
Bromodomain Protein BRD4-Mediated Mutant p53 Transcription Promotes TNBC Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
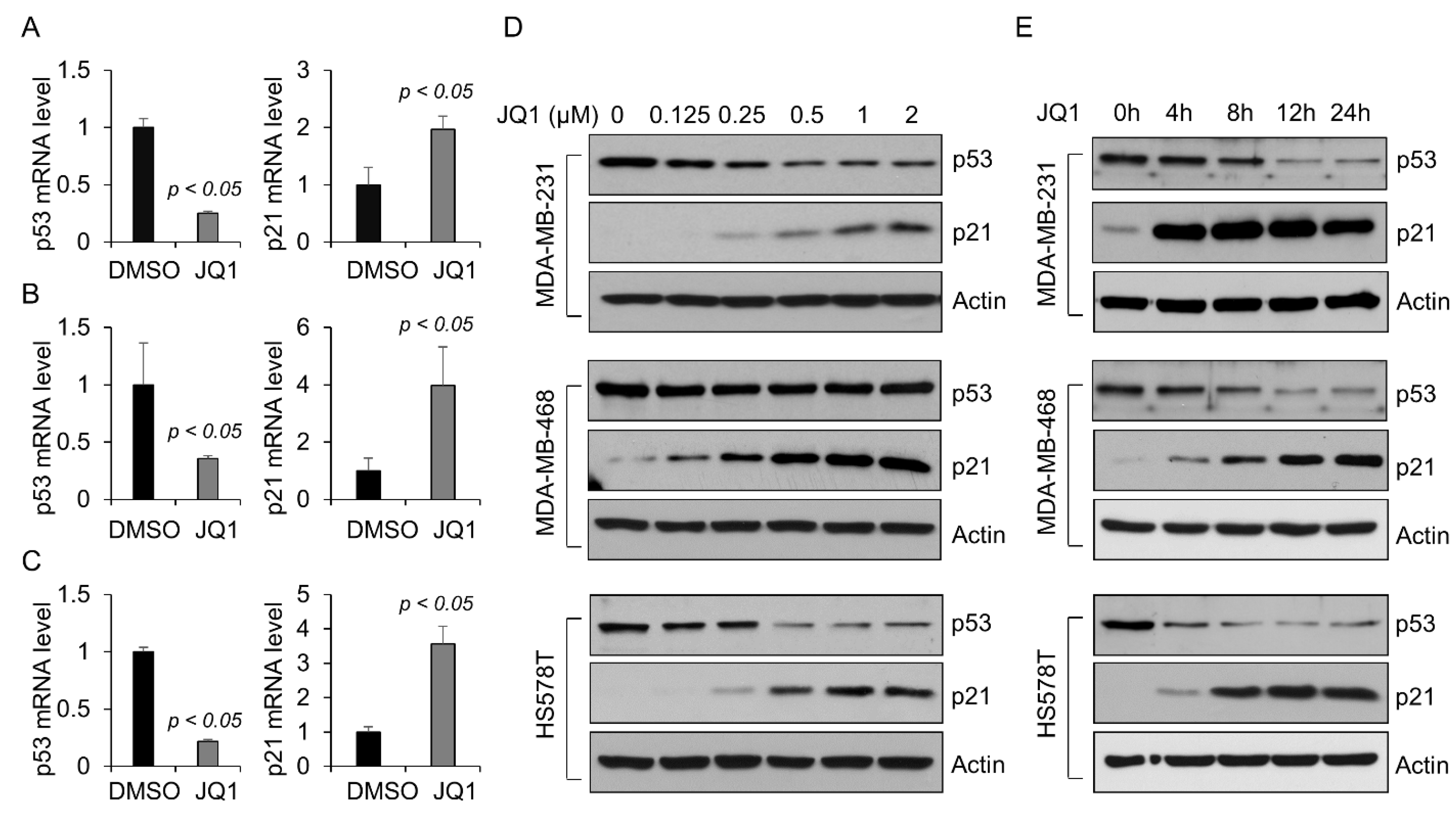
2.1. BET Bromodomain Inhibitor JQ1 Decreases the Level of Mutant p53 Protein in TNBC Cells
2.2. BRD4 Regulates the Transcription of Mutant p53 in Triple-Negative Breast Cancer Cells
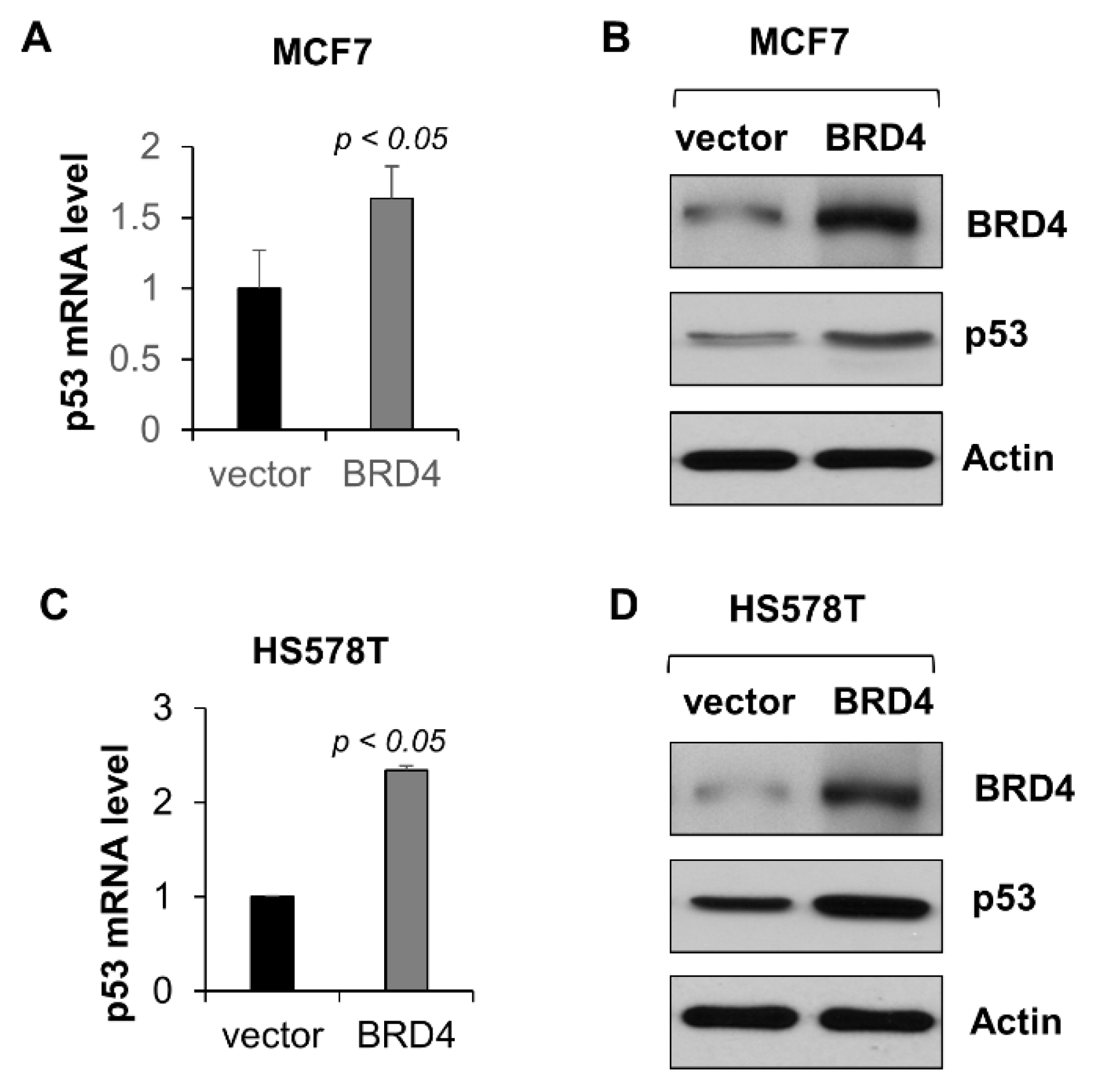
2.3. BRD4 Regulates the Transcription of Wild-Type p53 in MCF-7 Cells
2.4. Inhibition of BRD4 with JQ1 Results in Proliferative Defects in TNBC Cells Carrying Mutant p53
2.5. Knockdown of BRD4 Inhibits Tumor Growth of TNBC with Mutant p53
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. The Cancer Genome Atlas (TCGA) Data Mining
4.3. BRD4 Knockdown by Lentivirus Carrying BRD4 shRNA
4.4. Overexpression of BRD4
4.5. Western Blot Analysis
4.6. Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
4.7. Chromatin Immunoprecipitation (CHIP)-qPCR Assay
4.8. MTS Assay
4.9. Cell Cycle Analysis
4.10. Colony Formation Assay
4.11. Mice Xenograft Studies
4.12. Immunohistochemistry Staining
4.13. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; El-Deiry, W.S. p73 or p53 directly regulates human p53 transcription to maintain cell cycle checkpoints. Cancer Res. 2006, 66, 6982–6989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, V.; Martensen, S.A.; Reisman, D.; Evron, E.; Odenwald, W.F.; Jaffee, E.; Marks, J.; Sukumar, S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000, 405, 974–978. [Google Scholar] [CrossRef]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.L.; Bae, I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Seward, S.; Semaan, A.; Qazi, A.M.; Gruzdyn, O.V.; Chamala, S.; Bryant, C.C.; Kumar, S.; Cameron, D.; Sethi, S.; Ali-Fehmi, R.; et al. EZH2 blockade by RNA interference inhibits growth of ovarian cancer by facilitating re-expression of p21(waf1/cip1) and by inhibiting mutant p53. Cancer Lett. 2013, 336, 53–60. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J., 3rd; Singer, D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockwood, W.W.; Zejnullahu, K.; Bradner, J.E.; Varmus, H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl Acad. Sci. USA 2012, 109, 19408–19413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Ottinger, M.; Pliquet, D.; Christalla, T.; Frank, R.; Stewart, J.P.; Schulz, T.F. The interaction of the gammaherpesvirus 68 orf73 protein with cellular BET proteins affects the activation of cell cycle promoters. J. Virol. 2009, 83, 4423–4434. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Fan, L.X.; Peters, D.J.; Trudel, M.; Bradner, J.E.; Li, X. Therapeutic targeting of BET bromodomain protein, Brd4, delays cyst growth in ADPKD. Hum. Mol. Genet. 2015, 24, 3982–3993. [Google Scholar] [CrossRef] [Green Version]
- Wasielewski, M.; Elstrodt, F.; Klijn, J.G.; Berns, E.M.; Schutte, M. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res. Treat. 2006, 99, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Xu, Y.; Vakoc, C.R. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026674. [Google Scholar] [CrossRef]
- Qi, J. Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: Chemical modulation of chromatin structure. Cold Spring Harb. Perspect. Biol. 2014, 6, a018663. [Google Scholar] [CrossRef] [PubMed]
- Vikhanskaya, F.; Lee, M.K.; Mazzoletti, M.; Broggini, M.; Sabapathy, K. Cancer-derived p53 mutants suppress p53-target gene expression--potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007, 35, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [Green Version]
- Stewart, H.J.; Horne, G.A.; Bastow, S.; Chevassut, T.J. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer Med. 2013, 2, 826–835. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [Green Version]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Kalampokas, E.; Kalampokas, T.; Spartalis, E.; Daskalopoulou, A.; Valsami, S.; Kontos, M.; Nonni, A.; et al. Histone Deacetylases as New Therapeutic Targets in Triple-negative Breast Cancer: Progress and Promises. Cancer Genom. Proteom. 2017, 14, 299–313. [Google Scholar]
- Bhadury, J.; Nilsson, L.M.; Muralidharan, S.V.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmaki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Borbely, G.; Haldosen, L.A.; Dahlman-Wright, K.; Zhao, C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 2015, 6, 33623–33635. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [Green Version]
- da Motta, L.L.; Ledaki, I.; Purshouse, K.; Haider, S.; De Bastiani, M.A.; Baban, D.; Morotti, M.; Steers, G.; Wigfield, S.; Bridges, E.; et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2017, 36, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [Green Version]
- Andrikopoulou, A.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.A.; Zagouri, F. The emerging role of BET inhibitors in breast cancer. Breast 2020, 53, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Luo, Y.; Starremans, P.G.; McNamara, C.A.; Pei, Y.; Zhou, J. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat. Cell Biol. 2005, 7, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Wilson, M.D.; Spyrou, C.; Brown, G.D.; Hadfield, J.; Odom, D.T. ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods 2009, 48, 240–248. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.X.; Agborbesong, E.; Li, L.X.; Li, X. Bromodomain Protein BRD4-Mediated Mutant p53 Transcription Promotes TNBC Progression. Int. J. Mol. Sci. 2022, 23, 15163. https://doi.org/10.3390/ijms232315163
Zhou JX, Agborbesong E, Li LX, Li X. Bromodomain Protein BRD4-Mediated Mutant p53 Transcription Promotes TNBC Progression. International Journal of Molecular Sciences. 2022; 23(23):15163. https://doi.org/10.3390/ijms232315163
Chicago/Turabian StyleZhou, Julie Xia, Ewud Agborbesong, Linda Xiaoyan Li, and Xiaogang Li. 2022. "Bromodomain Protein BRD4-Mediated Mutant p53 Transcription Promotes TNBC Progression" International Journal of Molecular Sciences 23, no. 23: 15163. https://doi.org/10.3390/ijms232315163
APA StyleZhou, J. X., Agborbesong, E., Li, L. X., & Li, X. (2022). Bromodomain Protein BRD4-Mediated Mutant p53 Transcription Promotes TNBC Progression. International Journal of Molecular Sciences, 23(23), 15163. https://doi.org/10.3390/ijms232315163