Pirot, N.; Vozenin, M.-C.; Petit, B.; Roger, P.; Batteux, F.; Le Quellec, A.; Jorgensen, C.; et al. Antifibrotic, Antioxidant, and Immunomodulatory Effects of Mesenchymal Stem Cells in HOCl-Induced Systemic Sclerosis. Arthritis Rheumatol. 2016, 68, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Abraham, D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J. Clin. Investig. 2007, 117, 557–567. [Google Scholar] [CrossRef]
- Bonniaud, P.; Margetts, P.J.; Ask, K.; Flanders, K.; Gauldie, J.; Kolb, M. TGF-β and Smad3 Signaling Link Inflammation to Chronic Fibrogenesis. J. Immunol. 2005, 175, 5390–5395. [Google Scholar] [CrossRef]
- Stochmal, A.; Czuwara, J.; Zaremba, M.; Rudnicka, L. Altered serum level of metabolic and endothelial factors in patients with systemic sclerosis. Arch. Dermatol. Res. 2020, 312, 453–458. [Google Scholar] [CrossRef]
- Luo, L.-F.; Shi, Y.; Zhou, Q.; Xu, S.-Z.; Lei, T.-C. Insufficient expression of the melanocortin-1 receptor by human dermal fibroblasts contributes to excess collagen synthesis in keloid scars. Exp. Dermatol. 2013, 22, 764–766. [Google Scholar] [CrossRef]
- Nirodi, C.S.; Devalaraja, R.; Nanney, L.B.; Arrindell, S.; Russell, S.; Trupin, J.; Richmond, A. Chemokine and chemokine receptor expression in keloid and normal fibroblasts. Wound Repair Regen. 2000, 8, 371–382. [Google Scholar] [CrossRef]
- Krieg, T.; Abraham, D.; Lafyatis, R. Fibrosis in connective tissue disease: The role of the myofibroblast and fibroblast-epithelial cell interactions. Thromb. Haemost. 2007, 9 (Suppl. S2), S4. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.M.; McFarland, K.L.; Combs, K.A.; Supp, D.M. Partial epithelial-mesenchymal transition in keloid scars: Regulation of keloid keratinocyte gene expression by transforming growth factor-beta1. Burns. Trauma 2016, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Cao, R.; Wang, L.; Liu, Y.; Pan, B.; Yin, Y.; Lv, X.; Zhuang, Q.; Sun, X.; Xiao, R. Epithelial-mesenchymal transition in keloid tissues and TGF-beta1-induced hair follicle outer root sheath keratinocytes. Wound Repair Regen. 2015, 23, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Overed-Sayer, C.; Rapley, L.; Mustelin, T.; Clarke, D.L. Are mast cells instrumental for fibrotic diseases? Front. Pharmacol. 2014, 4, 174. [Google Scholar] [CrossRef]
- Wilgus, T.A.; Ud-Din, S.; Bayat, A. A review of the evidence for and against a role for mast cells in cutaneous scarring and fibrosis. Int. J. Mol. Sci. 2020, 21, 9673. [Google Scholar] [CrossRef]
- Ud-Din, S.; Wilgus, T.A.; Bayat, A. Mast cells in skin scarring: A review of animal and human research. Front. Immunol. 2020, 11, 552205. [Google Scholar] [CrossRef]
- Moretti, L.; Stalfort, J.; Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022, 298, 101530. [Google Scholar] [CrossRef]
- Bitto, A.; Bagnato, G.L.; Pizzino, G.; Roberts, W.N.; Irrera, N.; Minutoli, L.; Russo, G.; Squadrito, F.; Saitta, A.; Altavilla, D. Simvastatin prevents vascular complications in the chronic reactive oxygen species murine model of systemic sclerosis. Free Radic. Res. 2016, 50, 514–522. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peiró, T.; Alonso-Carpio, M.; Ribera, P.; Almudéver, P.; Roger, I.; Montero, P.; Marín, S.; Milara, J.; Cortijo, J. Increased Expression of Galectin-3 in Skin Fibrosis: Evidence from In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2022, 23, 15319. https://doi.org/10.3390/ijms232315319
Peiró T, Alonso-Carpio M, Ribera P, Almudéver P, Roger I, Montero P, Marín S, Milara J, Cortijo J. Increased Expression of Galectin-3 in Skin Fibrosis: Evidence from In Vitro and In Vivo Studies. International Journal of Molecular Sciences. 2022; 23(23):15319. https://doi.org/10.3390/ijms232315319
Chicago/Turabian StylePeiró, Teresa, Miriam Alonso-Carpio, Pilar Ribera, Patricia Almudéver, Inés Roger, Paula Montero, Severiano Marín, Javier Milara, and Julio Cortijo. 2022. "Increased Expression of Galectin-3 in Skin Fibrosis: Evidence from In Vitro and In Vivo Studies" International Journal of Molecular Sciences 23, no. 23: 15319. https://doi.org/10.3390/ijms232315319
APA StylePeiró, T., Alonso-Carpio, M., Ribera, P., Almudéver, P., Roger, I., Montero, P., Marín, S., Milara, J., & Cortijo, J. (2022). Increased Expression of Galectin-3 in Skin Fibrosis: Evidence from In Vitro and In Vivo Studies. International Journal of Molecular Sciences, 23(23), 15319. https://doi.org/10.3390/ijms232315319