
Mitochondrial Dysfunction and Oxidative Stress in Hereditary Ectopic Calcification Diseases


Abstract
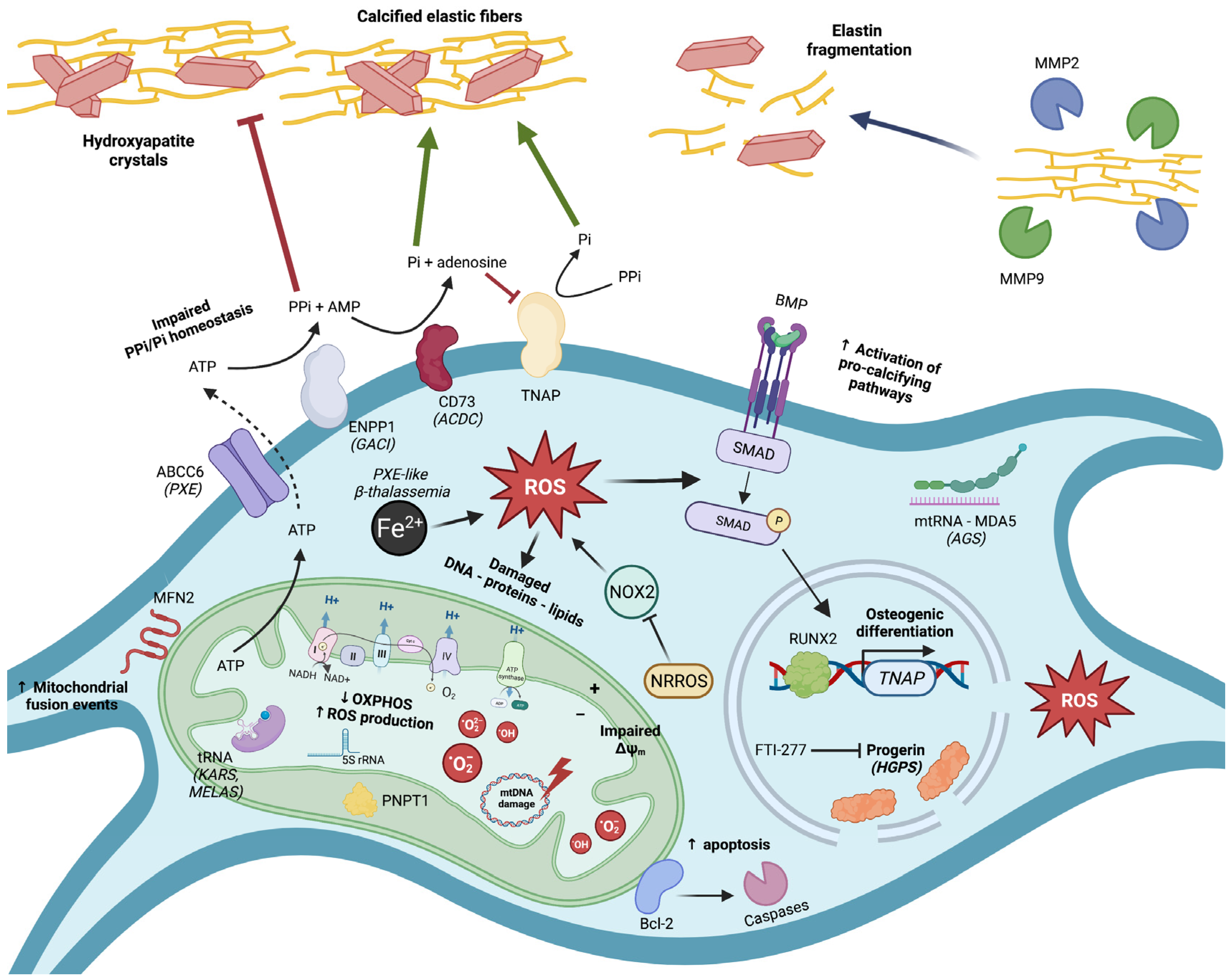
:1. A Brief Introduction into Ectopic Calcification
2. Mitochondrial Dysfunction and Oxidative Stress in Pseudoxanthoma Elasticum (PXE)
Conclusion and Outstanding Questions
3. Mitochondrial Dysfunction and Oxidative Stress in β-Thalassemia-Associated EC
Conclusion and Outstanding Questions
4. Mitochondrial Dysfunction and Oxidative Stress in Hutchinson–Gilford Progeria Syndrome (HGPS)
Conclusion and Outstanding Questions
5. Mitochondrial Dysfunction and Oxidative Stress in Hereditary Central Nervous System (CNS) Calcification Diseases
Conclusion and Outstanding Questions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Vilder, E.Y.; Vanakker, O.M. From variome to phenome: Pathogenesis, diagnosis and management of ectopic mineralization disorders. World J. Clin. Cases 2015, 3, 556–574. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T. Biomineralization—An active or passive process? Connect. Tissue Res. 2012, 53, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Letavernier, E.; Bouderlique, E.; Zaworski, J.; Martin, L.; Daudon, M. Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications. Int. J. Mol. Sci. 2019, 20, 6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendig, D.; Schulz, V.; Arndt, M.; Szliska, C.; Kleesiek, K.; Götting, C. Role of serum fetuin-A, a major inhibitor of systemic calcification, in pseudoxanthoma elasticum. Clin. Chem. 2006, 52, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralph, D.; van de Wetering, K.; Uitto, J.; Li, Q. Inorganic Pyrophosphate Deficiency Syndromes and Potential Treatments for Pathologic Tissue Calcification. Am. J. Pathol. 2022, 192, 762–770. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Ronchetti, I.; Boraldi, F.; Annovi, G.; Cianciulli, P.; Quaglino, D. Fibroblast involvement in soft connective tissue calcification. Front. Genet. 2013, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Hosen, M.J.; Coucke, P.J.; Le Saux, O.; De Paepe, A.; Vanakker, O.M. Perturbation of specific pro-mineralizing signalling pathways in human and murine pseudoxanthoma elasticum. Orphanet J. Rare Dis. 2014, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Bakhshian Nik, A.; Hutcheson, J.D.; Aikawa, E. Extracellular Vesicles As Mediators of Cardiovascular Calcification. Front. Cardiovasc. Med. 2017, 4, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzer, P.; Hannan, F.M.; Lanzer, J.D.; Janzen, J.; Raggi, P.; Furniss, D.; Schuchardt, M.; Thakker, R.; Fok, P.W.; Saez-Rodriguez, J.; et al. Medial Arterial Calcification: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1145–1165. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2018, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Fujii, H.; Kono, K.; Watanabe, K.; Goto, S.; Nishi, S. Influence of oxidative stress on vascular calcification in the setting of coexisting chronic kidney disease and diabetes mellitus. Sci. Rep. 2020, 10, 20708. [Google Scholar] [CrossRef] [PubMed]
- Phadwal, K.; Vrahnas, C.; Ganley, I.G.; MacRae, V.E. Mitochondrial Dysfunction: Cause or Consequence of Vascular Calcification? Front. Cell Dev. Biol. 2021, 9, 611922. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, K.H.; Guo, Y.; Liao, H.; Tang, J.; Xiao, R.P. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ. Res. 2007, 101, 1113–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Peña-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Liberman, M.; Bassi, E.; Martinatti, M.K.; Lario, F.C.; Wosniak, J., Jr.; Pomerantzeff, P.M.; Laurindo, F.R. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Branchetti, E.; Sainger, R.; Poggio, P.; Grau, J.B.; Patterson-Fortin, J.; Bavaria, J.E.; Chorny, M.; Lai, E.; Gorman, R.C.; Levy, R.J.; et al. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e66–e74. [Google Scholar] [CrossRef] [Green Version]
- Rutsch, F.; Nitschke, Y.; Terkeltaub, R. Genetics in arterial calcification: Pieces of a puzzle and cogs in a wheel. Circ. Res. 2011, 109, 578–592. [Google Scholar] [CrossRef]
- Finger, R.P.; Charbel Issa, P.; Ladewig, M.S.; Gotting, C.; Szliska, C.; Scholl, H.P.; Holz, F.G. Pseudoxanthoma elasticum: Genetics, clinical manifestations and therapeutic approaches. Surv. Ophthalmol. 2009, 54, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Verschuere, S.; Van Gils, M.; Nollet, L.; Vanakker, O.M. From membrane to mineralization: The curious case of the ABCC6 transporter. FEBS Lett. 2020, 594, 4109–4133. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Nakano, A.; Jiang, Q.J.; Pulkkinen, L.; Uitto, J. Tissue-specific expression of the ABCC6 gene. J. Investig. Dermatol. 2005, 125, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Endo, M.; Dibra, F.; Wang, K.; Uitto, J. Pseudoxanthoma elasticum is a metabolic disease. J. Investig. Dermatol. 2009, 129, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Boraldi, F.; Annovi, G.; Vermeer, C.; Schurgers, L.J.; Trenti, T.; Tiozzo, R.; Guerra, D.; Quaglino, D. Matrix gla protein and alkaline phosphatase are differently modulated in human dermal fibroblasts from PXE patients and controls. J. Investig. Dermatol. 2013, 133, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, N.; Martin, L.; Calvas, P.; Le Bert, M.; Hovnanian, A. Pseudoxanthoma elasticum: A clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J. Med. Genet. 2005, 42, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Verwer, M.C.; Hazenberg, C.; Spiering, W.; de Borst, G.J. Peripheral Interventions in Patients with Pseudoxanthoma Elasticum (PXE). Eur. J. Vasc. Endovasc. Surg. 2022; in press. [Google Scholar]
- Kauw, F.; Kranenburg, G.; Kappelle, L.J.; Hendrikse, J.; Koek, H.L.; Visseren, F.L.J.; Mali, W.P.T.; de Jong, P.A.; Spiering, W. Cerebral disease in a nationwide Dutch pseudoxanthoma elasticum cohort with a systematic review of the literature. J. Neurol. Sci. 2017, 373, 167–172. [Google Scholar] [CrossRef]
- Bao, L.L.; Yang, J.S.; Xiao, J.; Guo, Z.T. Pseudoxanthoma elasticum. A report of 5 cases in one family. Chin. Med. J. 1991, 104, 237–243. [Google Scholar]
- Martin, L.J.; Lau, E.; Singh, H.; Vergnes, L.; Tarling, E.J.; Mehrabian, M.; Mungrue, I.; Xiao, S.; Shih, D.; Castellani, L.; et al. ABCC6 localizes to the mitochondria-associated membrane. Circ. Res. 2012, 111, 516–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinkó, E.; Iliás, A.; Ujhelly, O.; Homolya, L.; Scheffer, G.L.; Bergen, A.A.; Sarkadi, B.; Váradi, A. Subcellular localization and N-glycosylation of human ABCC6, expressed in MDCKII cells. Biochem. Biophys. Res. Commun. 2003, 308, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Fulop, K.; Yamaguchi, Y.; Ilias, A.; Szabo, Z.; Brampton, C.N.; Pomozi, V.; Huszar, K.; Aranyi, T.; Varadi, A. Expression and in vivo rescue of human ABCC6 disease-causing mutants in mouse liver. PLoS ONE 2011, 6, e24738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomozi, V.; Le Saux, O.; Brampton, C.; Apana, A.; Ilias, A.; Szeri, F.; Martin, L.; Monostory, K.; Paku, S.; Sarkadi, B.; et al. ABCC6 is a basolateral plasma membrane protein. Circ. Res. 2013, 112, e148–e151. [Google Scholar] [CrossRef] [PubMed]
- Ferré, M.; Reynier, P.; Chevrollier, A.; Prunier-Mirebeau, D.; Lefthériotis, G.; Henrion, D.; Bonneau, D.; Procaccio, V.; Martin, L. Is ABCC6 a genuine mitochondrial protein? BMC Res. Notes 2013, 6, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lofaro, F.D.; Boraldi, F.; Garcia-Fernandez, M.; Estrella, L.; Valdivielso, P.; Quaglino, D. Relationship Between Mitochondrial Structure and Bioenergetics in Pseudoxanthoma elasticum Dermal Fibroblasts. Front. Cell Dev. Biol. 2020, 8, 610266. [Google Scholar] [CrossRef] [PubMed]
- Pasquali-Ronchetti, I.; Garcia-Fernandez, M.I.; Boraldi, F.; Quaglino, D.; Gheduzzi, D.; De Vincenzi Paolinelli, C.; Tiozzo, R.; Bergamini, S.; Ceccarelli, D.; Muscatello, U. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: Possible role in the pathogenesis of clinical manifestations. J. Pathol. 2006, 208, 54–61. [Google Scholar] [CrossRef]
- Boraldi, F.; Annovi, G.; Guerra, D.; Paolinelli Devincenzi, C.; Garcia-Fernandez, M.I.; Panico, F.; De Santis, G.; Tiozzo, R.; Ronchetti, I.; Quaglino, D. Fibroblast protein profile analysis highlights the role of oxidative stress and vitamin K recycling in the pathogenesis of pseudoxanthoma elasticum. Proteom. Clin. Appl. 2009, 3, 1084–1098. [Google Scholar] [CrossRef]
- Garcia-Fernandez, M.I.; Gheduzzi, D.; Boraldi, F.; Paolinelli, C.D.; Sanchez, P.; Valdivielso, P.; Morilla, M.J.; Quaglino, D.; Guerra, D.; Casolari, S.; et al. Parameters of oxidative stress are present in the circulation of PXE patients. Biochim. Biophys. Acta 2008, 1782, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Jiang, Q.; Uitto, J. Pseudoxanthoma elasticum: Oxidative stress and antioxidant diet in a mouse model (Abcc6−/−). J. Investig. Dermatol. 2008, 128, 1160–1164. [Google Scholar] [CrossRef] [Green Version]
- Nollet, L.; Van Gils, M.; Willaert, A.; Coucke, P.J.; Vanakker, O.M. Minocycline attenuates excessive DNA damage response and reduces ectopic calcification in pseudoxanthoma elasticum. J. Investig. Dermatol. 2021, 142, 1629–1638.e6. [Google Scholar] [CrossRef]
- Huang, J.; Ralph, D.; Boraldi, F.; Quaglino, D.; Uitto, J.; Li, Q. Inhibition of the DNA Damage Response Attenuates Ectopic Calcification in Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2022, 142, 2140–2148.e1. [Google Scholar] [CrossRef]
- Tiemann, J.; Wagner, T.; Lindenkamp, C.; Plümers, R.; Faust, I.; Knabbe, C.; Hendig, D. Linking ABCC6 Deficiency in Primary Human Dermal Fibroblasts of PXE Patients to p21-Mediated Premature Cellular Senescence and the Development of a Proinflammatory Secretory Phenotype. Int. J. Mol. Sci. 2020, 21, 9665. [Google Scholar] [CrossRef]
- Bouderlique, E.; Nollet, L.; Letavernier, E.; Vanakker, O. Minocycline Counteracts Ectopic Calcification in a Murine Model of Pseudoxanthoma Elasticum: A Proof-of-Concept Study. Int. J. Mol. Sci. 2022, 23, 1838. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, R.; Hendig, D.; Szliska, C.; Kleesiek, K.; Götting, C. Pseudoxanthoma elasticum: Genetic variations in antioxidant genes are risk factors for early disease onset. Clin. Chem. 2007, 53, 1734–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiemann, J.; Wagner, T.; Vanakker, O.M.; van Gils, M.; Cabrera, J.B.; Ibold, B.; Faust, I.; Knabbe, C.; Hendig, D. Cellular and Molecular Biomarkers Indicate Premature Aging in Pseudoxanthoma Elasticum Patients. Aging Dis. 2020, 11, 536–546. [Google Scholar] [CrossRef]
- Baccarani-Contri, M.; Bacchelli, B.; Boraldi, F.; Quaglino, D.; Taparelli, F.; Carnevali, E.; Francomano, M.A.; Seidenari, S.; Bettoli, V.; De Sanctis, V.; et al. Characterization of pseudoxanthoma elasticum-like lesions in the skin of patients with beta-thalassemia. J. Am. Acad. Dermatol. 2001, 44, 33–39. [Google Scholar] [CrossRef]
- Barteselli, G.; Dell’arti, L.; Finger, R.P.; Charbel Issa, P.; Marcon, A.; Vezzola, D.; Mapelli, C.; Cassinerio, E.; Cappellini, M.D.; Ratiglia, R.; et al. The spectrum of ocular alterations in patients with beta-thalassemia syndromes suggests a pathology similar to pseudoxanthoma elasticum. Ophthalmology 2014, 121, 709–718. [Google Scholar] [CrossRef]
- Hamlin, N.; Beck, K.; Bacchelli, B.; Cianciulli, P.; Pasquali-Ronchetti, I.; Le Saux, O. Acquired Pseudoxanthoma elasticum-like syndrome in beta-thalassaemia patients. Br. J. Haematol. 2003, 122, 852–854. [Google Scholar] [CrossRef]
- Boraldi, F.; Garcia-Fernandez, M.; Paolinelli-Devincenzi, C.; Annovi, G.; Schurgers, L.; Vermeer, C.; Cianciulli, P.; Ronchetti, I.; Quaglino, D. Ectopic calcification in beta-thalassemia patients is associated with increased oxidative stress and lower MGP carboxylation. Biochim. Biophys. Acta 2013, 1832, 2077–2084. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Douet, V.; VanWart, C.M.; Heller, M.B.; Le Saux, O. A mouse model of beta-thalassemia shows a liver-specific down-regulation of Abcc6 expression. Am. J. Pathol. 2011, 178, 774–783. [Google Scholar] [CrossRef] [Green Version]
- Douet, V.; VanWart, C.M.; Heller, M.B.; Reinhard, S.; Le Saux, O. HNF4alpha and NF-E2 are key transcriptional regulators of the murine Abcc6 gene expression. Biochim. Biophys. Acta 2006, 1759, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.C. The NF-E2 transcription factor. Int. J. Biochem. Cell Biol. 1998, 30, 429–432. [Google Scholar] [CrossRef]
- Jiang, Q.; Matsuzaki, Y.; Li, K.; Uitto, J. Transcriptional regulation and characterization of the promoter region of the human ABCC6 gene. J. Investig. Dermatol. 2006, 126, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Boraldi, F.; Lofaro, F.D.; Romano, O.; Grilli, A.; Losi, L.; Moscarelli, P.; Bicciato, S.; Quaglino, D. Exome sequencing and bioinformatic approaches reveals rare sequence variants involved in cell signalling and elastic fibre homeostasis: New evidence in the development of ectopic calcification. Cell. Signal. 2019, 59, 131–140. [Google Scholar] [CrossRef]
- Kunji, E.R.S.; King, M.S.; Ruprecht, J.J.; Thangaratnarajah, C. The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology 2020, 35, 302–327. [Google Scholar] [CrossRef]
- Fibach, E.; Dana, M. Oxidative Stress in β-Thalassemia. Mol. Diagn. Ther. 2019, 23, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Kaya, B. Pseudoxanthoma elasticum-like syndrome in a patient with sickle cell anaemia. Br. J. Haematol. 2010, 148, 342. [Google Scholar] [CrossRef] [PubMed]
- Jampol, L.M.; Acheson, R.; Eagle, R.C., Jr.; Serjeant, G.; O’Grady, R. Calcification of Bruch’s membrane in angioid streaks with homozygous sickle cell disease. Arch. Ophthalmol. 1987, 105, 93–98. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.B.; Brown, W.T.; Collins, F.S. Hutchinson-Gilford Progeria Syndrome. In GeneReviews(®); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Hennekam, R.C. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. Part A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R. ATP-based therapy prevents vascular calcification and extends longevity in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 23698–23704. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Rivera-Torres, J.; Osorio, F.G.; Acin-Perez, R.; Enriquez, J.A.; Lopez-Otin, C.; Andres, V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 2013, 127, 2442–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Torres, J.; Acín-Perez, R.; Cabezas-Sánchez, P.; Osorio, F.G.; Gonzalez-Gómez, C.; Megias, D.; Cámara, C.; López-Otín, C.; Enríquez, J.A.; Luque-García, J.L.; et al. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteom. 2013, 91, 466–477. [Google Scholar] [CrossRef]
- Li, Q.; Kingman, J.; Sundberg, J.P.; Levine, M.A.; Uitto, J. Etidronate prevents, but does not reverse, ectopic mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−). Oncotarget 2016, 9, 30721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Sundberg, J.P.; Levine, M.A.; Terry, S.F.; Uitto, J. The effects of bisphosphonates on ectopic soft tissue mineralization caused by mutations in the ABCC6 gene. Cell Cycle 2015, 14, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Li, Q.; Chou, D.W.; Uitto, J. Atorvastatin counteracts aberrant soft tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−). J. Mol. Med. 2013, 91, 1177–1184. [Google Scholar] [CrossRef] [Green Version]
- Viteri, G.; Chung, Y.W.; Stadtman, E.R. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech. Ageing Dev. 2010, 131, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.T.; Park, J.T.; Choi, K.; Choi, H.J.C.; Jung, C.W.; Kim, G.R.; Lee, Y.S.; Park, S.C. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell 2017, 16, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Hall, A.; Galanos, P.; Rizza, S.; Yamamoto, T.; Gram, H.H.; Munk, S.H.N.; Shoaib, M.; Sørensen, C.S.; Bohr, V.A.; et al. Lamin A/C impairments cause mitochondrial dysfunction by attenuating PGC1α and the NAMPT-NAD+ pathway. Nucleic Acids Res. 2022, 50, 9948–9965. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B., Sr.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, F.G.; Caschera, L.; Teixeira, S.R.; Viaene, A.N.; Pinelli, L.; Mankad, K.; Alves, C.; Ortiz-Gonzalez, X.R.; Andronikou, S.; Vossough, A. Intracranial calcifications in childhood: Part 1. Pediatr. Radiol. 2020, 50, 1424–1447. [Google Scholar] [CrossRef]
- Donzuso, G.; Mostile, G.; Nicoletti, A.; Zappia, M. Basal ganglia calcifications (Fahr’s syndrome): Related conditions and clinical features. Neurol. Sci. 2019, 40, 2251–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J. Mitochondriopathies. Eur. J. Neurol. 2004, 11, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Bamborschke, D.; Kreutzer, M.; Koy, A.; Koerber, F.; Lucas, N.; Huenseler, C.; Herkenrath, P.; Lee-Kirsch, M.A.; Cirak, S. PNPT1 mutations may cause Aicardi-Goutières-Syndrome. Brain Dev. 2021, 43, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef] [Green Version]
- Vedrenne, V.; Gowher, A.; De Lonlay, P.; Nitschke, P.; Serre, V.; Boddaert, N.; Altuzarra, C.; Mager-Heckel, A.M.; Chretien, F.; Entelis, N.; et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am. J. Hum. Genet. 2012, 91, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Calvo-Garrido, J.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet. 2019, 15, e1008240. [Google Scholar] [CrossRef] [Green Version]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rötig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Smith, C.; McColl, B.W.; Patir, A.; Barrington, J.; Armishaw, J.; Clarke, A.; Eaton, J.; Hobbs, V.; Mansour, S.; Nolan, M.; et al. Biallelic mutations in NRROS cause an early onset lethal microgliopathy. Acta Neuropathol. 2020, 139, 947–951. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Tan, N.B.; Howell, K.B.; Barresi, S.; Freeman, J.L.; Vecchio, D.; Piccione, M.; Radio, F.C.; Calame, D.; Zong, S.; et al. Bi-allelic LoF NRROS Variants Impairing Active TGF-β1 Delivery Cause a Severe Infantile-Onset Neurodegenerative Condition with Intracranial Calcification. Am. J. Hum. Genet. 2020, 106, 559–569. [Google Scholar] [CrossRef]
- Noubade, R.; Wong, K.; Ota, N.; Rutz, S.; Eidenschenk, C.; Valdez, P.A.; Ding, J.; Peng, I.; Sebrell, A.; Caplazi, P.; et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 2014, 509, 235–239. [Google Scholar] [CrossRef]
- Qin, Y.; Garrison, B.S.; Ma, W.; Wang, R.; Jiang, A.; Li, J.; Mistry, M.; Bronson, R.T.; Santoro, D.; Franco, C.; et al. A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System. Cell 2018, 174, 156–171.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macintosh, J.; Derksen, A.; Poulin, C.; Braverman, N.; Vanderver, A.; Thiffault, I.; Albrecht, S.; Bernard, G. Novel biallelic variants in NRROS associated with a lethal microgliopathy, brain calcifications, and neurodegeneration. Neurogenetics 2022, 23, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ardissone, A.; Tonduti, D.; Legati, A.; Lamantea, E.; Barone, R.; Dorboz, I.; Boespflug-Tanguy, O.; Nebbia, G.; Maggioni, M.; Garavaglia, B.; et al. KARS-related diseases: Progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature. Orphanet J. Rare Dis. 2018, 13, 45. [Google Scholar] [CrossRef]
- Ruzzenente, B.; Assouline, Z.; Barcia, G.; Rio, M.; Boddaert, N.; Munnich, A.; Rötig, A.; Metodiev, M.D. Inhibition of mitochondrial translation in fibroblasts from a patient expressing the KARS p.(Pro228Leu) variant and presenting with sensorineural deafness, developmental delay, and lactic acidosis. Hum. Mutat. 2018, 39, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Melis, D.; Carvalho, D.; Barbaro-Dieber, T.; Espay, A.J.; Gambello, M.J.; Gener, B.; Gerkes, E.; Hitzert, M.M.; Hove, H.B.; Jansen, S.; et al. Primrose syndrome: Characterization of the phenotype in 42 patients. Clin. Genet. 2020, 97, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Cordeddu, V.; Redeker, B.; Stellacci, E.; Jongejan, A.; Fragale, A.; Bradley, T.E.; Anselmi, M.; Ciolfi, A.; Cecchetti, S.; Muto, V.; et al. Mutations in ZBTB20 cause Primrose syndrome. Nat. Genet. 2014, 46, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.J.; Chen, C.; Zhang, S.; Liu, M.; Wei, C.; Wang, K.; Ma, X.; Song, Y.; Wang, R.; Zhang, H.; et al. Zbtb20 deficiency causes cardiac contractile dysfunction in mice. FASEB J. 2020, 34, 13862–13876. [Google Scholar] [CrossRef]
- Sharma, R.; Reinstadler, B.; Engelstad, K.; Skinner, O.S.; Stackowitz, E.; Haller, R.G.; Clish, C.B.; Pierce, K.; Walker, M.A.; Fryer, R.; et al. Circulating markers of NADH-reductive stress correlate with mitochondrial disease severity. J. Clin. Investig. 2021, 131, e136055. [Google Scholar] [CrossRef]
- Cheema, N.J.; Cameron, J.M.; Hood, D.A. Effect of rapamycin on mitochondria and lysosomes in fibroblasts from patients with mtDNA mutations. Am. J. Physiol. Cell Physiol. 2021, 321, C176–C186. [Google Scholar] [CrossRef]
- Mori, D.; Matsui, I.; Shimomura, A.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Oka, T.; Kubota, K.; Yonemoto, S.; et al. Protein carbamylation exacerbates vascular calcification. Kidney Int. 2018, 94, 72–90. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Disease | Causal Gene(s) | Clinical EC Phenotype | Mitochondrial Dysfunction/Oxidative Stress |
|---|---|---|---|
| Pseudoxanthoma elasticum (PXE) | ABCC6 (or ENPP1) | EC in reticular dermis (skin), Bruch’s membrane (eye), tunica media (artery) and papilla (kidney) | Swollen mitochondria, ↓ cristae, ↓ OCR, ↑ fusion events, ↑ ∆ψm, ↑ ROS, ↑ MDA/AOPP, ↓ TAS, ↓ Bcl-2, ↑ protein oxidation |
| β-thalassemia/sickle cell anemia | HBB | PXE-like EC in skin, eyes (angioid streaks), and arteries | ↑ LOOH/AOPP/MDA, ↑ SOD and GPX↓ NF-E2—ABCC6 association, ↑ ROS (due to ↑ Hb degradation + iron overload) |
| Hutchinson–Gilford progeria syndrome (HGPS) | LMNA | Cardiovascular calcification (tunica media) | ↓ ATP production, ↓ ATP synthase (complex V), ↓ cytochrome c, ↑ ROS, ↑ protein oxidation, ↓ OCR, ↑ SOD |
| PNPT1-associated Aicardi–Goutières syndrome (AGS) | PNPT1 | Basal ganglia calcification | ↓ Mitochondrial import of 5S rRNA, ↓ mitochondrial translation, ↓ complex III/IV activity |
| NRROS-associated neurodegeneration | NRROS | Puntacte calcifications in subcortical and periventricular white matter | Globular mitochondria with concentrically arranged cristae, hypothesized ↑ ROS due to ↑ NOX2 |
| KARS-related progressive leukoencephalopathy | KARS | Brain (pons, thalamus, cerebellum, and white matter) and spinal cord calcifications | ↓ Mitochondrial translation, ↓ complex I/II/IV protein levels |
| Primrose syndrome | ZBTB20 | Calcification of the external ear and brain parenchyma | Not established but abnormal acylcarnitine and urine organic acid profiles (↑ excretion of dicarboxylic acids, ethylmalonic and glutaric acids) |
| Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) | Mitochondrial tRNA (MTTL1) or complex I genes | Bilateral basal ganglia calcification | Altered mitochondrial metabolome (acylcarnitine, β-OH fatty acids), ↓ ∆ψm, ↓ basal respiration, ↑ SOD, ↑ ROS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nollet, L.L.; Vanakker, O.M. Mitochondrial Dysfunction and Oxidative Stress in Hereditary Ectopic Calcification Diseases. Int. J. Mol. Sci. 2022, 23, 15288. https://doi.org/10.3390/ijms232315288
Nollet LL, Vanakker OM. Mitochondrial Dysfunction and Oxidative Stress in Hereditary Ectopic Calcification Diseases. International Journal of Molecular Sciences. 2022; 23(23):15288. https://doi.org/10.3390/ijms232315288
Chicago/Turabian StyleNollet, Lukas L., and Olivier M. Vanakker. 2022. "Mitochondrial Dysfunction and Oxidative Stress in Hereditary Ectopic Calcification Diseases" International Journal of Molecular Sciences 23, no. 23: 15288. https://doi.org/10.3390/ijms232315288
APA StyleNollet, L. L., & Vanakker, O. M. (2022). Mitochondrial Dysfunction and Oxidative Stress in Hereditary Ectopic Calcification Diseases. International Journal of Molecular Sciences, 23(23), 15288. https://doi.org/10.3390/ijms232315288