
Expression and Characterization of Monomeric Recombinant Isocitrate Dehydrogenases from Corynebacterium glutamicum and Azotobacter vinelandii for NADPH Regeneration


Abstract
:1. Introduction
2. Results
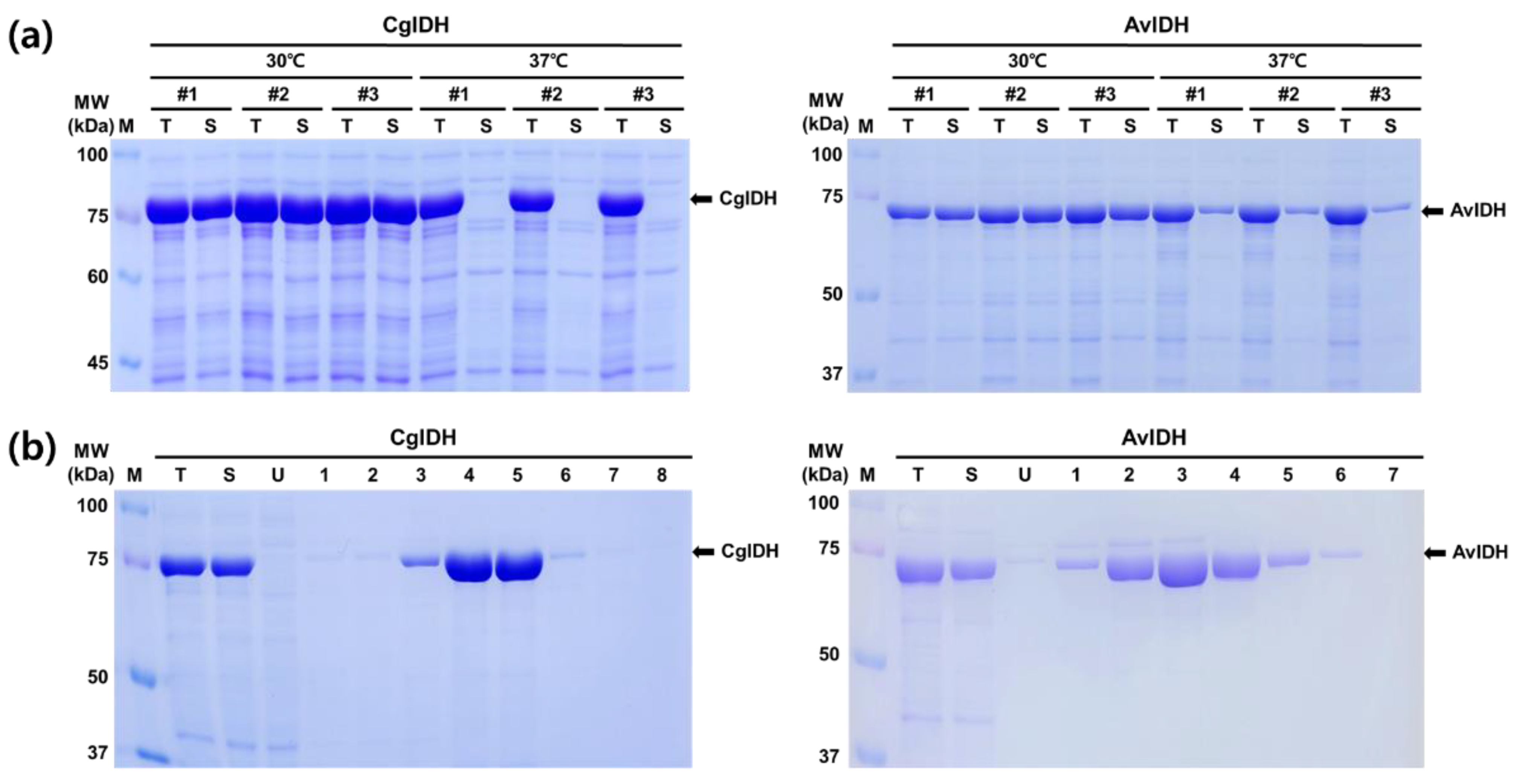
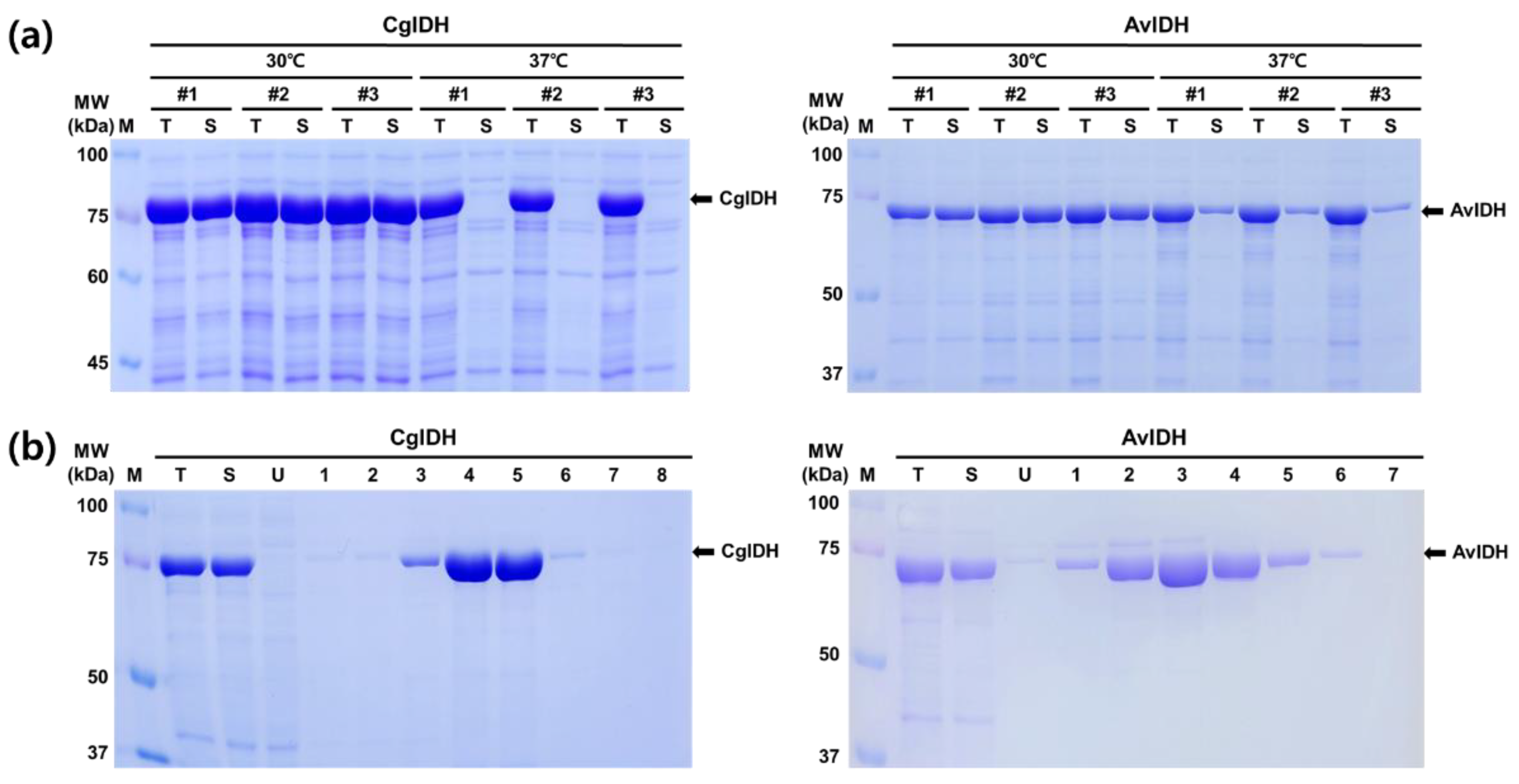
2.1. Expression and Purification of CgIDH and AvIDH
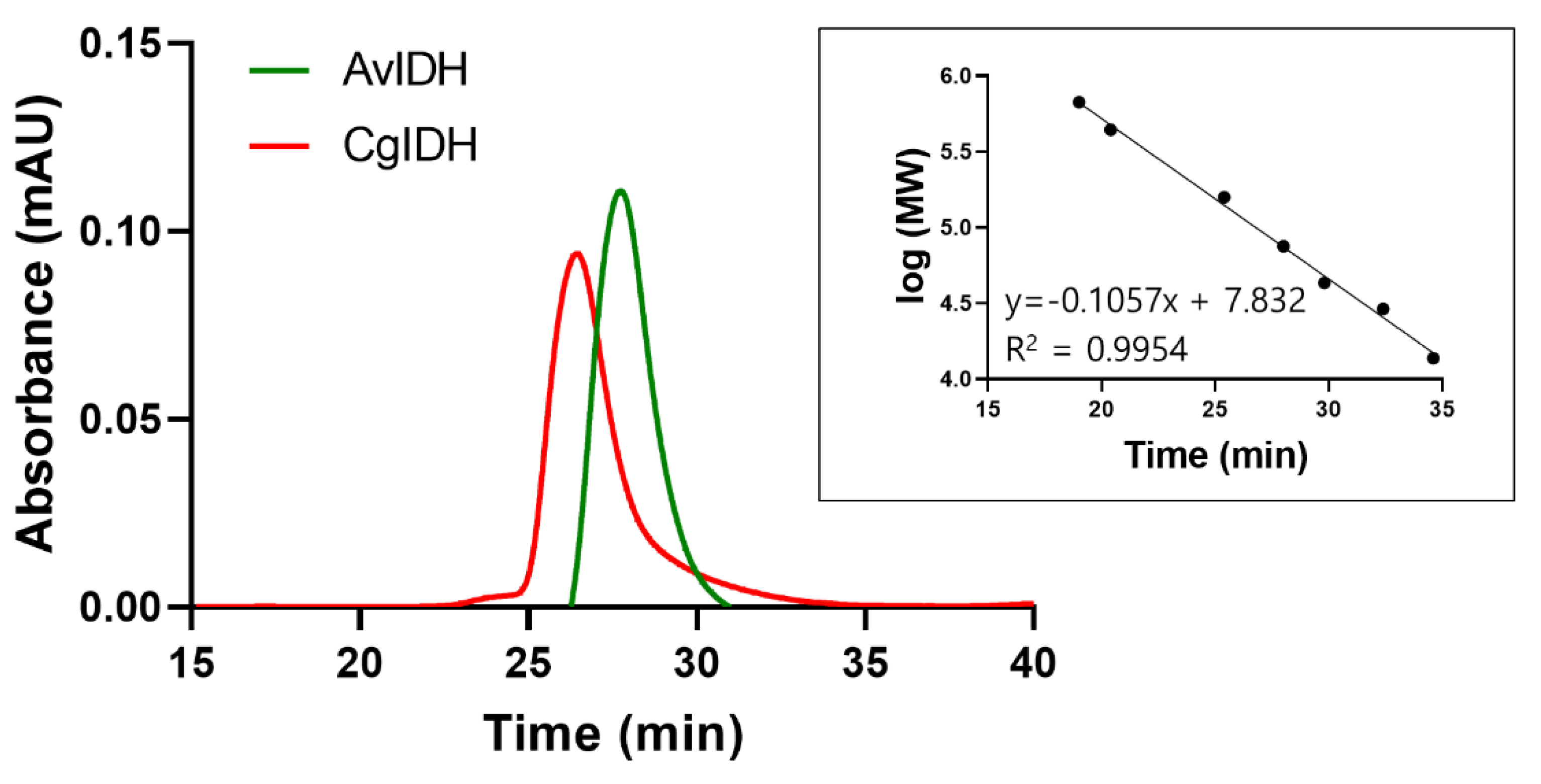
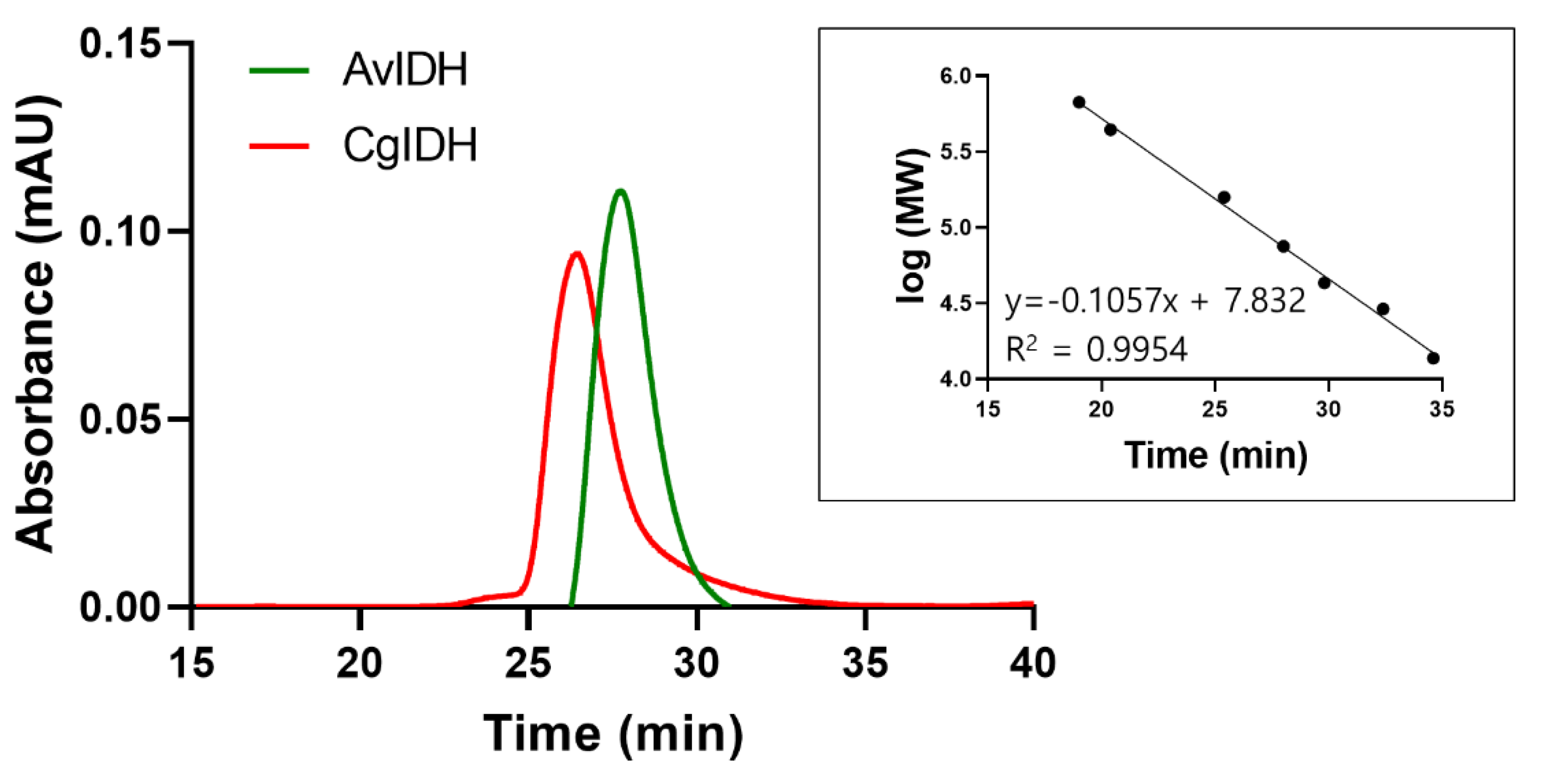
2.2. Quaternary Structure Analyses of CgIDH and AvIDH
2.3. Kinetic Parameters and Biochemical Properties of CgIDH and AvIDH
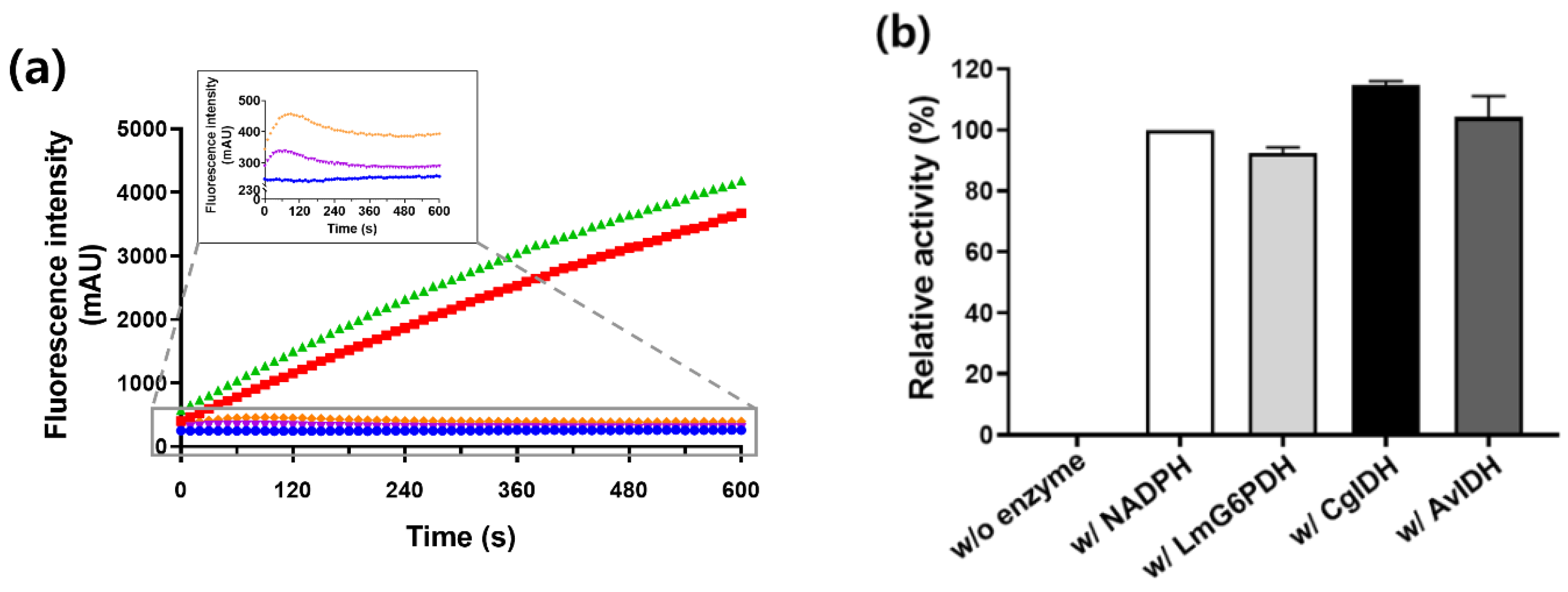
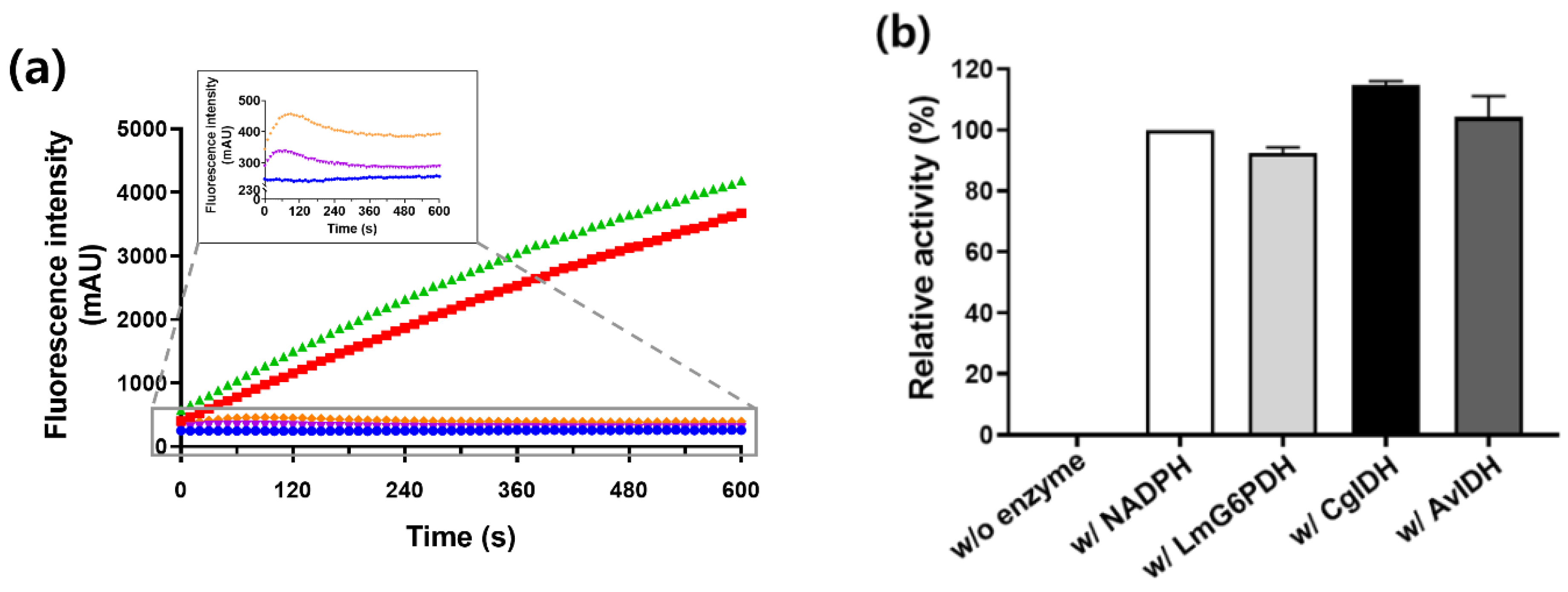
2.4. Regeneration of NADPH Using Monomeric CgIDH and AvIDH
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Cloning
4.2. Protein Expression and Purification
4.3. Size Exclusion Chromatography
4.4. Enzyme Assay and Kinetic Parameters
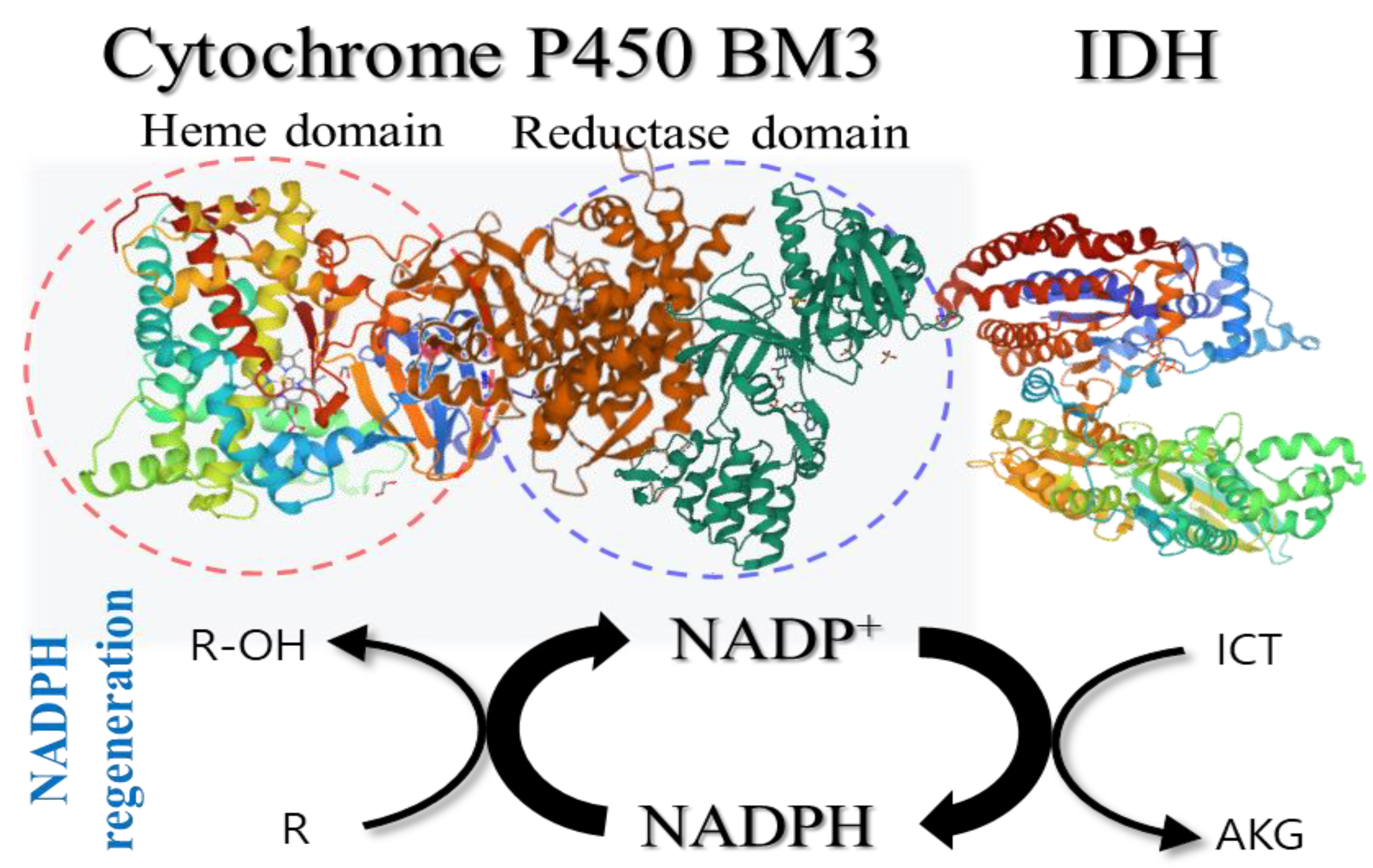
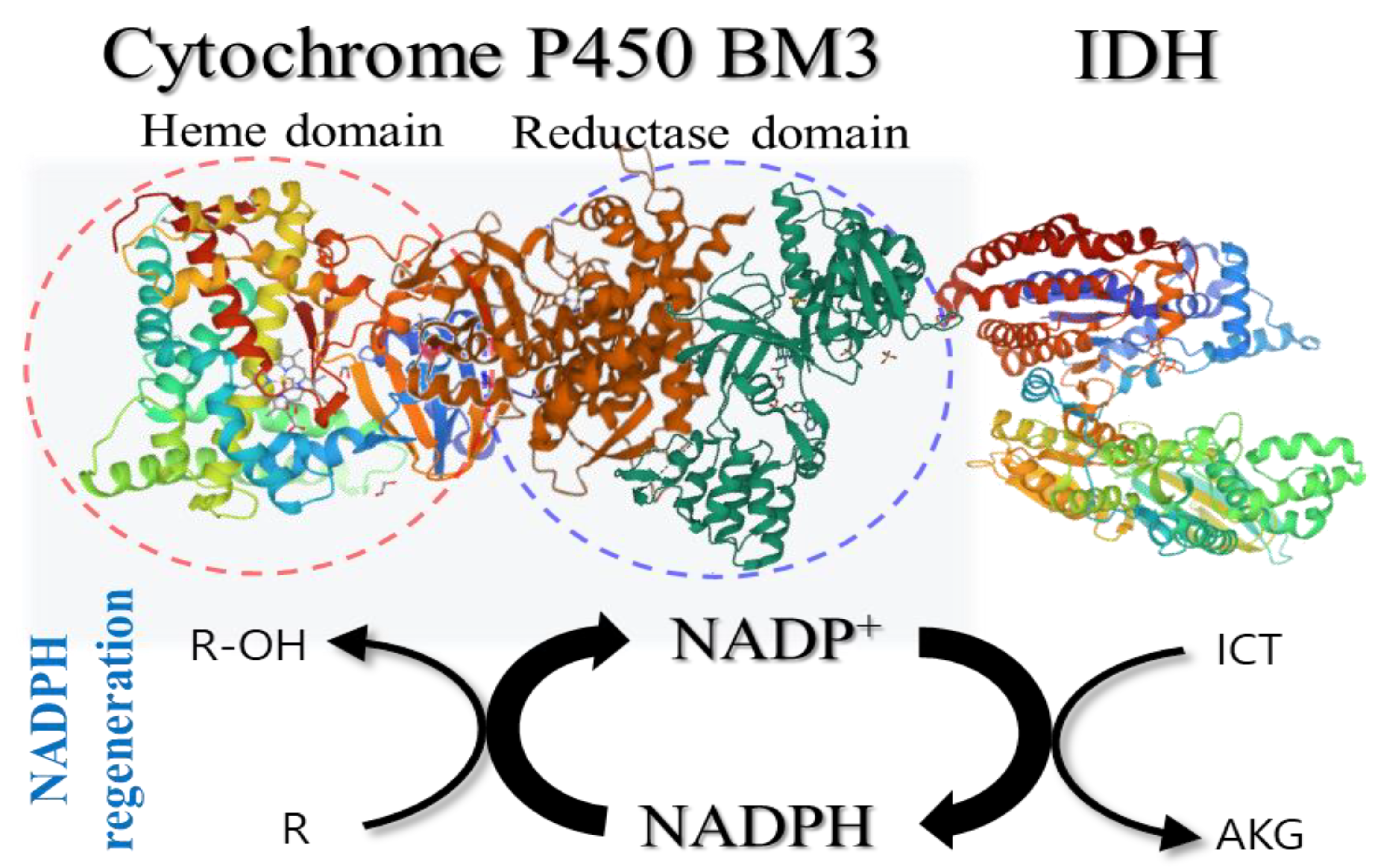
4.5. Coupling Reactions of IDH with NADPH-Dependent P450 BM3
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fryszkowska, A.; Devine, P.N. Biocatalysis in drug discovery and development. Curr. Opin. Chem. Biol. 2020, 55, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Fundamentals of green chemistry: Efficiency in reaction design. Chem. Soc. Rev. 2012, 41, 1437–1451. [Google Scholar] [CrossRef] [Green Version]
- Vidal, L.S.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD (P) H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta. Proteins. Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef]
- Faber, K. Biotransformations in Organic Chemistry, 6th ed.; Biocatalytic Applications; Springer: Berlin/Heidelberg, Germany, 2011; pp. 31–313. ISBN 978-3-642-17393-6. [Google Scholar]
- Wang, X.; Saba, T.; Yiu, H.H.P.; Howe, R.F.; Anderson, J.A.; Shi, J. Cofactor NAD (P) H regeneration inspired by heterogeneous pathways. Chem. Soc. Rev. 2017, 2, 621–654. [Google Scholar] [CrossRef] [Green Version]
- Spaans, S.K.; Weusthuis, R.A.; Van Der Oost, J.; Kengen, S.W. NADPH-generating systems in bacteria and archaea. Front. Microbiol. 2015, 6, 742. [Google Scholar] [CrossRef]
- Johannes, T.W.; Woodyer, R.D.; Zhao, H. Efficient regeneration of NADPH using an engineered phosphite dehydrogenase. Biotechnol. Bioeng. 2007, 96, 18–26. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, Y.; Zhang, L.; Chen, X.; Shen, Y. Enhancing NADPH regeneration and increasing hydroxylation efficiency with P450 monooxygenase through strengthening expression of glucose-6-phosphate dehydrogenase in industrial filamentous fungi. Biocatal. Agric. Biotechnol. 2017, 11, 307–311. [Google Scholar] [CrossRef]
- Zhai, X.-H.; Ma, Y.-H.; Lai, D.-Y.; Zhou, S.; Chen, Z.-M. Development of a whole-cell biocatalyst with NADPH regeneration system for biosulfoxidation. J. Ind. Microbiol. Biotechnol. 2013, 40, 797–803. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, H.; Yu, H.; Deng, T.; Wang, Z.; Zhang, H.; Wu, J.; Yang, L. Computational design of highly stable and soluble alcohol dehydrogenase for NADPH regeneration. Bioresour. Bioprocess. 2021, 8, 1–13. [Google Scholar] [CrossRef]
- Chen, R.; Wei, Q.; Wei, X.; Liu, Y.; Zhang, X.; Chen, X.; Yin, X.; Xie, T. Stable and efficient immobilization of bi-enzymatic NADPH cofactor recycling system under consecutive microwave irradiation. PLoS. ONE 2020, 15, e0242564. [Google Scholar] [CrossRef]
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszyńska, D. Immobilization as a strategy for improving enzyme properties-application to oxidoreductases. Molecules 2014, 19, 8995–9018. [Google Scholar] [CrossRef] [Green Version]
- Ellis, G.A.; Klein, W.P.; Lasarte-Aragones, G.; Thakur, M.; Walper, S.A.; Medintz, I.L. Artificial multienzyme scaffolds: Pursuing in vitro substrate channeling with an overview of current progress. ACS. Catal. 2019, 9, 10812–10869. [Google Scholar] [CrossRef] [Green Version]
- Mourelle-Insua, Á.; Aalbers, F.S.; Lavandera, I.; Gotor-Fernández, V.; Fraaije, M.W. What to sacrifice? Fusions of cofactor regenerating enzymes with Baeyer-Villiger monooxygenases and alcohol dehydrogenases for self-sufficient redox biocatalysis. Tetrahedron 2019, 75, 1832–1839. [Google Scholar] [CrossRef]
- Beyer, N.; Kulig, J.K.; Bartsch, A.; Hayes, M.A.; Janssen, D.B.; Fraaije, M.W. P450BM3 fused to phosphite dehydrogenase allows phosphite-driven selective oxidations. Appl. Microbiol. Biotechnol. 2017, 101, 2319–2331. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Upadhyay, V.; Singh, A.; Panda, A.K. Structure-function relationship of inclusion bodies of a multimeric protein. Front. Microbiol. 2020, 11, 876. [Google Scholar] [CrossRef]
- Eikmanns, B.J.; Rittmann, D.; Sahm, H. Cloning, sequence analysis, expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J. Bacteriol. 1995, 177, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahara, T.; Takada, Y.; Takeuchi, Y.; Yamaoka, N.; Fukunaga, N. Cloning, sequencing, and expression of a gene encoding the monomeric isocitrate dehydrogenase of the nitrogen-fixing bacterium, Azotobacter vinelandii. Biosci. Biotechnol. Biochem. 2002, 66, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Leyland, M.L.; Kelly, D.J. Purification and characterization of a monomeric isocitrate dehydrogenase with dual coenzyme specificity from the photosynthetic bacterium Rhodomicrobium vannielii. Eur. J. Biochem. 1991, 202, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cadoux-Hudson, T.; Schofield, C.J. Isocitrate dehydrogenase variants in cancer—Cellular consequences and therapeutic opportunities. Curr. Opin. Chem. Biol. 2020, 57, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Meinhold, P.; Peters, M.W.; Hartwick, A.; Hernandez, A.R.; Arnold, F.H. Engineering cytochrome P450 BM3 for terminal alkane hydroxylation. Adv. Synth. Catal. 2006, 348, 763–772. [Google Scholar] [CrossRef]
- Park, W.-J.; You, S.-H.; Choi, H.-A.; Chu, Y.-J.; Kim, G.-J. Over-expression of recombinant proteins with N-terminal His-tag via subcellular uneven distribution in Escherichia coli. Acta. Biochim. Biophys. Sin. 2015, 47, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.-B.; Wang, P.; Wang, A.; Wang, W.-C.; Tang, W.-G.; Zhu, G.-P. Expression and characterization of a novel isocitrate dehydrogenase from Streptomyces diastaticus No. 7 strain M1033. Mol. Biol. Rep. 2013, 40, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.-M.; Wang, P.; Li, X.; Zhao, X.-Y.; Xu, L.; Song, P.; Zhu, G.-P. Biochemical characterization of NADP+-dependent isocitrate dehydrogenase from Microcystis aeruginosa PCC7806. Mol. Biol. Rep. 2013, 40, 2995–3002. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yang, H. A highly specific monomeric isocitrate dehydrogenase from Corynebacterium glutamicum. Arch. Biochem. Biophys. 2000, 383, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Yasutake, Y.; Tanaka, I.; Takada, Y. Elucidation of stability determinants of cold-adapted monomeric isocitrate dehydrogenase from a psychrophilic bacterium, Colwellia maris, by construction of chimeric enzymes. Microbiology 2005, 151, 1083–1094. [Google Scholar] [CrossRef] [Green Version]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases in biotechnology and synthetic biology. Trends. Biotechnol. 2019, 37, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.H.; Park, B.-Y.; Kim, S.-Y.; Park, S.-H.; Jung, H.-J.; Park, M.; Park, K.D.; Ahn, T.; Kang, H.-S.; Yun, C.-H. Regioselective hydroxylation of omeprazole enantiomers by bacterial CYP102A1 mutants. Drug. Metab. Dispos. 2014, 42, 1493–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.J.; Cheon, Y.H.; Kim, H.S. Directed evolution of a novel N-carbamylase/D-hydantoinase fusion enzyme for functional expression with enhanced stability. Biotechnol. Bioeng. 2000, 68, 211–217. [Google Scholar] [CrossRef]
- Sivashanmugam, A.; Murray, V.; Cui, C.; Zhang, Y.; Wang, J.; Li, Q. Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein. Sci. 2009, 18, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, D.; Bornscheuer, U.T. Protein engineering of microbial enzymes. Curr. Opin. Microbiol. 2010, 13, 274–282. [Google Scholar] [CrossRef]
- Seo, P.-W.; Jo, E.-S.; You, S.-H.; Cheong, D.-E.; Kim, G.-J.; Kim, J.-S. Structure-Guided Generation of a Redox-Independent Blue Fluorescent Protein from mBFP. J. Mol. Biol. 2019, 431, 3191–3202. [Google Scholar] [CrossRef]
- Choi, G.-S.; Kim, J.-Y.; Kim, J.-H.; Ryu, Y.-W.; Kim, G.-J. Construction and characterization of a recombinant esterase with high activity and enantioselectivity to (S)-ketoprofen ethyl ester. Protein. Expr. Purif. 2003, 29, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.J.; Lee, D.E.; Kim, H.-S. Functional expression and characterization of the two cyclic amidohydrolase enzymes, allantoinase and a novel phenylhydantoinase, from Escherichia coli. J. Bacteriol. 2000, 182, 7021–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.-H.; Kim, K.-H.; Calcutt, M.W.; Guengerich, F.P. Kinetic analysis of oxidation of coumarins by human cytochrome P450 2A6. J. Biol. Chem. 2005, 280, 12279–12291. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | NADP+ | Isocitrate | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Km (μM) * | kcat (s−1) | kcat/Km (μM−1s−1) | Km (μM) | kcat (s−1) | kcat/Km (μM−1s−1) | ||
| Previously reported CgIDH | 4.0 | 87.0 | 21.8 | 5.0 | 87.0 | 17.4 | [25] |
| Previously reported AvIDH | 5.8 | 92.6 | 15.9 | 7.9 | 73.0 | 9.3 | [26] |
| CgIDH | 10.3 | 43.7 | 4.3 | 15.0 | 49.4 | 3.3 | This study |
| AvIDH | 5.9 | 38.7 | 6.6 | 13.9 | 44.0 | 3.2 | This study |
| Enzyme | Optimum Temperature (°C) | Optimum pH | |||||
| CgIDH | 40 | 7.5 | |||||
| AvIDH | 40 | 7.5 | |||||
| Metal Ions * | Relative Activity (%) | |
|---|---|---|
| CgIDH | AvIDH | |
| EDTA | ND | ND |
| Mn2+ | 100 ± 6.1 | 100 ± 6.0 |
| Mg2+ | 43.7 ± 2.9 | 50.0 ± 3.5 |
| Ca2+ | 2.1 ± 0.3 | 1.3 ± 0.2 |
| Cu2+ | 2.6 ± 0.5 | 2.1 ± 0.2 |
| Co2+ | 6.4 ± 0.9 | 7.8 ± 0.3 |
| Ni2+ | 2.7 ± 0.2 | 2.2 ± 0.1 |
| Zn2+ | 8.8 ± 0.8 | 11.7 ± 0.1 |
| Names | Content & Sequence | RE * |
|---|---|---|
| Plasmids | ||
| pET24a | Kanr, T7 promoter, lacO, PBR322 ori | |
| pET24a-CgIDH | idh gene from C. glutamicum subcloned into pET24a | |
| pET24a-AvIDH | idh gene from A. vinelandii subcloned into pET24a | |
| pET24a-LmG6PDH | g6pdh gene from L. mesenteroides subcloned into pET24a | |
| pET24a-BM3 | CYP102A1 gene from B. megaterium subcloned into pET24a | |
| Primers | ||
| CgIDH F | 5′-GAAGGAGATATACATATGGCTAAGATCATCTGGACCCG-3′ | NdeI |
| CgIDH R | 5′-GTGGTGGTGGTGCTCGAGCTTCTTCAGTGCGTCAACGATCTC-3′ | XhoI |
| AvIDH F | 5′-GAAGGAGATATACATATGTCCACACCGAAGATTATCTATACGC-3′ | NdeI |
| AvIDH R | 5′-GTGGTGGTGGTGCTCGAGTGCAAGAGGTGCCAGAGCC-3′ | XhoI |
| LmG6PDH F | 5′-TAAGAAGGAGATATACATATGGTTTCAGAAATCAAGACGTTAGTAACTTTC-3′ | NdeI |
| LmG6PDH R | 5′-GTGGTGGTGGTGCTCGAGACCTTTAAACACCCAAGCATCACC-3′ | XhoI |
| BM3 F | 5′-TAACAATTCCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGA GATATACATATGAC-3′ | XbaI |
| BM3 R | 5′-GCTGCCTCCTTTGCTGCTGCCTCCTTAGCAGCAGCTTCCCCAGCCC ACACGTCTTTTGC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-D.; Yoo, S.-K.; Yoo, H.-S.; Yun, C.-H.; Kim, G.-J. Expression and Characterization of Monomeric Recombinant Isocitrate Dehydrogenases from Corynebacterium glutamicum and Azotobacter vinelandii for NADPH Regeneration. Int. J. Mol. Sci. 2022, 23, 15318. https://doi.org/10.3390/ijms232315318
Lee H-D, Yoo S-K, Yoo H-S, Yun C-H, Kim G-J. Expression and Characterization of Monomeric Recombinant Isocitrate Dehydrogenases from Corynebacterium glutamicum and Azotobacter vinelandii for NADPH Regeneration. International Journal of Molecular Sciences. 2022; 23(23):15318. https://doi.org/10.3390/ijms232315318
Chicago/Turabian StyleLee, Hun-Dong, Su-Kyoung Yoo, Ho-Seok Yoo, Chul-Ho Yun, and Geun-Joong Kim. 2022. "Expression and Characterization of Monomeric Recombinant Isocitrate Dehydrogenases from Corynebacterium glutamicum and Azotobacter vinelandii for NADPH Regeneration" International Journal of Molecular Sciences 23, no. 23: 15318. https://doi.org/10.3390/ijms232315318
APA StyleLee, H.-D., Yoo, S.-K., Yoo, H.-S., Yun, C.-H., & Kim, G.-J. (2022). Expression and Characterization of Monomeric Recombinant Isocitrate Dehydrogenases from Corynebacterium glutamicum and Azotobacter vinelandii for NADPH Regeneration. International Journal of Molecular Sciences, 23(23), 15318. https://doi.org/10.3390/ijms232315318