
Opposing Roles of DCs and iNKT Cells in the Induction of Foxp3 Expression by MLN CD25+CD4+ T Cells during IFNγ-Driven Colitis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
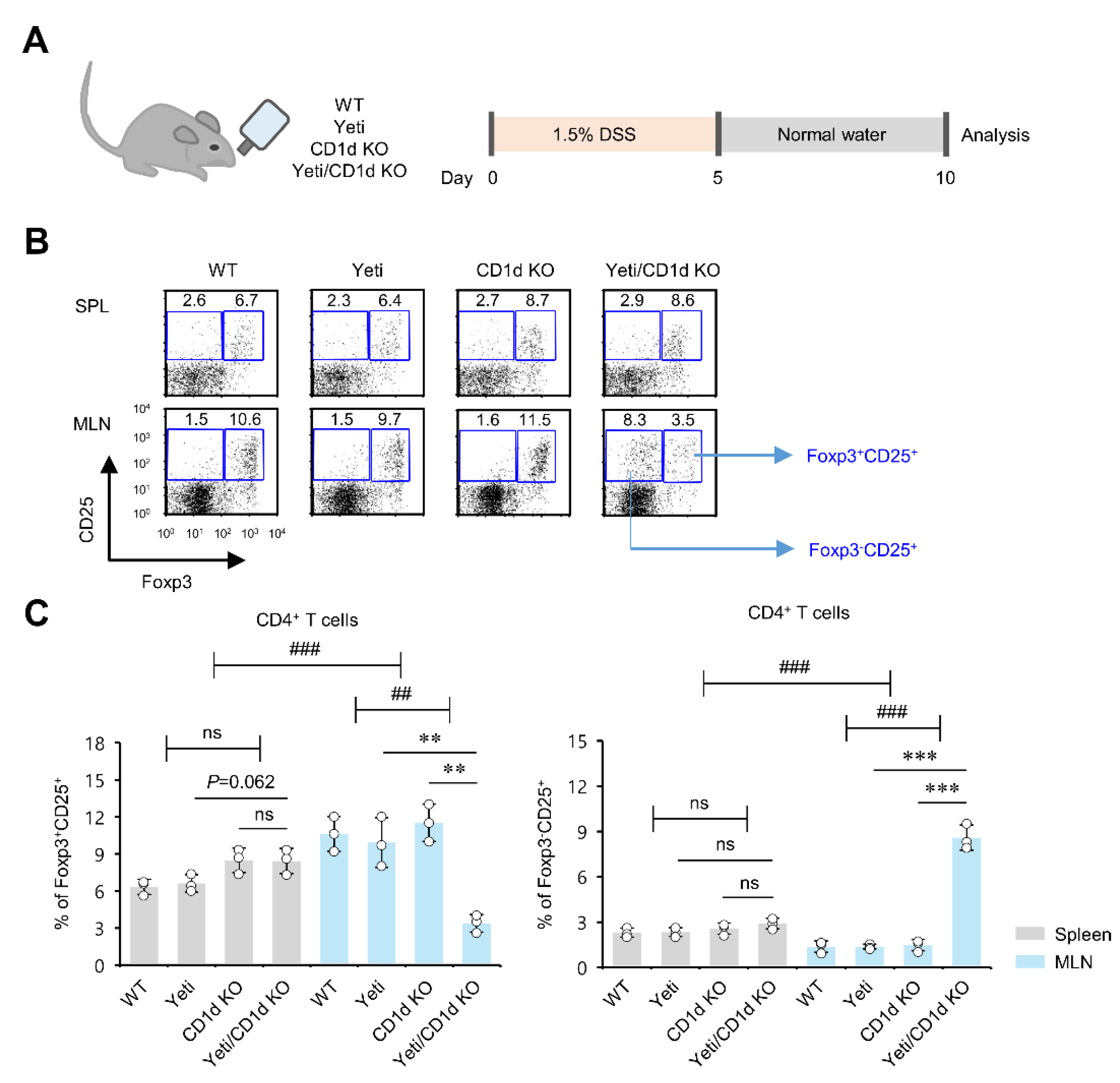
2.1. Dysregulated IFNγ Expression in the Absence of iNKT Cells Causes Alterations in Foxp3 Expression by CD25+CD4+ T Cells in MLNs but Not Spleen during DSS-Induced Colitis
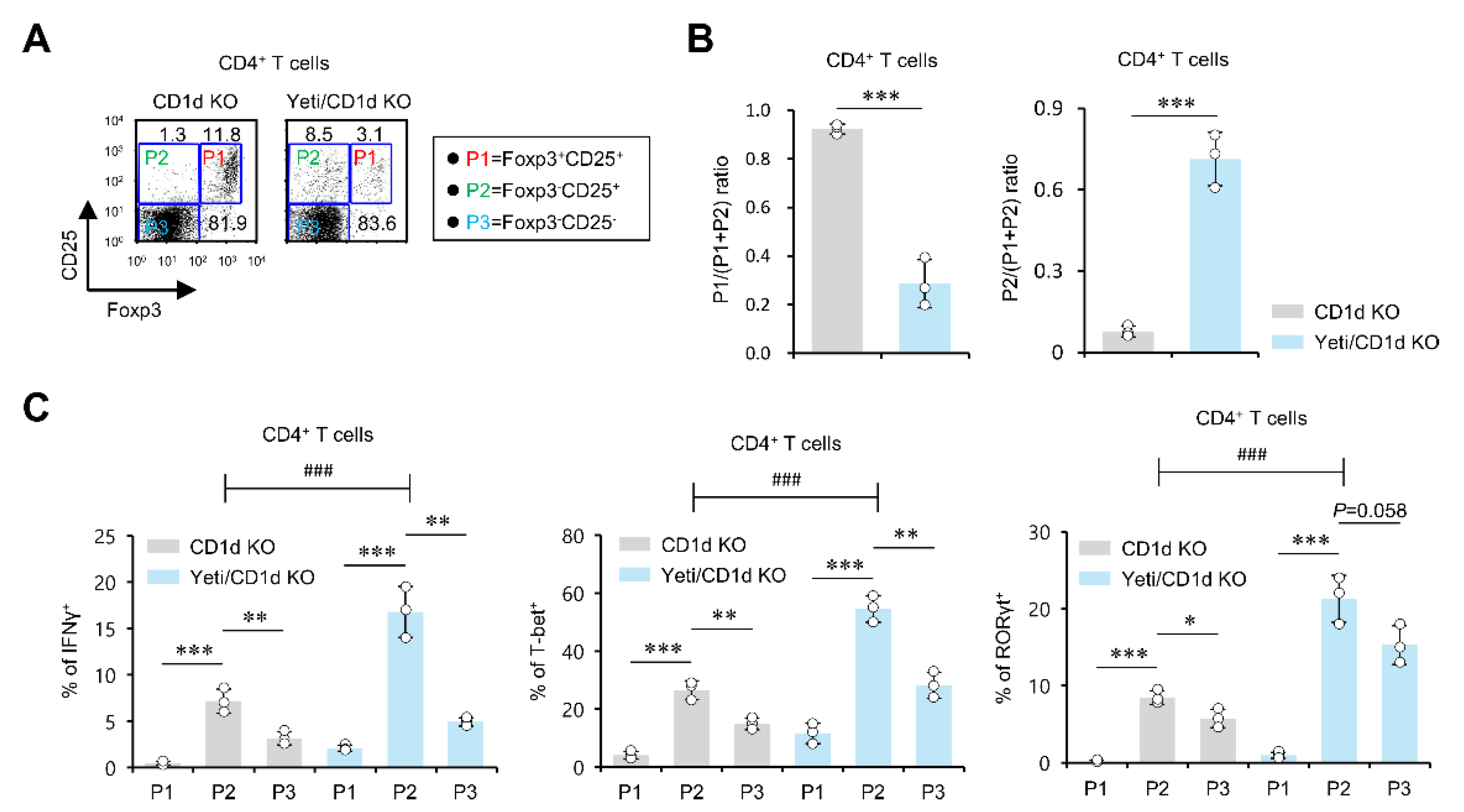
2.2. The MLN Foxp3−CD25+CD4+ T Cells from DSS-Treated Yeti/CD1d KO Mice Exhibit a Pathogenic Effector Phenotype
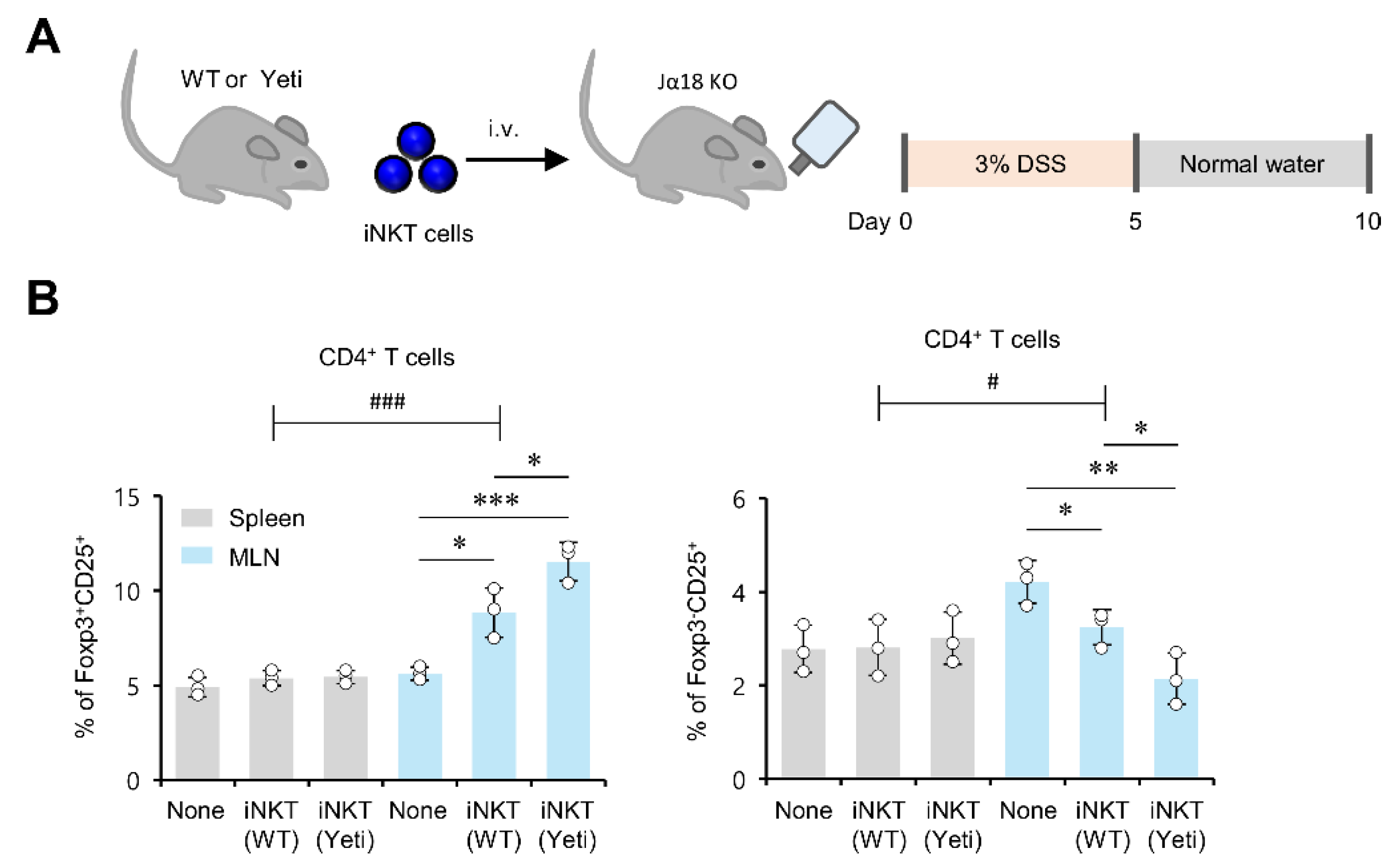
2.3. MLN iNKT Cells Play a Critical Role in Regulating Foxp3 Expression by CD25+CD4+ T Cells during DSS-Induced Colitis
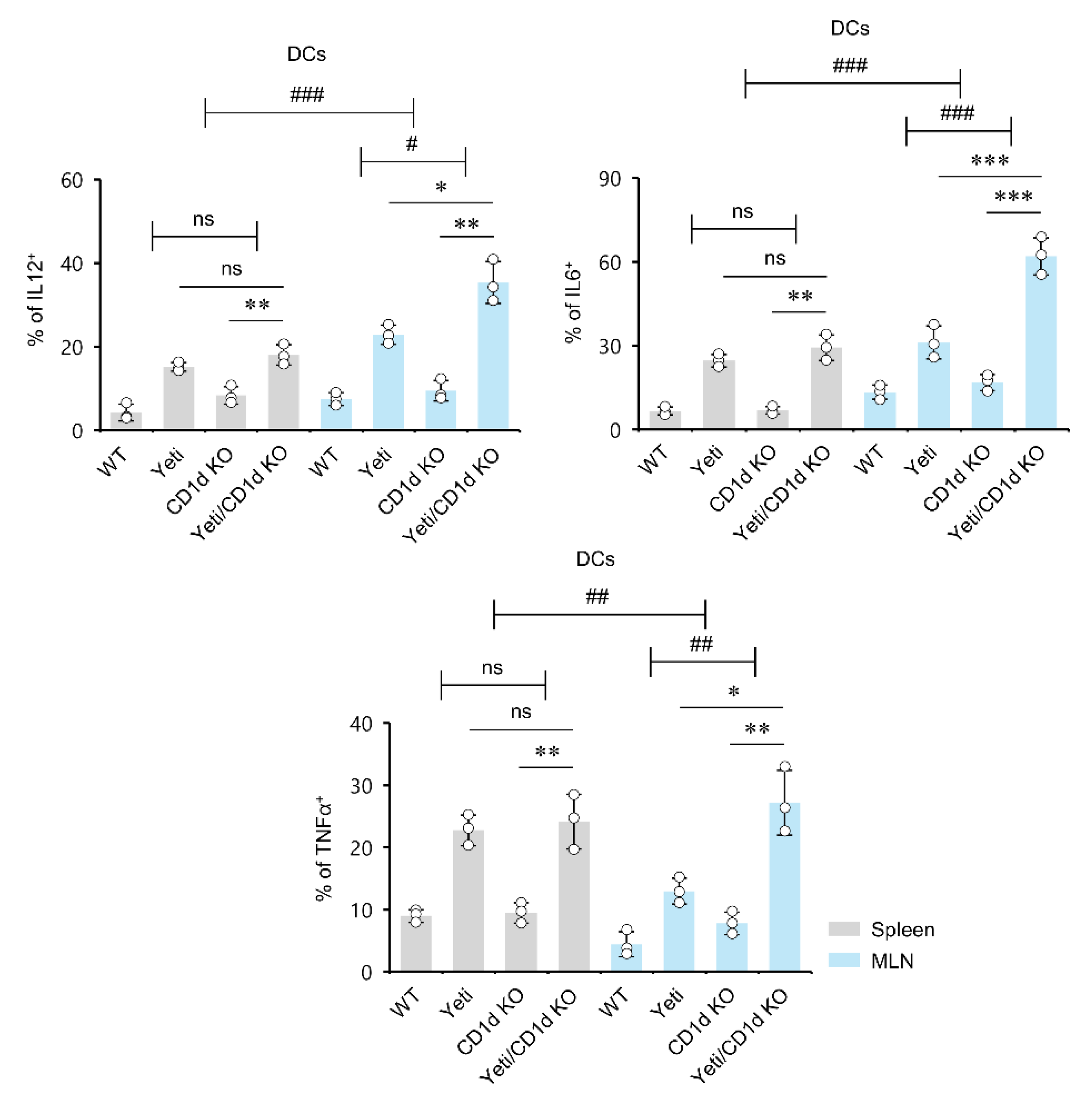
2.4. Dysregulated IFNγ Production in the Absence of iNKT Cells Induces MLN DCs to Produce Pro-Inflammatory Cytokines during DSS-Induced Colitis
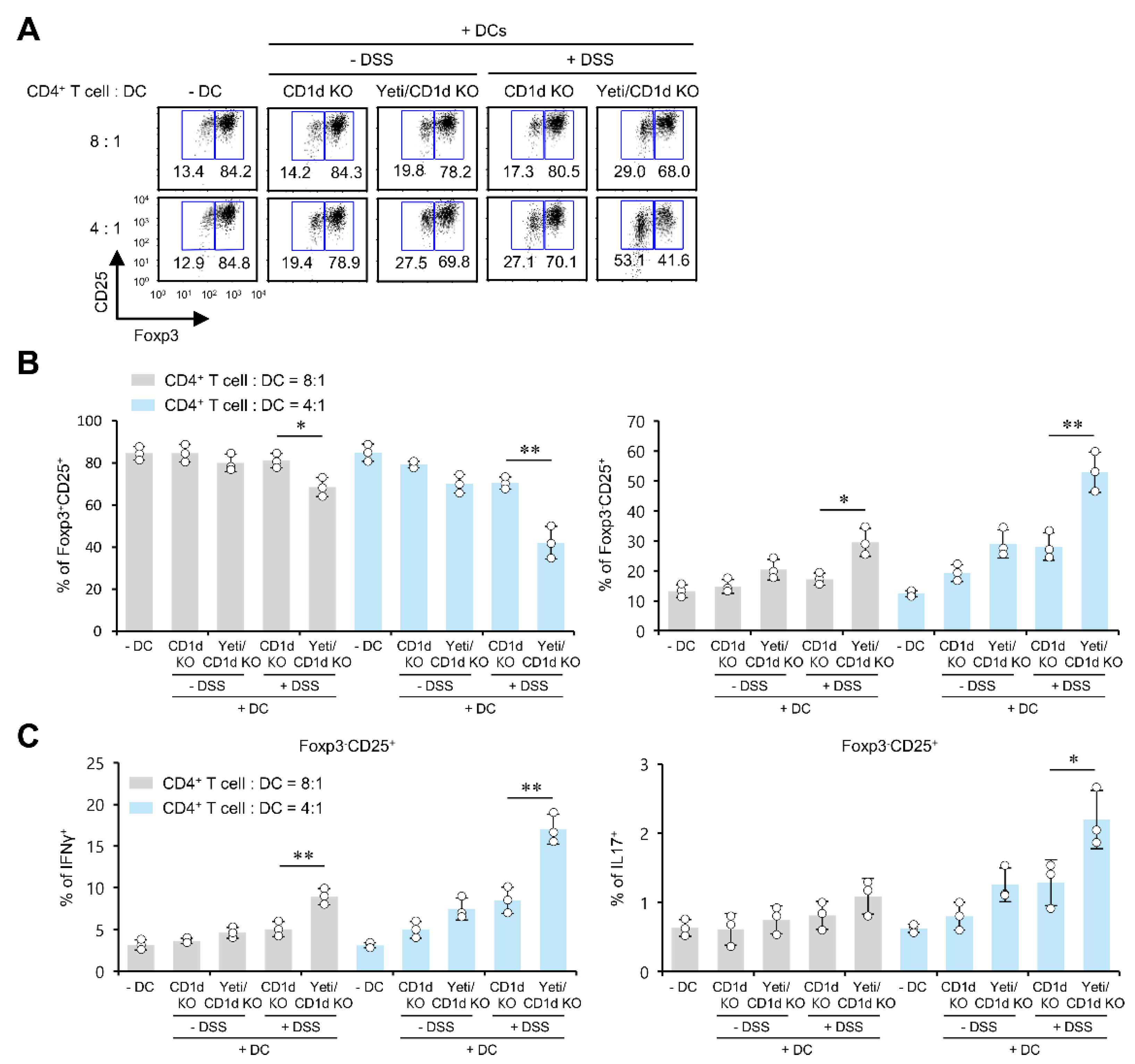
2.5. Pro-Inflammatory DCs from Yeti/CD1d KO Mice Induce the Differentiation of Foxp3−CD25+CD4+ Effector Cells and Antagonize Treg Cell Differentiation
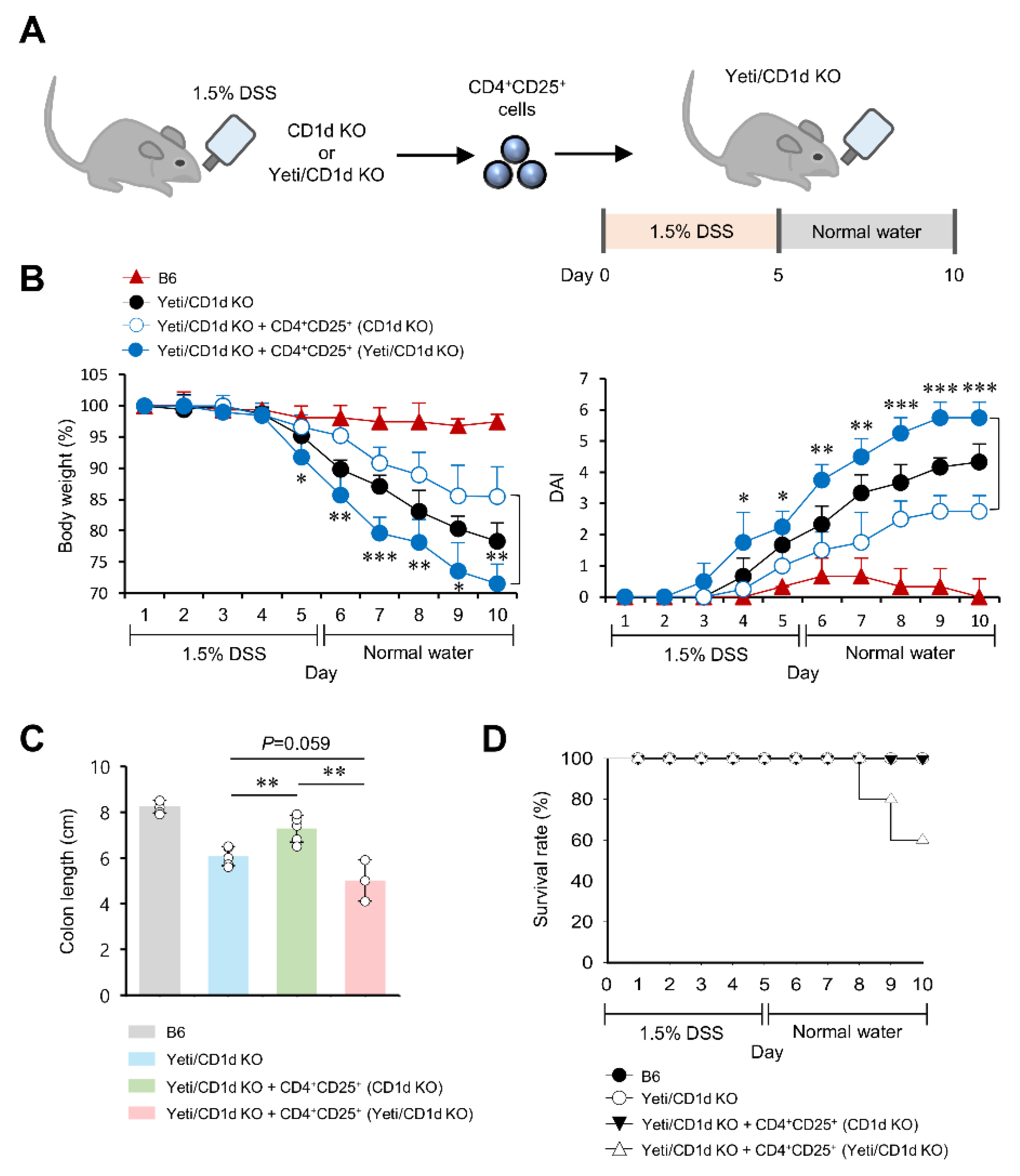
2.6. CD25+CD4+ Effector T Cells from DSS-Treated Yeti/CD1d KO Mice Are Pathogenic in DSS-Induced Colitis
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Mice and Reagents
4.3. Induction of Colonic Inflammation
4.4. Cell Culture and Cell Enrichment by Magnetically Activated Cell Sorting (MACS)
4.5. In Vitro Treg Cell Differentiation
4.6. Flow Cytometry
4.7. Intracellular Cytokine Staining
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pollard, K.M.; Cauvi, D.M.; Toomey, C.B.; Morris, K.V.; Kono, D.H. Interferon-gamma and systemic autoimmunity. Discov. Med. 2013, 16, 123–131. [Google Scholar] [PubMed]
- Mavropoulos, A.; Sully, G.; Cope, A.P.; Clark, A.R. Stabilization of IFN-gamma mRNA by MAPK p38 in IL-12- and IL-18-stimulated human NK cells. Blood 2005, 105, 282–288. [Google Scholar] [CrossRef]
- Salerno, F.; Guislain, A.; Freen-Van Heeren, J.J.; Nicolet, B.P.; Young, H.A.; Wolkers, M.C. Critical role of post-transcriptional regulation for IFN-gamma in tumor-infiltrating T cells. Oncoimmunology 2019, 8, e1532762. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.L.; Berthet, C.; Coppola, V.; Kastenmuller, W.; Buschman, M.D.; Schaughency, P.M.; Shirota, H.; Scarzello, A.J.; Subleski, J.J.; Anver, M.R.; et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. J. Autoimmun 2014, 53, 33–45. [Google Scholar] [CrossRef]
- Ebrahimi Daryani, N.; Sadr, M.; Soltani, S.; Saghazadeh, A.; Moossavi, S.; Shahkarami, S.; Farhadi, E.; Rezaei, N. Association of T Helper 1 Cytokine Gene Single Nucleotide Polymorphisms with Ulcerative Colitis and Crohn’s Disease. Dig. Dis. 2019, 37, 21–32. [Google Scholar] [CrossRef]
- Reinhardt, R.L.; Liang, H.E.; Bao, K.; Price, A.E.; Mohrs, M.; Kelly, B.L.; Locksley, R.M. A novel model for IFN-gamma-mediated autoinflammatory syndromes. J. Immunol. 2015, 194, 2358–2368. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, H.J.; Cheon, J.H.; Wu, L.; Van Kaer, L.; Hong, S. iNKT Cells Suppress Pathogenic NK1.1(+)CD8(+) T Cells in DSS-Induced Colitis. Front. Immunol. 2018, 9, 2168. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.W.; Van Kaer, L.; Hong, S. CD1d-Dependent iNKT Cells Control DSS-Induced Colitis in a Mouse Model of IFNgamma-Mediated Hyperinflammation by Increasing IL22-Secreting ILC3 Cells. Int. J. Mol. Sci. 2021, 22, 1250. [Google Scholar] [CrossRef]
- Van Kaer, L.; Postoak, J.L.; Song, W.; Wu, L. Innate and Innate-like Effector Lymphocytes in Health and Disease. J. Immunol. 2022, 209, 199–207. [Google Scholar] [CrossRef]
- Van Kaer, L.; Postoak, J.L.; Wang, C.; Yang, G.; Wu, L. Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE. Cell Mol. Immunol. 2019, 16, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Wu, L. Therapeutic Potential of Invariant Natural Killer T Cells in Autoimmunity. Front. Immunol. 2018, 9, 519. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Joyce, S. Survivre et vivre: When iNKT cells met a Hippo. J. Exp. Med. 2020, 217, e20200343. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Kim, J.H.; Kim, S.H.; Kim, H.R.; Zhang, K.; Pan, Y.; Ko, M.K.; Kim, B.M.; Chu, H.; Lee, H.R.; et al. Skin-resident natural killer T cells participate in cutaneous allergic inflammation in atopic dermatitis. J. Allergy Clin. Immunol. 2021, 147, 1764–1777. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, S.W.; Park, S.H.; Van Kaer, L.; Hong, S. Selective Expansion of Double-Negative iNKT Cells Inhibits the Development of Atopic Dermatitis in Valpha14 TCR Transgenic NC/Nga Mice by Increasing Memory-Type CD8(+) T and Regulatory CD4(+) T Cells. J. Investig. Dermatol. 2021, 141, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, T.C.; Park, Y.H.; Lee, S.W.; Jeon, J.; Park, S.H.; Van Kaer, L.; Hong, S. Repeated alpha-GalCer Administration Induces a Type 2 Cytokine-Biased iNKT Cell Response and Exacerbates Atopic Skin Inflammation in Valpha14(Tg) NC/Nga Mice. Biomedicines 2021, 9, 1619. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.W.; Hong, S. Regulation of Allergic Immune Responses by Microbial Metabolites. Immune Netw. 2018, 18, e15. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.W.; Im, W.; Kim, M.; Van Kaer, L.; Hong, S. iNKT Cell Activation Exacerbates the Development of Huntington’s Disease in R6/2 Transgenic Mice. Mediat. Inflamm. 2019, 2019, 3540974. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, H.J.; Van Kaer, L.; Hong, S.; Hong, S. Graphene oxide polarizes iNKT cells for production of TGFbeta and attenuates inflammation in an iNKT cell-mediated sepsis model. Sci. Rep. 2018, 8, 10081. [Google Scholar] [CrossRef]
- Lynch, L.; Michelet, X.; Zhang, S.; Brennan, P.J.; Moseman, A.; Lester, C.; Besra, G.; Vomhof-Dekrey, E.E.; Tighe, M.; Koay, H.F.; et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat. Immunol. 2015, 16, 85–95. [Google Scholar] [CrossRef]
- Iwamura, C.; Shinoda, K.; Endo, Y.; Watanabe, Y.; Tumes, D.J.; Motohashi, S.; Kawahara, K.; Kinjo, Y.; Nakayama, T. Regulation of memory CD4 T-cell pool size and function by natural killer T cells in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 16992–16997. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Park, H.J.; Pei, Y.; Yeo, Y.; Hong, S. Topical application of zwitterionic chitosan suppresses neutrophil-mediated acute skin inflammation. Int. J. Biol. Macromol. 2020, 158, 1184–1193. [Google Scholar] [CrossRef]
- Ju, A.; Lee, S.W.; Lee, Y.E.; Han, K.C.; Kim, J.C.; Shin, S.C.; Park, H.J.; EunKyeong Kim, E.; Hong, S.; Jang, M. A carrier-free multiplexed gene editing system applicable for suspension cells. Biomaterials 2019, 217, 119298. [Google Scholar] [CrossRef]
- Zhang, H.L.; Zheng, Y.J.; Pan, Y.D.; Xie, C.; Sun, H.; Zhang, Y.H.; Yuan, M.Y.; Song, B.L.; Chen, J.F. Regulatory T-cell depletion in the gut caused by integrin beta7 deficiency exacerbates DSS colitis by evoking aberrant innate immunity. Mucosal Immunol. 2016, 9, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.M.; Rudensky, A.Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 2007, 8, 277–284. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A.; Van Kaer, L.; Fu Dong, S. CD4+CD25+ Tregs and NKT cells: Regulators regulating regulators. Trends Immunol. 2006, 27, 322–327. [Google Scholar] [CrossRef]
- Laffont, S.; Siddiqui, K.R.; Powrie, F. Intestinal inflammation abrogates the tolerogenic properties of MLN CD103+ dendritic cells. Eur. J. Immunol. 2010, 40, 1877–1883. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, H.J.; Kim, N.; Hong, S. Natural Killer Dendritic Cells Enhance Immune Responses Elicited by alpha-Galactosylceramide-Stimulated Natural Killer T Cells. BioMed Res. Int. 2013, 2013, 460706. [Google Scholar] [CrossRef]
- Tresoldi, E.; Dell’Albani, I.; Stabilini, A.; Jofra, T.; Valle, A.; Gagliani, N.; Bondanza, A.; Roncarolo, M.G.; Battaglia, M. Stability of human rapamycin-expanded CD4+CD25+ T regulatory cells. Haematologica 2011, 96, 1357–1365. [Google Scholar] [CrossRef]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef]
- Murai, M.; Krause, P.; Cheroutre, H.; Kronenberg, M. Regulatory T-cell stability and plasticity in mucosal and systemic immune systems. Mucosal Immunol. 2010, 3, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Nish, S.A.; Schenten, D.; Wunderlich, F.T.; Pope, S.D.; Gao, Y.; Hoshi, N.; Yu, S.; Yan, X.; Lee, H.K.; Pasman, L.; et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. eLife 2014, 3, e01949. [Google Scholar] [CrossRef] [PubMed]
- Oldenhove, G.; Bouladoux, N.; Wohlfert, E.A.; Hall, J.A.; Chou, D.; Dos Santos, L.; O’Brien, S.; Blank, R.; Lamb, E.; Natarajan, S.; et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 2009, 31, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Game, D.S.; Davies, D.; Lombardi, G.; Lechler, R.I. Activated CD1d-restricted natural killer T cells secrete IL-2: Innate help for CD4+CD25+ regulatory T cells? Eur. J. Immunol. 2005, 35, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Kim, H.Y.; Albacker, L.A.; Lee, H.H.; Baumgarth, N.; Akira, S.; Savage, P.B.; Endo, S.; Yamamura, T.; Maaskant, J.; et al. Influenza infection in suckling mice expands an NKT cell subset that protects against airway hyperreactivity. J. Clin. Investig. 2011, 121, 57–69. [Google Scholar] [CrossRef]
- Chuang, Y.T.; Leung, K.; Chang, Y.J.; DeKruyff, R.H.; Savage, P.B.; Cruse, R.; Benoit, C.; Elewaut, D.; Baumgarth, N.; Umetsu, D.T. A natural killer T-cell subset that protects against airway hyperreactivity. J. Allergy Clin. Immunol. 2019, 143, 565–576.e7. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, H.J.; Van Kaer, L.; Hong, S. Roles and therapeutic potential of CD1d-Restricted NKT cells in inflammatory skin diseases. Front. Immunol. 2022, 13, 979370. [Google Scholar] [CrossRef]
- Ostanin, D.V.; Bao, J.; Koboziev, I.; Gray, L.; Robinson-Jackson, S.A.; Kosloski-Davidson, M.; Price, V.H.; Grisham, M.B. T cell transfer model of chronic colitis: Concepts, considerations, and tricks of the trade. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G135–G146. [Google Scholar] [CrossRef]
- Chi, X.; Jin, W.; Bai, X.; Zhao, X.; Shao, J.; Li, J.; Sun, Q.; Su, B.; Wang, X.; Yang, X.O.; et al. RORalpha is critical for mTORC1 activity in T cell-mediated colitis. Cell Rep. 2021, 36, 109682. [Google Scholar] [CrossRef]
- Lee, Y.J.; Wang, H.; Starrett, G.J.; Phuong, V.; Jameson, S.C.; Hogquist, K.A. Tissue-Specific Distribution of iNKT Cells Impacts Their Cytokine Response. Immunity 2015, 43, 566–578. [Google Scholar] [CrossRef]
- Wei, Y.; Zeng, B.; Chen, J.; Cui, G.; Lu, C.; Wu, W.; Yang, J.; Wei, H.; Xue, R.; Bai, L.; et al. Enterogenous bacterial glycolipids are required for the generation of natural killer T cells mediated liver injury. Sci. Rep. 2016, 6, 36365. [Google Scholar] [CrossRef] [PubMed]
- Saez de Guinoa, J.; Jimeno, R.; Gaya, M.; Kipling, D.; Garzon, M.J.; Dunn-Walters, D.; Ubeda, C.; Barral, P. CD1d-mediated lipid presentation by CD11c(+) cells regulates intestinal homeostasis. EMBO J. 2018, 37, e97537. [Google Scholar] [CrossRef] [PubMed]
- Jacobse, J.; Li, J.; Rings, E.; Samsom, J.N.; Goettel, J.A. Intestinal Regulatory T Cells as Specialized Tissue-Restricted Immune Cells in Intestinal Immune Homeostasis and Disease. Front. Immunol. 2021, 12, 716499. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, S.W.; Park, Y.H.; Kim, T.C.; Van Kaer, L.; Hong, S. CD1d-independent NK1.1+ Treg cells are IL2-inducible Foxp3+ T cells co-expressing immunosuppressive and cytotoxic molecules. Front. Immunol. 2022, 13, 951592. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.W.; Park, H.J.; Van Kaer, L.; Hong, S. Opposing Roles of DCs and iNKT Cells in the Induction of Foxp3 Expression by MLN CD25+CD4+ T Cells during IFNγ-Driven Colitis. Int. J. Mol. Sci. 2022, 23, 15316. https://doi.org/10.3390/ijms232315316
Lee SW, Park HJ, Van Kaer L, Hong S. Opposing Roles of DCs and iNKT Cells in the Induction of Foxp3 Expression by MLN CD25+CD4+ T Cells during IFNγ-Driven Colitis. International Journal of Molecular Sciences. 2022; 23(23):15316. https://doi.org/10.3390/ijms232315316
Chicago/Turabian StyleLee, Sung Won, Hyun Jung Park, Luc Van Kaer, and Seokmann Hong. 2022. "Opposing Roles of DCs and iNKT Cells in the Induction of Foxp3 Expression by MLN CD25+CD4+ T Cells during IFNγ-Driven Colitis" International Journal of Molecular Sciences 23, no. 23: 15316. https://doi.org/10.3390/ijms232315316
APA StyleLee, S. W., Park, H. J., Van Kaer, L., & Hong, S. (2022). Opposing Roles of DCs and iNKT Cells in the Induction of Foxp3 Expression by MLN CD25+CD4+ T Cells during IFNγ-Driven Colitis. International Journal of Molecular Sciences, 23(23), 15316. https://doi.org/10.3390/ijms232315316