
Activated Hepatic Stellate Cells in Hepatocellular Carcinoma: Their Role as a Potential Target for Future Therapies



Abstract
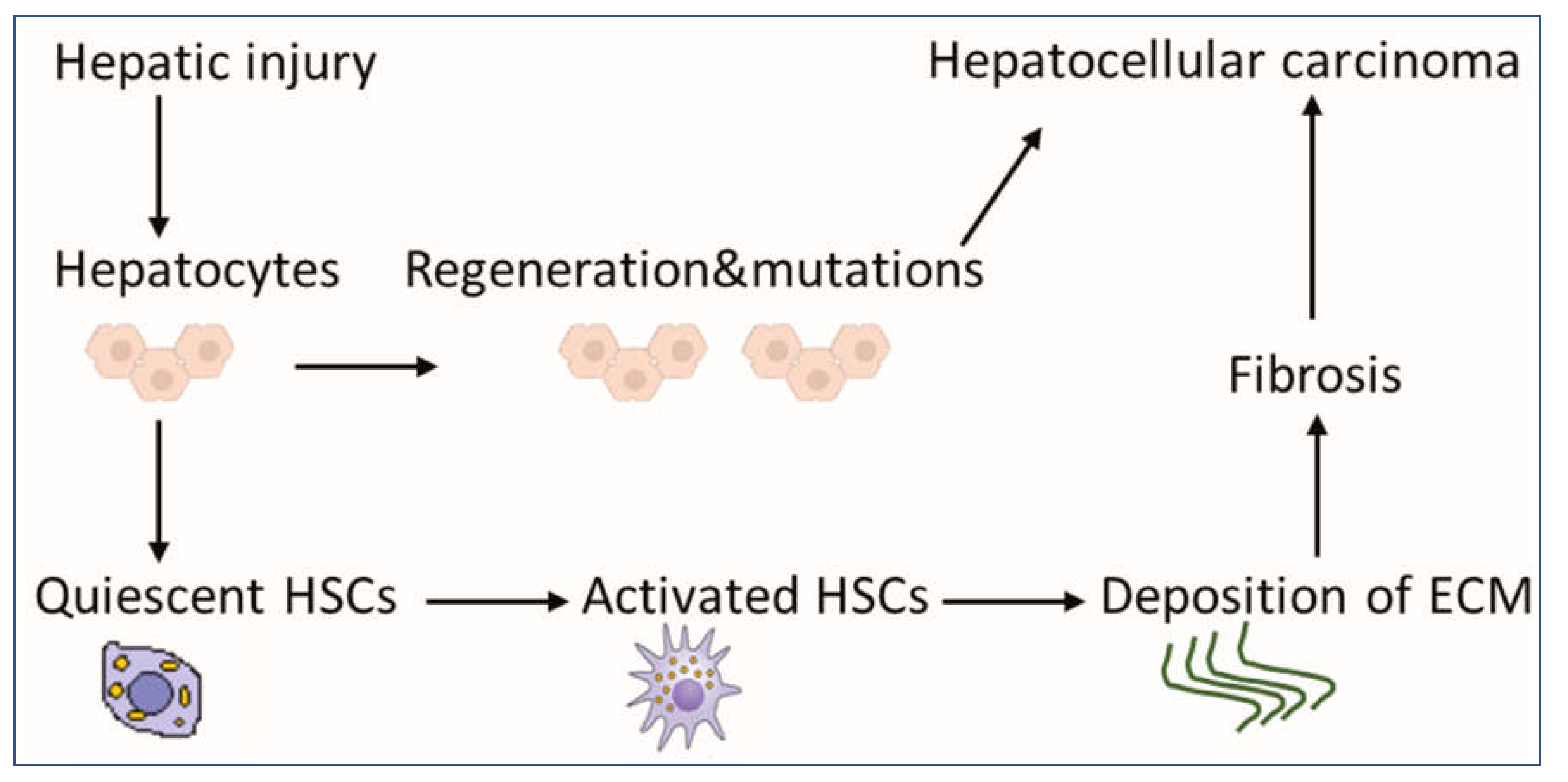
1. Introduction
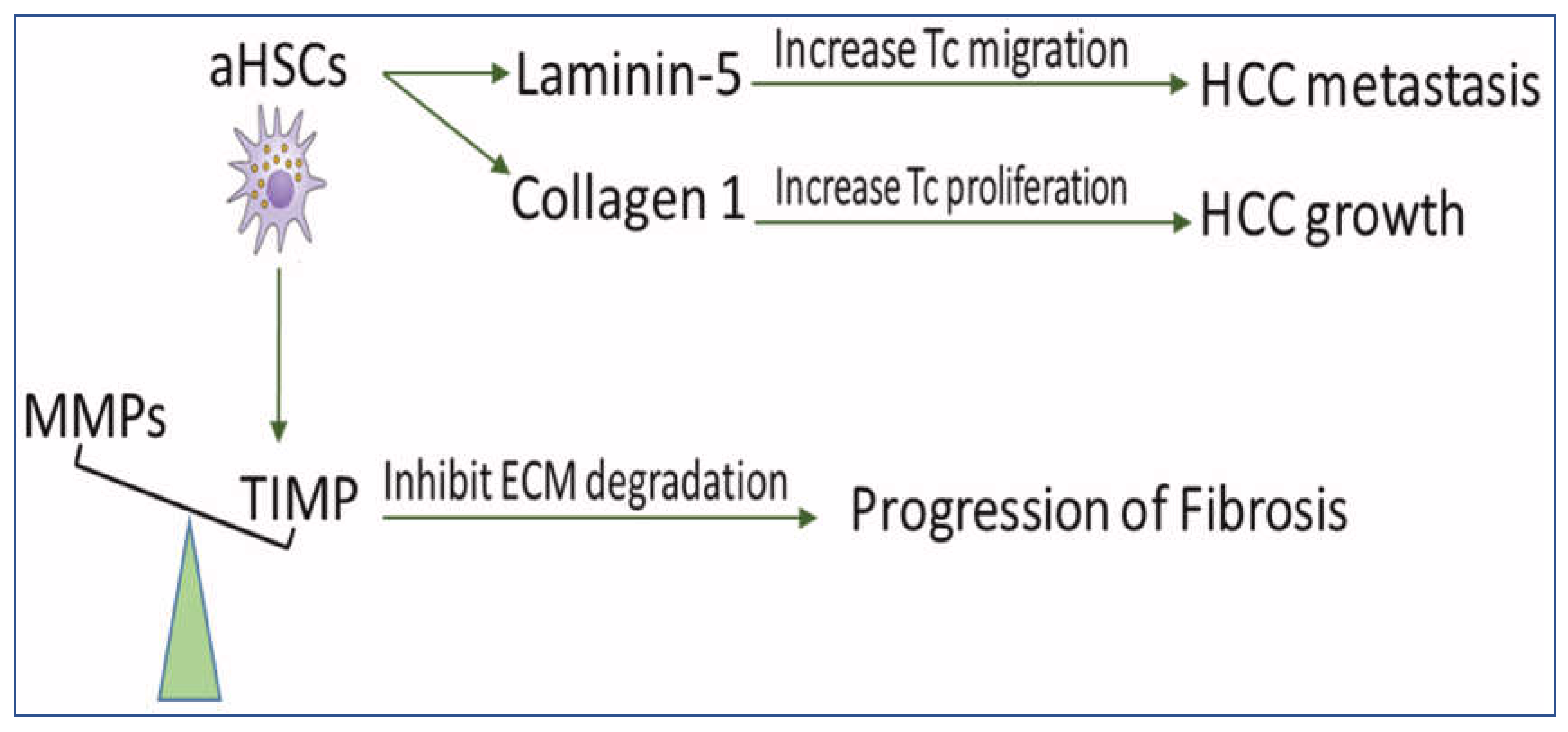
2. The Role of aHSCs in HCC
2.1. The Suppression of the Antitumor Immune Response by aHSCs

{kind=link}
{kind=link}
| Mediator | Immune cell | Response | Possible Effect |
|---|---|---|---|
| PD-L1 | T lymphocytes | TL apoptosis, attenuation of TL infiltration, and suppression of TL-mediated cytotoxicity [20] | HCC growth |
| Dendritic-cell-derived immunoglobulin receptor 2 | Dendritic cells | Inhibition of DC-induced antigen-specific TL responses [41] | |
| COX2–PGE2–EP4 | MDSCs | MDSC accumulation [25] | |
| Transforming growth factor-β | NK cells | Inhibition of NK cell function [38] | |
| Interleukin-6 and tumor necrosis factor-α | Th17 | Th17 expansion and Th17 differentiation [28] | HCC metastasis |
| CCL2/CCR2 | Macrophages | Stimulation of M2 macrophages phenotypic transformation [35] |
2.2. HSCs Upregulate the Deposition of ECM for the Development of Fibrosis and HCC
3. Activation and Deactivation of HSCs as a Result of Therapy
3.1. Activation of HSCs by Conventional Therapy
3.2. Pharmacological Approaches to Deactivate HSCs
4. Suggestions for the Application of HSCs in the Treatment of HCC
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| aHSCs | activated hepatic stellate cells |
| ECM | extracellular matrix |
| TLs | T lymphocytes |
| PD-L1 | programmed death-ligand 1 |
| MDSCs | myeloid-derived suppressor cells |
| Th17 | T helper 17 cells |
| NF-kB | nuclear factor-kB |
| IL6 | interleukin-6 |
| NK | natural killer |
| IL-10 | interleukin-10 |
| TGFβ | transforming growth factor-β |
| DCs | dendritic cells |
| MMPs | metallo-proteinases |
| TIMPs | tissue inhibitors of MMP |
| Ln-5 | laminin-5 |
| TACE | transarterial chemoembolization |
| MAPK | mitogen-activated protein kinase |
| RFA | radiofrequency ablation |
| NK-1R | neurokinin-1 receptor |
| SP | substance P |
| DIgR2 | dendritic-cell-derived immunoglobulin receptor 2 |
References
- Ramani, A.; Tapper, E.B.; Griffin, C.; Shankar, N.; Parikh, N.D.; Asrani, S.K. Hepatocellular Carcinoma-Related Mortality in the USA, 1999–2018. Am. J. Dig. Dis. 2022, 67, 4100–4111. [Google Scholar] [CrossRef] [PubMed]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. 2021, 149, 1–61. Adv. Cancer Res. [CrossRef]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Kanel, G.C.; Korula, J. General Aspects of the Liver and Liver Diseases. Atlas Liver Pathol. 2011, 3–15. [Google Scholar] [CrossRef]
- Barry, A.; Baldeosingh, R.; Lamm, R.; Patel, K.; Zhang, K.; Dominguez, D.A.; Kirton, K.J.; Shah, A.P.; Dang, H. Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 2020, 8, 709. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363. [Google Scholar] [CrossRef]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y. Hepatic stellate cells in liver development, regeneration, and cancer Find the latest version: Review series Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 2013, 123, 1902–1910. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef]
- Mossenta, M.; Busato, D.; Dal Bo, M.; Macor, M.; Toffoli, G. Novel Nanotechnology Approaches to Overcome Drug Resistance in the Treatment of Hepatocellular Carcinoma: Glypican 3 as a Useful Target for Innovative Therapies. Int. J. Mol. Sci. 2022, 23, 10038. [Google Scholar] [CrossRef]
- Sarveazad, A.; Agah, S.; Babahajian, A.; Amini, N.; Bahardoust, M. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J. Res. Med Sci. Off. J. Isfahan Univ. Med. Sci. 2019, 24, 86. [Google Scholar] [CrossRef]
- Hemminki, K.; Försti, A.; Hemminki, O.; Liska, V.; Hemminki, A. Long-term survival trends for primary liver and pancreatic cancers in the Nordic countries. JHEP Reports 2022, 4, 100602. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Wang, H.; Burke, L.J.; Bridle, K.R.; Li, X.; Zhao, C.-X.; Crawford, D.H.G.; Roberts, M.; Liang, X. Therapeutic modulators of hepatic stellate cells for hepatocellular carcinoma. Int. J. Cancer 2020, 147, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Miao, H.; Fu, R.; Zhang, J.; Zheng, W. Hepatic Stellate Cell: A Potential Target for Hepatocellular Carcinoma. Curr. Mol. Pharmacol. 2020, 13, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, L.; Yin, Z.; Su, W.; Ren, G.; Zhou, C.; You, J.; Fan, J.; Wang, X. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int. J. Cancer 2011, 129, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Zheng, X.; Jin, W.; Wang, S.; Ding, H. Progression on the Roles and Mechanisms of Tumor-Infiltrating T Lymphocytes in Patients With Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 1480. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, Y.; Xu, W.; Zheng, X.; Yi, X.; Huang, L.; Wang, Y.; Wu, K. Activated Hepatic Stellate Cells (HSCs) Exert Immunosuppressive Effects in Hepatocellular Carcinoma by Producing Complement C3. OncoTargets Ther. 2020, ume 13, 1497–1505. [Google Scholar] [CrossRef]
- Charles, R.; Chou, H.-S.; Wang, L.; Fung, J.; Lu, L.; Qian, S. Human Hepatic Stellate Cells Inhibit T-Cell Response Through B7-H1 Pathway. Transplantation 2013, 96, 17–24. [Google Scholar] [CrossRef]
- Schildberg, F.A.; Wojtalla, A.; Siegmund, S.V.; Endl, E.; Diehl, L.; Abdullah, Z.; Kurts, C.; Knolle, P.A. Murine hepatic stellate cells veto CD8 T cell activation by a CD54-dependent mechanism. Hepatology 2011, 54, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Su, W.; Kuang, P.; Zhang, L.; Liu, J.; Yin, Z.; Wang, X. The role of hepatic stellate cells in the regulation of T-cell function and the promotion of hepatocellular carcinoma. Int. J. Oncol. 2012, 41, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Su, Y.; Hua, X.; Xie, C.; Liu, J.; Huang, Y.; Zhou, L.; Zhang, M.; Li, X.; Gao, Z. Levels of hepatic Th17 cells and regulatory T cells upregulated by hepatic stellate cells in advanced HBV-related liver fibrosis. J. Transl. Med. 2017, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.-N.; Yuan, Y.-X.; Zhu, B.; Jia, Q. Myeloid-Derived Suppressor Cells: A Multifaceted Accomplice in Tumor Progression. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Xu, Y. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget 2016, 7, 8866–8878. [Google Scholar] [CrossRef]
- Lee, H.L.; Jang, J.W.; Lee, S.W.; Yoo, S.H.; Kwon, J.H.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Han, N.I.; Yoon, S.K. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci. Rep. 2019, 9, 3260. [Google Scholar] [CrossRef]
- Li, J.; Lau, G.K.-K.; Chen, P.L.; Dong, S.-S.; Lan, H.Y.; Huang, X.-R.; Li, Y.; Luk, J.; Yuan, Y.; Guan, X.-Y. Interleukin 17A Promotes Hepatocellular Carcinoma Metastasis via NF-kB Induced Matrix Metalloproteinases 2 and 9 Expression. PLoS ONE 2011, 6, e21816. [Google Scholar] [CrossRef]
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.-X.; He, H.-W.; Cai, X.-Y.; Zhou, J.; Cheng, Y.-F.; Fan, J.; et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3–11. [Google Scholar] [CrossRef]
- Ichikawa, S.; Mucida, D.; Tyznik, A.J.; Kronenberg, M.; Cheroutre, H. Hepatic Stellate Cells Function as Regulatory Bystanders. J. Immunol. 2011, 186, 5549–5555. [Google Scholar] [CrossRef]
- Ricketts, T.D.; Prieto-Dominguez, N.; Gowda, P.S.; Ubil, E. Mechanisms of Macrophage Plasticity in the Tumor Environment: Manipulating Activation State to Improve Outcomes. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yin, L.; Ouyang, X.; Zeng, K.; Xiao, Y.; Li, Y. M2 Macrophages Promote HCC Cells Invasion and Migration via miR-149-5p/MMP9 Signaling. J. Cancer 2020, 11, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Yeung, O.W.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.-X.; Ng, K.T.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef]
- Xi, S.; Zheng, X.; Li, X.; Jiang, Y.; Wu, Y.; Gong, J.; Jie, Y.; Li, Z.; Cao, J.; Sha, L.; et al. Activated Hepatic Stellate Cells Induce Infiltration and Formation of CD163+ Macrophages via CCL2/CCR2 Pathway. Front. Med. 2021, 8, 627927. [Google Scholar] [CrossRef]
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 5381. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Li, Z. Natural killer cells in hepatocellular carcinoma: Current status and perspectives for future immunotherapeutic approaches. Front. Med. 2017, 11, 509–521. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Tacke, F. Role of lymphocytes in liver cancer. OncoImmunology 2013, 2, e26468. [Google Scholar] [CrossRef]
- Glässner, A.; Eisenhardt, M.; Krämer, B.; Körner, C.; Coenen, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Investig. 2012, 92, 967–977. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural Killer Cells Ameliorate Liver Fibrosis by Killing Activated Stellate Cells in NKG2D-Dependent and Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Dependent Manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Xia, T.-H. Tumor-specific hepatic stellate cells (tHSCs) induces DIgR2 expression in dendritic cells to inhibit T cells. Oncotarget 2017, 8, 55084–55093. Available online: www.impactjournals.com/oncotarget/ (accessed on 15 August 2017). [CrossRef]
- HCC Monitor. New evidence supports a key role of the immune system in HCC, HCC monitor. Target. Oncol. 2016, 2, 3. Available online: https://www.targetedonc.com (accessed on 15 August 2017).
- Zois, C.D.; Baltayiannis, G.H.; Karayiannis, P.; Tsianos, E.V. Systematic review: Hepatic fibrosis-regression with therapy. Aliment. Pharmacol. Ther. 2008, 28, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Arriazu, E.; de Galarreta, M.R.; Cubero, F.J.; Varela-Rey, M.; de Obanos, M.P.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular Matrix and Liver Disease. Antioxidants Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.K.; Yim, H.J. Reversal of liver cirrhosis: Current evidence and expectations. Korean J. Intern. Med. 2017, 32, 213–228. [Google Scholar] [CrossRef]
- Sun, M.; Kisseleva, Y. Reversibility of liver fibrosis. In Clinics and Research in Hepatology and Gastroenterology; Elsevier Masson SAS: Amsterdam, The Netherlands, 2015; pp. S60–S63. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Hepatic stellate cells and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2006, 21, S84–S87. [Google Scholar] [CrossRef]
- Arthur, M.J.; Mann, D.A.; Iredale, J.P. Tissue inhibitors of metalloproteinases, hepatic stellate cells and liver fibrosis. J. Gastroenterol. Hepatol. 1998, 13, S33–S38. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Mu, T.; Tong, N.; Cheng, P. Hepatic stellate cells specific liposomes with the Toll-like receptor 4 shRNA attenuates liver fibrosis. J. Cell. Mol. Med. 2021, 25, 1299–1313. [Google Scholar] [CrossRef]
- Arab, J.P.; Cabrera, D.; Sehrawat, T.S.; Jalan-Sakrikar, N.; Verma, V.K.; Simonetto, D.; Cao, S.; Yaqoob, U.; Leon, J.; Freire, M.; et al. Hepatic stellate cell activation promotes alcohol-induced steatohepatitis through Igfbp3 and SerpinA12. J. Hepatol. 2020, 73, 149–160. [Google Scholar] [CrossRef]
- Hartland, S.N.; Murphy, F.; Aucott, R.L.; Abergel, A.; Zhou, X.; Waung, J.; Patel, N.; Bradshaw, C.; Collins, J.; Mann, D.; et al. Active matrix metalloproteinase-2 promotes apoptosis of hepatic stellate cells via the cleavage of cellular N-cadherin. Liver Int. 2009, 29, 966–978. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, W.; Xiang, J.; Liu, P.; Ke, M.; Wang, B.; Lv, Y. Collagen I promotes hepatocellular carcinoma cell proliferation by regulating integrin β1/FAK signaling pathway in nonalcoholic fatty liver. Oncotarget 2017, 8, 95586–95595. Available online: www.impactjournals.com/oncotarget (accessed on 10 November 2017).
- Zhang, R.; Ma, M.; Lin, X.-H.; Liu, H.-H.; Chen, J.; Chen, J.; Gao, D.-M.; Cui, J.-F.; Ren, Z.-G.; Chen, R.-X. Extracellular matrix collagen I promotes the tumor progression of residual hepatocellular carcinoma after heat treatment. BMC Cancer 2018, 18, 901. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-P.; Chang, H.-L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.-Y.; Wu, A.T.H.; Yeh, C.-T.; Tsai, S.-H.; Lee, W.-H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786. [Google Scholar] [CrossRef]
- Yu, F.; Lin, Z.; Zheng, J.; Gao, S.; Lu, Z.; Dong, P. Suppression of collagen synthesis by Dicer gene silencing in hepatic stellate cells. Mol. Med. Rep. 2014, 9, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Azzariti, A.; Fransvea, E.; Porcelli, L.; Antonaci, S.; Paradiso, A. Laminin-5 offsets the efficacy of gefitinib (‘Iressa’) in hepatocellular carcinoma cells. Br. J. Cancer 2004, 91, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, A.E.; Caligiuri, A.; Lupo, L.; Giannelli, G. Hepatic Stellate Cells Induce Hepatocellular Carcinoma Cell Resistance to Sorafenib Through the Laminin-332/a3 Integrin Axis Recovery of Focal Adhesion Kinase Ubiquitination. Hepatology 2016, 64, 2103–2117. [Google Scholar] [CrossRef]
- Santamato, A.; Fransvea, E.; Dituri, F.; Caligiuri, A.; Quaranta, M.; Niimi, T.; Pinzani, M.; Antonaci, S.; Giannelli, G. Hepatic stellate cells stimulate HCC cell migration via laminin-5 production. Clin. Sci. 2011, 121, 159–168. [Google Scholar] [CrossRef]
- Wu, X.Z.; Chen, D.; Xie, G.R. Extracellular matrix remodeling in hepatocellular carcinoma: Effects of soil on seed? Med. Hypotheses 2006, 66, 1115–1120. [Google Scholar] [CrossRef]
- Qu, K.; Yan, Z.; Wu, Y.; Chen, Y.; Qu, P.; Xu, X.; Yuan, P.; Huang, X.; Xing, J.; Zhang, H.; et al. Transarterial chemoembolization aggravated peritumoral fibrosis via hypoxia-inducible factor-1α dependent pathway in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2015, 30, 925–932. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, B.; Liang, B.; Zhao, H.; Li, H.; Qian, J.; Liang, H.-M.; Feng, G.-S.; Zheng, C.-S. Hepatic Parenchymal Changes following Transcatheter Embolization and Chemoembolization in a Rabbit Tumor Model. PLoS ONE 2013, 8, e70757. [Google Scholar] [CrossRef]
- Das, D.; Fayazzadeh, E.; Li, X.; Koirala, N.; Wadera, A.; Lang, M.; Zernic, M.; Panick, C.; Nesbitt, P.; McLennan, G. Quiescent hepatic stellate cells induce toxicity and sensitivity to doxorubicin in cancer cells through a caspase-independent cell death pathway: Central role of apoptosis-inducing factor. J. Cell. Physiol. 2020, 235, 6167–6182. [Google Scholar] [CrossRef]
- Sung, Y.-C.; Liu, Y.-C.; Chao, P.-H.; Chang, C.-C.; Jin, P.-R.; Lin, T.-T.; Lin, J.-A.; Cheng, H.-T.; Wang, J.; Lai, C.P.; et al. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics 2018, 8, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, J.; Shi, H.; Wang, Z.; Zhang, G.; Cao, Y.; Jiang, C.; Ding, Y. Hepatic Stellate Cell Coculture Enables Sorafenib Resistance in Huh7 Cells through HGF/c-Met/Akt and Jak2/Stat3 Pathways. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.E.; Khan, A.; Harrington, K. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Sempoux, C.; Horsmans, Y.; Geubel, A.; Fraikin, J.; Van Beers, B.E.; Gigot, J.; Rahier, J. Severe radiation-induced liver disease following localized radiation therapy for biliopancreatic carcinoma: Activation of hepatic stellate cells as an early event. Hepatology 1997, 26, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhao, J.; Wang, Q.; Chen, P.; Hong, Y.; He, X.; Chen, D.; Liu, H.; Wang, Y.; Cai, X. The Invasive Potential of Hepatoma Cells Induced by Radiotherapy is Related to the Activation of Stellate Cells and Could be Inhibited by EGCG Through the TLR4 Signaling Pathway. Radiat. Res. 2022, 197, 365–375. [Google Scholar] [CrossRef]
- Kang, T.W.; Lim, H.K.; Cha, D.I. Aggressive tumor recurrence after radiofrequency ablation for hepatocellular carcinoma. Clin. Mol. Hepatol. 2017, 23, 95–101. [Google Scholar] [CrossRef]
- Rozenblum, N.; Zeira, E.; Bulvik, B.; Gourevitch, S.; Yotvat, H.; Galun, E.; Goldberg, S.N. Radiofrequency Ablation: Inflammatory Changes in the Periablative Zone Can Induce Global Organ Effects, including Liver Regeneration. Radiology 2015, 276, 416–425. [Google Scholar] [CrossRef]
- Cheng, R.; Xu, H.; Hong, Y. miR221 Regulates TGF-β1-induced HSC activation through Inhibiting Autophagy by directly targeting LAMP2. Mol Med Rep. 2021, 24, 5. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- El-Mezayen, N.S.; El-Hadidy, W.F.; El-Refaie, W.M.; Shalaby, T.; Khattab, M.M.; El-Khatib, A.S. Hepatic stellate cell-targeted imatinib nanomedicine versus conventional imatinib: A novel strategy with potent efficacy in experimental liver fibrosis. J. Control. Release 2017, 266, 226–237. [Google Scholar] [CrossRef]
- Li, Z.; Wang, F.; Li, Y.; Wang, X.; Lu, Q.; Wang, D.; Qi, C.; Li, C.; Li, Z.; Lian, B.; et al. Combined anti-hepatocellular carcinoma therapy inhibit drug-resistance and metastasis via targeting “substance P-hepatic stellate cells-hepatocellular carcinoma” axis. Biomaterials 2021, 276, 121003. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Wang, X.; Navarro-Corcuera, A.; Ilyas, S.; Jalan-Sakrikar, N.; Gan, C.; Tu, X.; Shi, Y.; Tu, K.; et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-β receptor I versus II. Cell Rep. 2022, 38, 110349. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Kang, Y.-K.; Hou, M.-M.; Numata, K.; Yeo, W.; Chopra, A.; Ikeda, M.; et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenib-experienced Asian cohort analysis. J. Hepatol. 2019, 71, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Zisser, A.; Ipsen, D.; Tveden-Nyborg, P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets? Biomedicines 2021, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Filliol, A.; Saito, Y.; Nair, A.; Dapito, D.H.; Yu, L.-X.; Ravichandra, A.; Bhattacharjee, S.; Affo, S.; Fujiwara, N.; Su, H.; et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature 2022, 610, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Chen, Z.; Jain, A.; Liu, H.; Zhao, Z.; Cheng, K. Targeted Drug Delivery to Hepatic Stellate Cells for the Treatment of Liver Fibrosis. J. Pharmacol. Exp. Ther. 2019, 370, 695–702. [Google Scholar] [CrossRef]
- Zabielska-Koczywąs, K.; Lechowski, R. The Use of Liposomes and Nanoparticles as Drug Delivery Systems to Improve Cancer Treatment in Dogs and Cats. Molecules 2017, 22, 2167. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, P.; Zhao, T.; Jia, M.; Yin, P.; Li, W.; Zhang, Z.-R.; Fu, Y.; Gong, T. Golgi Apparatus-Targeted Chondroitin-Modified Nanomicelles Suppress Hepatic Stellate Cell Activation for the Management of Liver Fibrosis. ACS Nano 2019, 13, 3910–3923. [Google Scholar] [CrossRef]


| Approach | Effect on HSCs | Possible Result | Ref. |
|---|---|---|---|
| TACE | Activate HSCs | Induce prominent hepatic fibrogenesis | [61] |
| Sorafenib | Activate HSCs | Resistance to sorafenib | [64] |
| Sorafenib and MAPK inhibitor | Prevent HSC activation | Anti-fibrotic effect | [63] |
| Radiotherapy | Activate HSCs | Increase HCC metastasis | [67] |
| Radiofrequency ablation | Activate HSCs | Tumor recurrence | [69] |
| Galunisertib and sorafenib | Deactivate HSCs | Prolonged overall survival in HCC patients | [71] |
| Imatinib–nanomedicine | Deactivate HSCs | Outstanding anti-fibrotic effects | [72] |
| Doxorubicin and capsaicin | Deactivate HSCs | Inhibit drug resistance and HCC metastasis | [73] |
| Nivolumab | Deactivate HSCs | Treat advanced HCC | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, E.; Trailin, A.; Ambrozkiewicz, F.; Liška, V.; Hemminki, K. Activated Hepatic Stellate Cells in Hepatocellular Carcinoma: Their Role as a Potential Target for Future Therapies. Int. J. Mol. Sci. 2022, 23, 15292. https://doi.org/10.3390/ijms232315292
Ali E, Trailin A, Ambrozkiewicz F, Liška V, Hemminki K. Activated Hepatic Stellate Cells in Hepatocellular Carcinoma: Their Role as a Potential Target for Future Therapies. International Journal of Molecular Sciences. 2022; 23(23):15292. https://doi.org/10.3390/ijms232315292
Chicago/Turabian StyleAli, Esraa, Andriy Trailin, Filip Ambrozkiewicz, Václav Liška, and Kari Hemminki. 2022. "Activated Hepatic Stellate Cells in Hepatocellular Carcinoma: Their Role as a Potential Target for Future Therapies" International Journal of Molecular Sciences 23, no. 23: 15292. https://doi.org/10.3390/ijms232315292
APA StyleAli, E., Trailin, A., Ambrozkiewicz, F., Liška, V., & Hemminki, K. (2022). Activated Hepatic Stellate Cells in Hepatocellular Carcinoma: Their Role as a Potential Target for Future Therapies. International Journal of Molecular Sciences, 23(23), 15292. https://doi.org/10.3390/ijms232315292