

Pathophysiology and Current Drug Treatments for Post-Stroke Depression: A Review
 and
and
Abstract
1. Introduction
2. Association between PSD and Post-Stroke Anxiety
2.1. Rehabilitation Services for PSD and PSA
2.2. Diagnostics for PSD and PSA
2.3. Risk Factors
3. Association between PSD and Post-Stroke Motor Activity
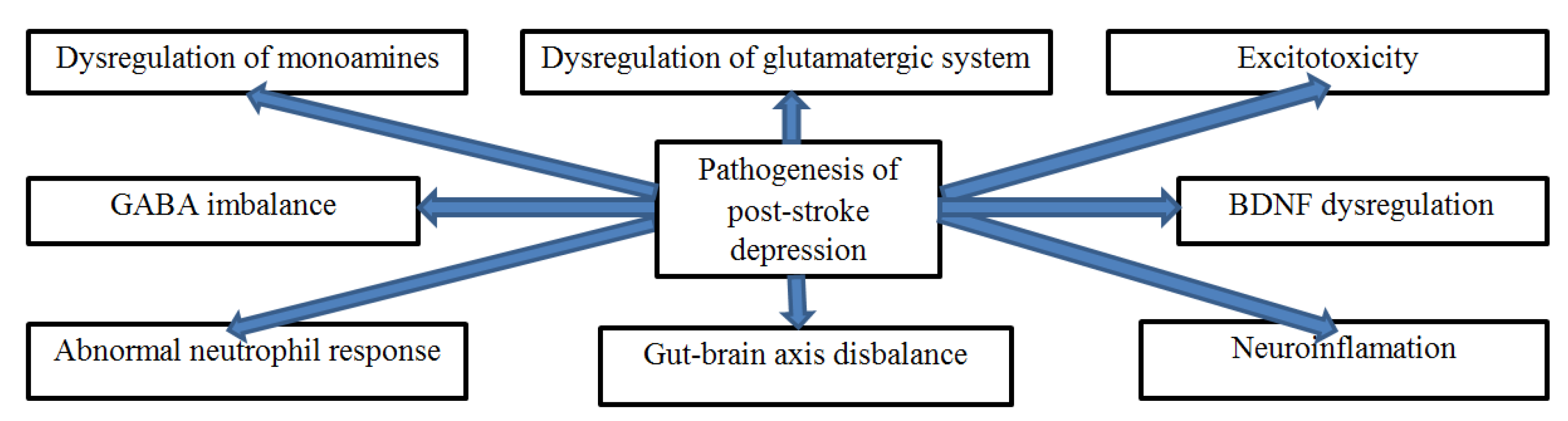
4. Pathogenesis of PSD
4.1. The Role of the Monoamine System
4.2. The Role of the Glutamatergic System
4.3. The Role of Excitotoxicity
4.4. The Role of the Gut-Brain Axis
4.5. The Role of Neuroinflammation
4.6. The Role of Abnormal Neutrophilic Response
4.7. The Role of HLA Dysregulation
4.8. Other Molecular Mechanisms
5. Current Drug Research
5.1. Research on Drugs That Are Based on Monoamine Theory
5.1.1. SSRIs
5.1.2. SNRIs
5.1.3. Norepinephrine-Dopamine Reuptake Inhibitors (NDRIs)
5.1.4. Monoamine Oxidase Inhibitors (MAOIs)
5.1.5. Tricyclic Antidepressants (TCAs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Number of Studies | Study Design | Study Approach | Results | Conclusion |
|---|---|---|---|---|---|
| Sun et al. 2017 [77] | 12 suitable trials, with data from 707 participants | Multiple treatments meta-analysis of RCTs | SSRIs | The results established that the acceptability scores for doxepin, citalopram, and fluoxetine were higher than paroxetine. | Paroxetine was the preferred drug at the onset of acute treatment for PSD, and fluoxetine was the least ideal choice. |
| Li and Zhang 2020 [78] | Data from 51 RCTs | Multiple treatments meta-analysis of RCTs | SSRIs, TCAs, mirtazapine | The results showed that escitalopram was the most favorable, while amitriptyline scored the lowest at two weeks. Citalopram was more effective than the other SSRIs at four weeks, while mirtazapine had the highest response rate among patients after eight weeks. | Escitalopram was associated with a quicker relief of depression, but mirtazapine was probably the best option for an 8-week treatment duration. |
| Zhou et al. 2020 [73] | Data of 10 RCTs with a total of 5370 patients | Multiple treatments meta-analysis of RCTs | SSRIs | The results showed that SSRI therapy was most effective in preventing PSD when used in the initial stages of the condition. | Early SSRIs therapy was effective in preventing post-stroke depression. However, SSRIs did not improve patients’ post-stroke functional independence. SSRIs were relatively safe. |
| Zhang et al. 2013 [85] | 95 patients | RCT | SNRI: Duloxetine | Duloxetine helps stroke patients prevent minor and major depression and also helps them rehabilitate rapidly from a stroke. | Prophylactic use also reduced the incidence of PSD and improved individuals’ quality of life and cognitive function. |
| Cravello et al. 2009 [86] | 50 patients | RCT | SNRI: Venlafaxine | The results showed that the patients had more significant improvement in alexithymia than those treated with fluoxetine. | Antidepressants such as SNRIs were effective in treating depression and improving emotional awareness in PSD patients. |
| Tsai et al. 2011 [87] | 92 patients | RCT | SNRI: Milnacipran | The results showed that milnacipran had a significant advantage in preventing PSD without substantial adverse effects. | Preventive use antidepressants such as SNRI (milnacipran) reduced incidences of PSD. |
| Patel et al. 2016 [92] | Data from 51 RCTs | Multiple treatments meta-analysis of RCTs | Bupropion | The results showed that bupropion was effective and highly tolerated. Patients using it had low rates of side effects. | The researchers concluded that bupropion since it had equivalent effectiveness to other antidepressants. |
| Liu et al. 2015 [93] | Data from 9 RCTs and 1,106 patients | Multiple treatments meta-analysis of RCTs | SNRIs: Fluoxetine Bupropion | The results showed no difference in effectiveness between bupropion and other antidepressants such as fluoxetine. | Bupropion hydrochloride sustained-release tablets have the same effectiveness and side effects as fluoxetine tablets in the treatment of depression. |
| Qin et al. 2018 [101] | Data from 14 RCTs included 949 patients | Multiple treatments meta-analysis of RCTs | MAOs SSRIs TCAs | The results showed insufficient evidence that MAOIs were more effective and tolerant than placebo and other antidepressants such as SSRIs. | MAOIs are less effective than SSRIs. |
| Deng et al. 2017 [103] | Data from 23 RCTs included 1542 patients | Multiple treatments meta-analysis of RCTs | NDRIs SSSIs TCAs | The results showed that TCAs are among the best approaches to managing post-stroke depression, just such as SSRIs and SNRIs. | NDRIs, SSRIs, and TCAs are associated with a considerably higher depression score reduction compared with the control groups. |
| Tan et al. 2015 [104] | Data from 48 RCTs included 3294 patients | Multiple treatments meta-analysis of RCTs | SSRIs TCAs | The results showed that TCAs had a higher efficacy index than citalopram as per the scores on the Hamilton Depression Scale. | They concluded that TCAs were more efficient than citalopram in treating PSD. |
| Arroll et al. 2005 [105] | Data from 12 RCTs included 2753 patients | Multiple treatments meta-analysis of RCTs | SSRIs TCAs | The results showed that when compared with a placebo, efficacy estimates showed that patients using TCAs had a relative improvement risk. | The researchers concluded that it was more effective to prescribe TCAs in primary care than a placebo. |
5.2. Glutamate-Based Antidepressants
5.2.1. NMDA Blockers
Ketamine
Memantine
Magnesium
D-cycloserine (DCS)
5.2.2. AMPA Antagonists
5.2.3. Metabotropic Glutamate Receptors
Drug Research on Group I mGluR in Depression
Drug Research on Group II mGluR in Depression
Drug Research on Group III mGluR in Depression
5.3. Blood Glutamate Scavengers
5.3.1. Pyruvate
5.3.2. Oxaloacetate
5.4. Microbiota Treatment in PSD
5.5. Anti-Inflammatory Treatments in PSD
5.6. Other Drugs
5.6.1. Mirtazapine
5.6.2. Agomelatine (AGM)
5.6.3. Psychostimulants
5.6.4. Nootropic Drugs
5.6.5. The Contribution of Cyclooxygenase-2 (COX-2) Inhibitors in Depression and Ischemic Brain Injury
The Link between COX-2 and mGluR7 in Depression and Cognition
6. Results of Non-Pharmacological Treatments
6.1. Cognitive-Behavioral Therapy
6.2. Electroconvulsive Therapy
6.3. Transcranial Direct Current Stimulation (tDCS)
6.4. Repetitive Transcranial Magnetic Stimulation (rTMS)
6.5. Vagus Nerve Stimulation (VNS)
7. Future Research in Drug Therapy of PSD
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robinson, R.G.; Jorge, R.E. Post-stroke depression: A review. Am. J. Psychiatry 2016, 173, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, P.B.; Ahmad, R.; Abdullahi, A.; Agrawal, A. New Insight into Cerebrovascular Diseases: An Updated Comprehensive Review; IntechOpen: London, UK, 2020. [Google Scholar]
- Lanctôt, K.L.; Lindsay, M.P.; Smith, E.E.; Sahlas, D.J.; Foley, N.; Gubitz, G.; Austin, M.; Ball, K.; Bhogal, S.; Blake, T. Canadian stroke best practice recommendations: Mood, cognition and fatigue following stroke, update 2019. Int. J. Stroke 2020, 15, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.W.; Wu, C.L.; Wang, X.C.; Liu, J.; Bi, J.Z.; Wang, D.Y. Monoamine neurotransmitters and fibroblast growth factor-2 in the brains of rats with post-stroke depression. Exp. Ther. Med. 2014, 8, 159–164. [Google Scholar] [CrossRef]
- Pedroso, V.S.P.; Souza, L.C.; Brunoni, A.R.; Teixeira, A.L. Post stroke depression: Clinics, etiopathogenesis and therapeutics. Arch. Clin. Psychiatry 2015, 42, 18–24. [Google Scholar] [CrossRef]
- Fang, M.; Zhong, L.; Jin, X.; Cui, R.; Yang, W.; Gao, S.; Lv, J.; Li, B.; Liu, T. Effect of inflammation on the process of stroke rehabilitation and poststroke depression. Front. Psychiatry 2019, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, J.D.; Ding, J.; Baum, L.; Sham, P.C. Immune dysregulation in depression: Evidence from genome-wide association. Brain Behav. Immun.-Health 2020, 7, 100108. [Google Scholar] [CrossRef]
- Li, M.; D’Arcy, C.; Li, X.; Zhang, T.; Joober, R.; Meng, X. What do DNA methylation studies tell us about depression? A systematic review. Transl. Psychiatry 2019, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, B.F.; Kutz, R.; Zlotnik, A.; Boyko, M. Blood glutamate scavenging as a novel glutamate-based therapeutic approach for post-stroke depression. Ther. Adv. Psychopharmacol. 2020, 10, 2045125320903951. [Google Scholar] [CrossRef]
- Conroy, S.K.; Brownlowe, K.B.; McAllister, T.W. Depression comorbid with stroke, traumatic brain injury, Parkinson’s disease, and multiple sclerosis: Diagnosis and treatment. Focus 2020, 18, 150–161. [Google Scholar] [CrossRef]
- Towfighi, A.; Ovbiagele, B.; El Husseini, N.; Hackett, M.L.; Jorge, R.E.; Kissela, B.M.; Mitchell, P.H.; Skolarus, L.E.; Whooley, M.A.; Williams, L.S. Poststroke depression: A scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2017, 48, e30–e43. [Google Scholar] [CrossRef]
- Schöttke, H.; Giabbiconi, C.-M. Post-stroke depression and post-stroke anxiety: Prevalence and predictors. Int. Psychogeriatr. 2015, 27, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Mpofu, E.; Athanasou, J. Reducing depressive or anxiety symptoms in post-stroke patients: Pilot trial of a constructive integrative psychosocial intervention. Int. J. Health Sci. 2017, 11, 53. [Google Scholar]
- Health, N.I.O. National Institute of Neurological Disorders and Stroke Website. Post-Stroke Rehabilitation Fact Sheet. 2020. Available online: https://www.ninds.nih.gov/post-stroke-rehabilitation-fact-sheet (accessed on 24 November 2022).
- Paolucci, S.; Iosa, M.; Coiro, P.; Venturiero, V.; Savo, A.; De Angelis, D.; Morone, G. Post-stroke depression increases disability more than 15% in ischemic stroke survivors: A case-control study. Front. Neurol. 2019, 10, 926. [Google Scholar] [CrossRef] [PubMed]
- Hackett, M.L.; Anderson, C.S. Treatment options for post-stroke depression in the elderly. Aging Health 2005, 1, 95–105. [Google Scholar] [CrossRef]
- Sharma, G.S.; Gupta, A.; Khanna, M.; Prakash, N.B. Post-Stroke Depression and Its Effect on Functional Outcomes during Inpatient Rehabilitation. J. Neurosci. Rural Pract. 2021, 12, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.H. The link between depression and physical symptoms. Prim. Care Companion J. Clin. Psychiatry 2004, 6, 12–16. [Google Scholar] [PubMed]
- Meader, N.; Moe-Byrne, T.; Llewellyn, A.; Mitchell, A.J. Screening for poststroke major depression: A meta-analysis of diagnostic validity studies. J. Neurol. Neurosurg. Psychiatry 2014, 85, 198–206. [Google Scholar] [CrossRef]
- Li, W.; Xiao, W.-M.; Chen, Y.-K.; Qu, J.-F.; Liu, Y.-L.; Fang, X.-W.; Weng, H.-Y.; Luo, G.-P. Anxiety in patients with acute ischemic stroke: Risk factors and effects on functional status. Front. Psychiatry 2019, 10, 257. [Google Scholar] [CrossRef]
- Coster, L.d.; Leentjens, A.F.; Lodder, J.; Verhey, F.R. The sensitivity of somatic symptoms in post-stroke depression: A discriminant analytic approach. Int. J. Geriatr. Psychiatry A J. Psychiatry Late Life Allied Sci. 2005, 20, 358–362. [Google Scholar] [CrossRef]
- Wei, C.; Zhang, F.; Chen, L.; Ma, X.; Zhang, N.; Hao, J. RETRACTED ARTICLE: Factors associated with post-stroke depression and fatigue: Lesion location and coping styles. J. Neurol. 2016, 263, 269–276. [Google Scholar] [CrossRef]
- Cramer, S.C. Drugs to enhance motor recovery after stroke. Stroke 2015, 46, 2998–3005. [Google Scholar] [CrossRef] [PubMed]
- Mariman, J.J.; Lorca, E.; Biancardi, C.; Burgos, P.; Álvarez-Ruf, J. Brain’s Energy after Stroke: From a Cellular Perspective toward Behavior. Front. Integr. Neurosci. 2022, 16, 826728. [Google Scholar] [CrossRef]
- Afzal, T.; Chardon, M.K.; Rymer, W.Z.; Suresh, N.L. Stretch reflex excitability in contralateral limbs of stroke survivors is higher than in matched controls. J. Neuroeng. Rehabil. 2019, 16, 154. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, F.; Zijlmans, J.; Duenas, G.; Cevallos, N. Post-stroke movement disorders: Report of 56 patients. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Fang, M.; Liu, X.-Y. The neurobiological pathogenesis of poststroke depression. Sci. World J. 2014, 2014, 521349. [Google Scholar] [CrossRef]
- Lai, Y.-J.; McCullough, L.D. Poststroke depression: Pathophysiology and treatment strategies. In Neurobiology of Depression: Road to Novel Therapeutic, 1st ed; Quevedo, J., Carvalho, A.F., Zarate, C.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 197–205. [Google Scholar] [CrossRef]
- Jiao, J.T.; Cheng, C.; Ma, Y.J.; Huang, J.; Dai, M.C.; Jiang, C.; Wang, C.; Shao, J.F. Association between inflammatory cytokines and the risk of post-stroke depression, and the effect of depression on outcomes of patients with ischemic stroke in a 2-year prospective study. Exp. Ther. Med. 2016, 12, 1591–1598. [Google Scholar] [CrossRef]
- Ostir, G.V.; Markides, K.S.; Peek, M.K.; Goodwin, J.S. The association between emotional well-being and the incidence of stroke in older adults. Psychosom. Med. 2001, 63, 210–215. [Google Scholar] [CrossRef]
- Robinson, R.G.; Bloom, F.E. Pharmacological treatment following experimental cerebral infarction: Implications for understanding psychological symptoms of human stroke. Biol. Psychiatry 1977, 12, 669–680. [Google Scholar]
- Lin, C.-H.; Hashimoto, K.; Lane, H.-Y. Glutamate-related biomarkers for neuropsychiatric disorders. Front. Psychiatry 2019, 10, 904. [Google Scholar] [CrossRef]
- Li, C.-T.; Yang, K.-C.; Lin, W.-C. Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: Evidence from clinical neuroimaging studies. Front. Psychiatry 2019, 9, 767. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Fagg, G.E. L-Glutamate, excitatory amino acid receptors and brain function. Trends Neurosci. 1985, 8, 207–210. [Google Scholar] [CrossRef]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, Y.; Liu, F.; Jia, N.; Gao, J.; Pang, X.; Deng, F. Diversiform Etiologies for Post-stroke Depression. Front. Psychiatry 2018, 9, 761. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-Y.; Zhao, Y.-D.; Li, J.; Chen, X.-Y.; Wang, R.-D.; Zeng, J.-W. Plasma levels of glutamate during stroke is associated with development of post-stroke depression. Psychoneuroendocrinology 2014, 47, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.-Y.; Qian, F.-Y.; Qian, J.-F.; Zhang, Z.-J. The combination of plasma glutamate and physical impairment after acute stroke as a potential indicator for the early-onset post-stroke depression. J. Psychosom. Res. 2017, 96, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Khoodoruth, M.A.S.; Estudillo-Guerra, M.A.; Pacheco-Barrios, K.; Nyundo, A.; Chapa-Koloffon, G.; Ouanes, S. Glutamatergic System in Depression and Its Role in Neuromodulatory Techniques Optimization. Front. Psychiatry 2022, 13, 886918. [Google Scholar] [CrossRef]
- Gropper, M.A.; Miller, R.D.; Eriksson, L.I.; Fleisher, L.A.; Wiener-Kronish, J.P.; Cohen, N.H.; Leslie, K. Miller’s Anesthesia, 2-Volume Set E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Mattson, M.P. Excitotoxicity. In Stress: Physiology, Biochemistry, and Pathology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 125–134. [Google Scholar]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F. Going the extra (synaptic) mile: Excitotoxicity as the road toward neurodegenerative diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Connolly, N.; Prehn, J.H. The metabolic response to excitotoxicity–lessons from single-cell imaging. J. Bioenerg. Biomembr. 2015, 47, 75–88. [Google Scholar] [CrossRef]
- Andrew, R.D.; Farkas, E.; Hartings, J.A.; Brennan, K.; Herreras, O.; Müller, M.; Kirov, S.; Ayata, C.; Ollen-Bittle, N.; Reiffurth, C. Questioning glutamate excitotoxicity in acute brain damage: The importance of spreading depolarization. Neurocritical Care 2022, 37, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Rama, R.; García, J.C. Excitotoxicity and oxidative stress in acute stroke. Ischemic Stroke Updat. 2016, 17–33. [Google Scholar]
- Naghavi, F.S.; Koffman, E.E.; Lin, B.; Du, J. Post-stroke neuronal circuits and mental illnesses. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 1–11. [Google Scholar] [PubMed]
- Vogel, C.H. Assessment and Approach to Treatment in Post-Stroke Depression. J. Am. Acad. Nurse Pract. 1995, 7, 493–497. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. Brain–gut–microbiota axis—Mood, metabolism and behaviour. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 69–70. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2015, 28, 203. [Google Scholar]
- Zhang, S.Q.; Tian, D.; Hu, C.Y.; Meng, Y.H. Chlorogenic Acid Ameliorates High-Fat and High-Fructose Diet-Induced Cognitive Impairment via Mediating the Microbiota–Gut–Brain Axis. J. Agric. Food Chem. 2022, 70, 2600–2615. [Google Scholar] [CrossRef]
- Xia, B.; Liu, X.; Li, X.; Wang, Y.; Wang, D.; Kou, R.; Zhang, L.; Shi, R.; Ye, J.; Bo, X. Sesamol ameliorates dextran sulfate sodium-induced depression-like and anxiety-like behaviors in colitis mice: The potential involvement of the gut–brain axis. Food Funct. 2022, 13, 2865–2883. [Google Scholar] [CrossRef]
- Zhu, G.; Zhao, J.; Zhang, H.; Chen, W.; Wang, G. Administration of bifidobacterium breve improves the brain function of aβ1-42-treated mice via the modulation of the gut microbiome. Nutrients 2021, 13, 1602. [Google Scholar] [CrossRef] [PubMed]
- Mi, Z.-H.; Li, Y.; Ma, Y.-L.; Jin, P.-M.; Song, L.-Z.-X.; Xu, T.-C. Clinical Application and Mechanism of Acupuncture in the Brain-Gut Interaction. Psychosom. Med. Res. 2021, 3, 108–121. [Google Scholar] [CrossRef]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The gut-brain axis: How microbiota and host inflammasome influence brain physiology and pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Rong, C.; Wang, F.; Liu, X.; Sun, Y.; Zhang, H.-T. GABAergic system in stress: Implications of GABAergic neuron subpopulations and the gut-vagus-brain pathway. Neural Plast. 2020, 2020, 8858415. [Google Scholar] [CrossRef] [PubMed]
- Ganci, M.; Suleyman, E.; Butt, H.; Ball, M. The role of the brain–gut–microbiota axis in psychology: The importance of considering gut microbiota in the development, perpetuation, and treatment of psychological disorders. Brain Behav. 2019, 9, e01408. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Weymann, K.B.; Wood, L.; Wang, Q.M. Inflammatory signaling in post-stroke fatigue and depression. Eur. Neurol. 2018, 80, 138–148. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A cell with many roles in inflammation or several cell types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Hu, J.; Zhou, W.; Zhou, Z.; Han, J.; Dong, W. Elevated neutrophil-to-lymphocyte and platelet-to-lymphocyte ratios predict post-stroke depression with acute ischemic stroke. Exp. Ther. Med. 2020, 19, 2497–2504. [Google Scholar] [CrossRef]
- Mosaad, Y. Clinical role of human leukocyte antigen in health and disease. Scand. J. Immunol. 2015, 82, 283–306. [Google Scholar] [CrossRef]
- Crux, N.B.; Elahi, S. Human leukocyte antigen (HLA) and immune regulation: How do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections? Front. Immunol. 2017, 8, 832. [Google Scholar] [CrossRef]
- Taneja, V. Cytokines pre-determined by genetic factors are involved in pathogenesis of rheumatoid arthritis. Cytokine 2015, 75, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Zheng, Y.; Froud, K. Brain-derived neurotrophic factor as a clinical biomarker in predicting the development of post-stroke depression: A review of evidence. Cureus 2021, 13, e15662. [Google Scholar] [CrossRef] [PubMed]
- Ikegame, T.; Bundo, M.; Murata, Y.; Kasai, K.; Kato, T.; Iwamoto, K. DNA methylation of the BDNF gene and its relevance to psychiatric disorders. J. Hum. Genet. 2013, 58, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Villa, R.F.; Ferrari, F.; Moretti, A. Post-stroke depression: Mechanisms and pharmacological treatment. Pharmacol. Ther. 2018, 184, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; He, Z.; Xu, Q.; Dong, J.; Xiao, T.; Liang, F.; Ma, X.; Wang, F.; Huang, J.H. The Relationship Between 5-Hydroxytryptamine and Its Metabolite Changes with Post-stroke Depression. Front. Psychiatry 2022, 13, 871754. [Google Scholar] [CrossRef]
- Elzib, H.; Pawloski, J.; Ding, Y.; Asmaro, K. Antidepressant pharmacotherapy and poststroke motor rehabilitation: A review of neurophysiologic mechanisms and clinical relevance. Brain Circ. 2019, 5, 62. [Google Scholar]
- Legg, L.A.; Rudberg, A.-S.; Hua, X.; Wu, S.; Hackett, M.L.; Tilney, R.; Lindgren, L.; Kutlubaev, M.A.; Hsieh, C.-F.; Barugh, A.J. Selective serotonin reuptake inhibitors (SSRIs) for stroke recovery. Cochrane Database Syst. Rev. 2021. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, S.; Liu, X.; Zhuang, W. Selective serotonin reuptake inhibitors for functional independence and depression prevention in early stage of post-stroke: A meta-analysis. Medicine 2020, 99, e19062. [Google Scholar] [CrossRef]
- Mead, G.E.; Hsieh, C.-F.; Lee, R.; Kutlubaev, M.; Claxton, A.; Hankey, G.J.; Hackett, M. Selective serotonin reuptake inhibitors for stroke recovery: A systematic review and meta-analysis. Stroke 2013, 44, 844–850. [Google Scholar] [CrossRef]
- Mortensen, J.K.; Larsson, H.; Johnsen, S.P.; Andersen, G. Post stroke use of selective serotonin reuptake inhibitors and clinical outcome among patients with ischemic stroke: A nationwide propensity score–matched follow-up study. Stroke 2013, 44, 420–426. [Google Scholar] [CrossRef][Green Version]
- Paolucci, S. Advances in antidepressants for treating post-stroke depression. Expert Opin. Pharmacother. 2017, 18, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liang, Y.; Jiao, Y.; Lin, J.; Qu, H.; Xu, J.; Zhao, C. Comparative efficacy and acceptability of antidepressant treatment in poststroke depression: A multiple-treatments meta-analysis. BMJ Open 2017, 7, e016499. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, C. Comparative efficacy of nine antidepressants in treating Chinese patients with post-stroke depression: A network meta-analysis. J. Affect. Disord. 2020, 266, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.; Gehret, J.; Young, G.; Kamen, L. Challenging Neuropathic Pain Syndromes: Evaluation and Evidence-Based Treatment; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Agius, M.; Bonnici, H. Antidepressants in use in clinical practice. Psychiatr. Danub. 2017, 29, 667–671. [Google Scholar]
- Chaves-Martins, Y. 3.26—Atypical Analgesics. In Comprehensive Pharmacology; Kenakin, T., Ed.; Elsevier: Oxford, UK, 2022; pp. 532–547. [Google Scholar]
- Locher, C.; Koechlin, H.; Zion, S.R.; Werner, C.; Pine, D.S.; Kirsch, I.; Kessler, R.C.; Kossowsky, J. Efficacy and safety of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and placebo for common psychiatric disorders among children and adolescents: A systematic review and meta-analysis. JAMA Psychiatry 2017, 74, 1011–1020. [Google Scholar] [CrossRef]
- Sansone, R.A.; Sansone, L.A. Serotonin norepinephrine reuptake inhibitors: A pharmacological comparison. Innov. Clin. Neurosci. 2014, 11, 37. [Google Scholar]
- Corallo, F.; Scarfì, C.; Arcadi, F.A.; Formica, C.; Di Cara, M.; Palmeri, R.; Romeo, L.; Lo Buono, V.; Bramanti, P.; Marino, S. Role of functional pharmacological therapy in post-stroke depression: A narrative review. J. Int. Med. Res. 2020, 48, 0300060520950557. [Google Scholar] [CrossRef]
- Zhang, L.-S.; Hu, X.-Y.; Yao, L.-Y.; Geng, Y.; Wei, L.-L.; Zhang, J.-H.; Chen, W. Prophylactic effects of duloxetine on post-stroke depression symptoms: An open single-blind trial. Eur. Neurol. 2013, 69, 336–343. [Google Scholar] [CrossRef]
- Cravello, L.; Caltagirone, C.; Spalletta, G. The SNRI venlafaxine improves emotional unawareness in patients with post-stroke depression. Hum. Psychopharmacol. Clin. Exp. 2009, 24, 331–336. [Google Scholar] [CrossRef]
- Tsai, C.-S.; Wu, C.-L.; Chou, S.-Y.; Tsang, H.-Y.; Hung, T.-H.; Su, J.-A. Prevention of poststroke depression with milnacipran in patients with acute ischemic stroke: A double-blind randomized placebo-controlled trial. Int. Clin. Psychopharmacol. 2011, 26, 263–267. [Google Scholar] [CrossRef]
- Stahl, S.M.; Pradko, J.F.; Haight, B.R.; Modell, J.G.; Rockett, C.B.; Learned-Coughlin, S. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim. Care Companion J. Clin. Psychiatry 2004, 6, 159. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, S. Review of the choice and use of antidepressant drugs. Prog. Neurol. Psychiatry 2013, 17, 18–26. [Google Scholar] [CrossRef]
- Tundo, A.; de Filippis, R.; Proietti, L. Pharmacologic approaches to treatment resistant depression: Evidences and personal experience. World J. Psychiatry 2015, 5, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Misane, M.D. Understanding depression. International Journal of Studies in Nursing 2020, 5, 1. [Google Scholar] [CrossRef]
- Patel, K.; Allen, S.; Haque, M.N.; Angelescu, I.; Baumeister, D.; Tracy, D.K. Bupropion: A systematic review and meta-analysis of effectiveness as an antidepressant. Ther. Adv. Psychopharmacol. 2016, 6, 99–144. [Google Scholar] [CrossRef]
- Liu, Z.; Qin, L.; Xu, G. Meta-analysis on effectiveness and safety of bupropion hydrochloride sustained-release tablets and fluoxetine tablets in treatment of depression. J. Jilin Univ. 2015, 41, 140–144. [Google Scholar] [CrossRef]
- Sung, S.-H.; Park, S.-C.; Han, K.-M.; Won, E.-S.; Lee, H.-Y.; Koo, J.-W.; Paik, J.-W.; Lee, K.-M.; Jeon, H.J.; Lee, M.-S. Evidence-based Korean pharmacological treatment guideline for depression, revised edition (II): Antidepressant efficacy compared with placebo, difference in efficacy of antidepressants, and appropriate time of efficacy judgment in antidepressant therapy. J. Korean Neuropsychiatr. Assoc. 2013, 52, 372–385. [Google Scholar] [CrossRef]
- Laban, T.S.; Saadabadi, A. Monoamine oxidase inhibitors (MAOI). In StatPearls [Internet]; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Khan, F. Poststroke depression. Aust. Fam. Physician 2004, 33, 831–834. [Google Scholar]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Remick, R.A.; Froese, C.; Keller, F.D. Common side effects associated with monoamine oxidase inhibitors. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1989, 13, 497–504. [Google Scholar] [CrossRef]
- Li, F.; Gu, D.X. Treatment for post-stroke depression. Chin. J. Clin. Rehabil. 2006, 10, 129–132. [Google Scholar]
- Thase, M.E.; Trivedi, M.H.; Rush, A.J. MAOIs in the contemporary treatment of depression. Neuropsychopharmacology 1995, 12, 185–219. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Chen, H.; Gao, W.; Zhao, L.; Zhao, M.; Qin, H.; Chen, W.; Chen, L.; Yang, M. Efficacy, acceptability, and tolerability of antidepressant treatments for patients with post-stroke depression: A network meta-analysis. Braz. J. Med. Biol. Res. 2018, 51, e7218. [Google Scholar] [CrossRef] [PubMed]
- Moraczewski, J.; Aedma, K.K. Tricyclic antidepressants. In StatPearls [Internet]; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Deng, L.; Sun, X.; Qiu, S.; Xiong, Y.; Li, Y.; Wang, L.; Wei, Q.; Wang, D.; Liu, M. Interventions for management of post-stroke depression: A Bayesian network meta-analysis of 23 randomized controlled trials. Sci. Rep. 2017, 7, 16466. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Huang, X.; Ding, L.; Hong, H. Efficacy and safety of citalopram in treating post-stroke depression: A meta-analysis. Eur. Neurol. 2015, 74, 188–201. [Google Scholar] [CrossRef]
- Arroll, B.; Macgillivray, S.; Ogston, S.; Reid, I.; Sullivan, F.; Williams, B.; Crombie, I. Efficacy and tolerability of tricyclic antidepressants and SSRIs compared with placebo for treatment of depression in primary care: A meta-analysis. Ann. Fam. Med. 2005, 3, 449–456. [Google Scholar] [CrossRef]
- Park, M.; Niciu, M.J.; Zarate, C.A. Novel glutamatergic treatments for severe mood disorders. Curr. Behav. Neurosci. Rep. 2015, 2, 198–208. [Google Scholar] [CrossRef]
- Zarate, C.A.; Niciu, M.J. Ketamine for depression: Evidence, challenges and promise. World Psychiatry 2015, 14, 348. [Google Scholar] [CrossRef]
- Ohgi, Y.; Futamura, T.; Hashimoto, K. Glutamate signaling in synaptogenesis and NMDA receptors as potential therapeutic targets for psychiatric disorders. Curr. Mol. Med. 2015, 15, 206–221. [Google Scholar] [CrossRef]
- Garay, R.; Zarate, C.A.; Cavero, I.; Kim, Y.-K.; Charpeaud, T.; Skolnick, P. The development of glutamate-based antidepressants is taking longer than expected. Drug Discov. Today 2018, 23, 1689. [Google Scholar] [CrossRef]
- Lazarevic, V.; Yang, Y.; Flais, I.; Svenningsson, P. Ketamine decreases neuronally released glutamate via retrograde stimulation of presynaptic adenosine A1 receptors. Mol. Psychiatry 2021, 26, 7425–7435. [Google Scholar] [CrossRef] [PubMed]
- Abdoulaye, I.A.; Wu, S.-S.; Chibaatar, E.; Yu, D.-F.; Le, K.; Cao, X.-J.; Guo, Y.-J. Ketamine induces lasting antidepressant effects by modulating the NMDAR/CaMKII-mediated synaptic plasticity of the hippocampal dentate gyrus in depressive stroke model. Neural Plast. 2021, 2021, 6635084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, B.; Yuan, Y.; Zhou, T.; Xiao, P.; Wu, Y.; Shang, Y.; Yuan, S.; Zhang, J. (R, S)-Ketamine Promotes Striatal Neurogenesis and Sensorimotor Recovery Through Improving Poststroke Depression–Mediated Decrease in Atrial Natriuretic Peptide. Biol. Psychiatry Glob. Open Sci. 2021, 1, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.L.; Norris, S.; Talbot, J.; Birmingham, M.; Hatchard, T.; Ortiz, A.; Owoeye, O.; Batten, L.A.; Blier, P. Single, repeated, and maintenance ketamine infusions for treatment-resistant depression: A randomized controlled trial. Am. J. Psychiatry 2019, 176, 401–409. [Google Scholar] [CrossRef]
- Serafini, G.; H Howland, R.; Rovedi, F.; Girardi, P.; Amore, M. The role of ketamine in treatment-resistant depression: A systematic review. Curr. Neuropharmacol. 2014, 12, 444–461. [Google Scholar] [CrossRef]
- López-Valdés, H.E.; Clarkson, A.N.; Ao, Y.; Charles, A.C.; Carmichael, S.T.; Sofroniew, M.V.; Brennan, K.C. Memantine enhances recovery from stroke. Stroke 2014, 45, 2093–2100. [Google Scholar] [CrossRef]
- Parsons, C.; Danysz, W.; Quack, G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist—A review of preclinical data. Neuropharmacology 1999, 38, 735–767. [Google Scholar] [CrossRef]
- Strzelecki, D.; Tabaszewska, A.; Barszcz, Z.; Józefowicz, O.; Kropiwnicki, P.; Rabe-Jabłońska, J. A 10-week memantine treatment in bipolar depression: A case report. Focus on depressive symptomatology, cognitive parameters and quality of life. Psychiatry Investig. 2013, 10, 421. [Google Scholar] [CrossRef]
- Kirkland, A.E.; Sarlo, G.L.; Holton, K.F. The role of magnesium in neurological disorders. Nutrients 2018, 10, 730. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, K.; Luan, X.; Liu, Z.; Cai, Y.; Wang, Q.; Zhu, B.; He, J. Association between serum magnesium levels and depression in stroke patients. Aging Dis. 2016, 7, 687. [Google Scholar] [CrossRef][Green Version]
- Volpe, S.L. Magnesium in disease prevention and overall health. Adv. Nutr. 2013, 4, 378S–383S. [Google Scholar] [CrossRef] [PubMed]
- Eby III, G.A.; Eby, K.L. Magnesium for treatment-resistant depression: A review and hypothesis. Med. Hypotheses 2010, 74, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Sheinin, A.; Shavit, S.; Benveniste, M. Subunit specificity and mechanism of action of NMDA partial agonist D-cycloserine. Neuropharmacology 2001, 41, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Schade, S.; Paulus, W. D-cycloserine in neuropsychiatric diseases: A systematic review. Int. J. Neuropsychopharmacol. 2016, 19, pyv102. [Google Scholar] [CrossRef]
- Schoevers, R.A.; Chaves, T.V.; Balukova, S.M.; Kortekaas, R. Oral ketamine for the treatment of pain and treatment-resistant depression. Br. J. Psychiatry 2016, 208, 108–113. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of metabotropic glutamate receptors in neurological disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef]
- Nakanishi, S. Molecular diversity of glutamate receptors and implications for brain function. Science 1992, 258, 597–603. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef]
- Hanson, J.E.; Smith, Y. Group I metabotropic glutamate receptors at GABAergic synapses in monkeys. J. Neurosci. 1999, 19, 6488–6496. [Google Scholar] [CrossRef][Green Version]
- Lüscher, C.; Huber, K.M. Group 1 mGluR-dependent synaptic long-term depression: Mechanisms and implications for circuitry and disease. Neuron 2010, 65, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Borroto-Escuela, D.O. Basimglurant for treatment of major depressive disorder: A novel negative allosteric modulator of metabotropic glutamate receptor 5. Expert Opin. Investig. Drugs 2015, 24, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.; Conn, P.J. Targeting metabotropic glutamate receptors for the treatment of depression and other stress-related disorders. Neuropharmacology 2021, 196, 108687. [Google Scholar] [CrossRef] [PubMed]
- Belozertseva, I.; Kos, T.; Popik, P.; Danysz, W.; Bespalov, A. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur. Neuropsychopharmacol. 2007, 17, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Takata, M.; Kitaichi, M.; Kassai, M.; Inoue, M.; Ishikawa, C.; Hirose, W.; Yoshida, K.; Shimizu, I. DSR-98776, a novel selective mGlu5 receptor negative allosteric modulator with potent antidepressant and antimanic activity. Eur. J. Pharmacol. 2015, 757, 11–20. [Google Scholar] [CrossRef]
- Hughes, Z.; Neal, S.; Smith, D.; Rizzo, S.S.; Pulicicchio, C.; Lotarski, S.; Lu, S.; Dwyer, J.; Brennan, J.; Olsen, M. Negative allosteric modulation of metabotropic glutamate receptor 5 results in broad spectrum activity relevant to treatment resistant depression. Neuropharmacology 2013, 66, 202–214. [Google Scholar] [CrossRef]
- Smith, D.F.; Jakobsen, S. Molecular neurobiology of depression: PET findings on the elusive correlation with symptom severity. Front. Psychiatry 2013, 4, 8. [Google Scholar] [CrossRef]
- Esterlis, I.; DellaGioia, N.; Pietrzak, R.H.; Matuskey, D.; Nabulsi, N.; Abdallah, C.G.; Yang, J.; Pittenger, C.; Sanacora, G.; Krystal, J.H. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: An [11C] ABP688 and PET imaging study in depression. Mol. Psychiatry 2018, 23, 824–832. [Google Scholar] [CrossRef]
- Hengartner, M.P.; Jakobsen, J.C.; Sørensen, A.; Plöderl, M. Efficacy of new-generation antidepressants assessed with the Montgomery-Asberg Depression Rating Scale, the gold standard clinician rating scale: A meta-analysis of randomised placebo-controlled trials. PLoS ONE 2020, 15, e0229381. [Google Scholar] [CrossRef]
- Rocher, J.-P.; Bonnet, B.; Bolea, C.; Lutjens, R.; Le Poul, E.; Poli, S.; Epping-Jordan, M.; Bessis, A.-S.; Ludwig, B.; Mutel, V. mGluR5 negative allosteric modulators overview: A medicinal chemistry approach towards a series of novel therapeutic agents. Curr. Top. Med. Chem. 2011, 11, 680–695. [Google Scholar] [CrossRef]
- Chaki, S.; Koike, H.; Fukumoto, K. Targeting of metabotropic glutamate receptors for the development of novel antidepressants. Chronic Stress 2019, 3, 2470547019837712. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zhang, J.-c.; Yao, W.; Ren, Q.; Ma, M.; Yang, C.; Chaki, S.; Hashimoto, K. Rapid and sustained antidepressant action of the mGlu2/3 receptor antagonist MGS0039 in the social defeat stress model: Comparison with ketamine. Int. J. Neuropsychopharmacol. 2017, 20, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.K.; Lee, J.A.; Jeong, S.; Seog, D.-H.; Lee, J.G.; Park, S.W. Effects of Chronic LY341495 on Hippocampal mTORC1 Signaling in Mice with Chronic Unpredictable Stress-Induced Depression. Int. J. Mol. Sci. 2022, 23, 6416. [Google Scholar] [CrossRef]
- Rafało-Ulińska, A.; Brański, P.; Pałucha-Poniewiera, A. Combined Administration of (R)-Ketamine and the mGlu2/3 Receptor Antagonist LY341495 Induces Rapid and Sustained Effects in the CUMS Model of Depression via a TrkB/BDNF-Dependent Mechanism. Pharmaceuticals 2022, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Nasca, C.; Xenos, D.; Barone, Y.; Caruso, A.; Scaccianoce, S.; Matrisciano, F.; Battaglia, G.; Mathé, A.A.; Pittaluga, A.; Lionetto, L. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc. Natl. Acad. Sci. 2013, 110, 4804–4809. [Google Scholar] [CrossRef]
- Dasgupta, A.; Lim, Y.J.; Kumar, K.; Baby, N.; Pang, K.L.K.; Benoy, A.; Behnisch, T.; Sajikumar, S. Group III metabotropic glutamate receptors gate long-term potentiation and synaptic tagging/capture in rat hippocampal area CA2. eLife 2020, 9, e55344. [Google Scholar] [CrossRef] [PubMed]
- Hallock, R.M.; Martyniuk, C.J.; Finger, T.E. Group III metabotropic glutamate receptors (mGluRs) modulate transmission of gustatory inputs in the brain stem. J. Neurophysiol. 2009, 102, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.Z.; Johnson, S.W. Group II metabotropic glutamate receptor modulation of excitatory transmission in rat subthalamic nucleus. J. Physiol. 2003, 553, 489–496. [Google Scholar] [CrossRef]
- Pałucha, A.; Tatarczyńska, E.; Brański, P.; Szewczyk, B.; Wierońska, J.; Kłak, K.; Chojnacka-Wojcik, E.; Nowak, G.; Pilc, A. Group III mGlu receptor agonists produce anxiolytic-and antidepressant-like effects after central administration in rats. Neuropharmacology 2004, 46, 151–159. [Google Scholar] [CrossRef]
- Podkowa, K.; Rzeźniczek, S.; Marciniak, M.; Acher, F.; Pilc, A.; Pałucha-Poniewiera, A. A novel mGlu4 selective agonist LSP4-2022 increases behavioral despair in mouse models of antidepressant action. Neuropharmacology 2015, 97, 338–345. [Google Scholar] [CrossRef]
- Kalinichev, M.; Le Poul, E.; Boléa, C.; Girard, F.; Campo, B.; Fonsi, M.; Royer-Urios, I.; Browne, S.E.; Uslaner, J.M.; Davis, M.J. Characterization of the novel positive allosteric modulator of the metabotropic glutamate receptor 4 ADX88178 in rodent models of neuropsychiatric disorders. J. Pharmacol. Exp. Ther. 2014, 350, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, K.A.; Soleimani, L.; Murrough, J.W. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr. Dis. Treat. 2013, 9, 1101. [Google Scholar] [PubMed]
- Gogliotti, R.G.; Senter, R.K.; Fisher, N.M.; Adams, J.; Zamorano, R.; Walker, A.G.; Blobaum, A.L.; Engers, D.W.; Hopkins, C.R.; Daniels, J.S. mGlu7 potentiation rescues cognitive, social, and respiratory phenotypes in a mouse model of Rett syndrome. Sci. Transl. Med. 2017, 9, eaai7459. [Google Scholar] [CrossRef] [PubMed]
- Fisher, N.M.; Gould, R.W.; Gogliotti, R.G.; McDonald, A.J.; Badivuku, H.; Chennareddy, S.; Buch, A.B.; Moore, A.M.; Jenkins, M.T.; Robb, W.H. Phenotypic profiling of mGlu7 knockout mice reveals new implications for neurodevelopmental disorders. Genes Brain Behav. 2020, 19, e12654. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.R.; Uslaner, J.M.; Flick, R.B.; Lee, A.; Groover, K.M.; Hutson, P.H. The mGluR7 allosteric agonist AMN082 produces antidepressant-like effects by modulating glutamatergic signaling. Pharmacol. Biochem. Behav. 2012, 101, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Pałucha-Poniewiera, A.; Pilc, A. A selective mGlu7 receptor antagonist MMPIP reversed antidepressant-like effects of AMN082 in rats. Behav. Brain Res. 2013, 238, 109–112. [Google Scholar] [CrossRef]
- Hovelso, N.; Sotty, F.; P Montezinho, L.; S Pinheiro, P.; F Herrik, K.; Mork, A. Therapeutic potential of metabotropic glutamate receptor modulators. Curr. Neuropharmacol. 2012, 10, 12–48. [Google Scholar] [CrossRef]
- Jantas, D.; Lech, T.; Gołda, S.; Pilc, A.; Lasoń, W. New evidences for a role of mGluR7 in astrocyte survival: Possible implications for neuroprotection. Neuropharmacology 2018, 141, 223–237. [Google Scholar] [CrossRef]
- Li, W.; Ju, K.; Li, Z.; He, K.; Chen, J.; Wang, Q.; Yang, B.; An, L.; Feng, G.; Sun, W. Significant association of GRM7 and GRM8 genes with schizophrenia and major depressive disorder in the Han Chinese population. Eur. Neuropsychopharmacol. 2016, 26, 136–146. [Google Scholar] [CrossRef]
- Gosnell, H.B.; Silberman, Y.; Grueter, B.A.; Duvoisin, R.M.; Raber, J.; Winder, D.G. mGluR8 modulates excitatory transmission in the bed nucleus of the stria terminalis in a stress-dependent manner. Neuropsychopharmacology 2011, 36, 1599–1607. [Google Scholar] [CrossRef]
- Boyko, M.; Gruenbaum, S.E.; Gruenbaum, B.F.; Shapira, Y.; Zlotnik, A. Brain to blood glutamate scavenging as a novel therapeutic modality: A review. J. Neural Transm. 2014, 121, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Kuts, R.; Tsenter, P.; Gruenbaum, B.F.; Grinshpun, Y.; Zvenigorodsky, V.; Shelef, I.; Natanel, D.; Brotfain, E.; Zlotnik, A. The effect of pyruvate on the development and progression of post-stroke depression: A new therapeutic approach. Neuropharmacology 2019, 155, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Moro, N.; Ghavim, S.S.; Harris, N.G.; Hovda, D.A.; Sutton, R.L. Pyruvate treatment attenuates cerebral metabolic depression and neuronal loss after experimental traumatic brain injury. Brain Res. 2016, 1642, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Goldshmit, Y.; Banyas, E.; Bens, N.; Yakovchuk, A.; Ruban, A. Blood glutamate scavengers and exercises as an effective neuroprotective treatment in mice with spinal cord injury. J. Neurosurg. Spine 2020, 33, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Arabaci, O.; Acari, A.; Ciftci, P.; Gozuacik, D. Glutamate Scavenging as a Neuroreparative Strategy in Ischemic Stroke. Front. Pharmacol. 2022, 13, 866738. [Google Scholar] [CrossRef]
- Ma, W.; Song, J.; Wang, H.; Shi, F.; Zhou, N.; Jiang, J.; Xu, Y.; Zhang, L.; Yang, L.; Zhou, M. Chronic paradoxical sleep deprivation-induced depressionlike behavior, energy metabolism and microbial changes in rats. Life Sci. 2019, 225, 88–97. [Google Scholar] [CrossRef]
- Lubberts, E.; Scher, J.U.; FitzGerald, O. Basic Science Session 2. Recent Advances in Our Understanding of Psoriatic Arthritis Pathogenesis. J. Rheumatol. 2022, 49, 16–19. [Google Scholar] [CrossRef]
- Mallone, F.; Lucchino, L.; Franzone, F.; Marenco, M.; Carlesimo, S.C.; Moramarco, A. High-dose vitamin B supplementation for persistent visual deficit in multiple sclerosis: A pilot study. Drug Discov. Ther. 2020, 14, 122–128. [Google Scholar] [CrossRef]
- Holm, K.J.; Markham, A. Mirtazapine. Drugs 1999, 57, 607–631. [Google Scholar] [CrossRef]
- Alam, A.; Voronovich, Z.; Carley, J.A. A review of therapeutic uses of mirtazapine in psychiatric and medical conditions. Prim. Care Companion CNS Disord. 2013, 15, 27255. [Google Scholar] [CrossRef]
- Niedermaier, N.; Bohrer, E.; Schulte, K.; Schlattmann, P.; Heuser, I. Prevention and treatment of poststroke depression with mirtazapine in patients with acute stroke. J. Clin. Psychiatry 2004, 65, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Gahr, M. Agomelatine in the treatment of major depressive disorder: An assessment of benefits and risks. Curr. Neuropharmacol. 2014, 12, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Bogolepova, A.; Chukanova, E.; MIu, S.; Chukanova, A.; IIu, G.; Semushkina, E. The use of valdoxan in the treatment of post-stroke depression. Zh. Nevrol. Psikhiatr. Im. S.S. Korsakova 2011, 111, 42–46. [Google Scholar] [CrossRef]
- Hardy, S.E. Methylphenidate for the treatment of depressive symptoms, including fatigue and apathy, in medically ill older adults and terminally ill adults. Am. J. Geriatr. Pharmacother. 2009, 7, 34–59. [Google Scholar] [CrossRef] [PubMed]
- Grade, C.; Redford, B.; Chrostowski, J.; Toussaint, L.; Blackwell, B. Methylphenidate in early poststroke recovery: A double-blind, placebo-controlled study. Arch. Phys. Med. Rehabil. 1998, 79, 1047–1050. [Google Scholar] [CrossRef]
- Urban, K.R.; Gao, W.-J. Psychostimulants as cognitive enhancers in adolescents: More risk than reward? Front. Public Health 2017, 5, 260. [Google Scholar] [CrossRef]
- Faraone, S.V. The pharmacology of amphetamine and methylphenidate: Relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci. Biobehav. Rev. 2018, 87, 255–270. [Google Scholar] [CrossRef]
- Cakic, V. Smart drugs for cognitive enhancement: Ethical and pragmatic considerations in the era of cosmetic neurology. J. Med. Ethics 2009, 35, 611–615. [Google Scholar] [CrossRef]
- Froestl, W.; Muhs, A.; Pfeifer, A. Cognitive enhancers (nootropics). Part 1: Drugs interacting with receptors. J. Alzheimer’s Dis. 2012, 32, 793–887. [Google Scholar] [CrossRef]
- Sugden, S.G.; Bourgeois, J.A. Modafinil monotherapy in poststroke depression. Psychosomatics 2004, 45, 80–81. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; González-Falcón, A.; García-Cabrera, M.; Leon, O.S.; Fiebich, B.L. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood–brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J. Neurochem. 2007, 100, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Gorelick, P.B. The Janus face of cyclooxygenase-2 in ischemic stroke: Shifting toward downstream targets. Stroke 2005, 36, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Müller, N. COX-2 inhibitors, aspirin, and other potential anti-inflammatory treatments for psychiatric disorders. Front. Psychiatry 2019, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Akhondzadeh, S.; Jafari, S.; Raisi, F.; Nasehi, A.A.; Ghoreishi, A.; Salehi, B.; Mohebbi-Rasa, S.; Raznahan, M.; Kamalipour, A. Clinical trial of adjunctive celecoxib treatment in patients with major depression: A double blind and placebo controlled trial. Depress. Anxiety 2009, 26, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Majd, M.; Hashemian, F.; Hosseini, S.M.; Shariatpanahi, M.V.; Sharifi, A. A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in treatment of drug-naive depressed women: A pilot study. Iran. J. Pharm. Res. IJPR 2015, 14, 891. [Google Scholar]
- Perrone, M.G.; Centonze, A.; Miciaccia, M.; Ferorelli, S.; Scilimati, A. Cyclooxygenase inhibition safety and efficacy in inflammation-based psychiatric disorders. Molecules 2020, 25, 5388. [Google Scholar] [CrossRef]
- Stachowicz, K. Deciphering the mechanisms of regulation of an excitatory synapse via cyclooxygenase-2. A review. Biochem. Pharmacol. 2021, 192, 114729. [Google Scholar] [CrossRef]
- Wium-Andersen, I.K.; Wium-Andersen, M.K.; Jørgensen, M.B.; Osler, M. Anti-inflammatory treatment and risk for depression after first-time stroke in a cohort of 147 487 Danish patients. J. Psychiatry Neurosci. 2017, 42, 320–330. [Google Scholar] [CrossRef]
- Strauss, K.I. COX2 inhibitors for acquired brain injuries: Is the time ripe? Crit. Care Med. 2010, 38, 723. [Google Scholar] [CrossRef]
- Strekalova, T.; Pavlov, D.; Trofimov, A.; Anthony, D.C.; Svistunov, A.; Proshin, A.; Umriukhin, A.; Lyundup, A.; Lesch, K.-P.; Cespuglio, R. Hippocampal Over-Expression of Cyclooxygenase-2 (COX-2) Is Associated with Susceptibility to Stress-Induced Anhedonia in Mice. Int. J. Mol. Sci. 2022, 23, 2061. [Google Scholar] [CrossRef]
- Stachowicz, K.; Sowa-Kućma, M.; Pańczyszyn-Trzewik, P.; Misztak, P.; Marciniak, M.; Bobula, B.; Tokarski, K. Behavioral consequences of co-administration of MTEP and the COX-2 inhibitor NS398 in mice. Part 2. Neurosci. Lett. 2021, 741, 135435. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Wong, D.; McKay, A.; Rajaratnam, S.M.; Spitz, G.; Williams, G.; Mansfield, D.; Ponsford, J.L. Cognitive behavioural therapy for post-stroke fatigue and sleep disturbance: A pilot randomised controlled trial with blind assessment. Neuropsychol. Rehabil. 2019, 29, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Lin, M.; Zhao, J.; Bi, S.; Ni, Z.; Shang, X. Different interventions for post-ischaemic stroke depression in different time periods: A single-blind randomized controlled trial with stratification by time after stroke. Clin. Rehabil. 2017, 31, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Kootker, J.A.; Rasquin, S.M.; Lem, F.C.; van Heugten, C.M.; Fasotti, L.; Geurts, A.C. Augmented cognitive behavioral therapy for poststroke depressive symptoms: A randomized controlled trial. Arch. Phys. Med. Rehabil. 2017, 98, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Ma, W.; Wang, G.-T.; Shen, W.-D. Efficacy and safety of electroacupuncture for post stroke depression: Study protocol for a randomized controlled trial. Trials 2018, 19, 152. [Google Scholar] [CrossRef] [PubMed]
- Semkovska, M.; Landau, S.; Dunne, R.; Kolshus, E.; Kavanagh, A.; Jelovac, A.; Noone, M.; Carton, M.; Lambe, S.; McHugh, C. Bitemporal versus high-dose unilateral twice-weekly electroconvulsive therapy for depression (EFFECT-Dep): A pragmatic, randomized, non-inferiority trial. Am. J. Psychiatry 2016, 173, 408–417. [Google Scholar] [CrossRef]
- Van Diermen, L.; Van Den Ameele, S.; Kamperman, A.M.; Sabbe, B.C.; Vermeulen, T.; Schrijvers, D.; Birkenhäger, T.K. Prediction of electroconvulsive therapy response and remission in major depression: Meta-analysis. Br. J. Psychiatry 2018, 212, 71–80. [Google Scholar] [CrossRef]
- Marchina, S.; Pisegna, J.M.; Massaro, J.M.; Langmore, S.E.; McVey, C.; Wang, J.; Kumar, S. Transcranial direct current stimulation for post-stroke dysphagia: A systematic review and meta-analysis of randomized controlled trials. J. Neurol. 2021, 268, 293–304. [Google Scholar] [CrossRef]
- Bornheim, S.; Croisier, J.-L.; Maquet, P.; Kaux, J.-F. Transcranial direct current stimulation associated with physical-therapy in acute stroke patients-A randomized, triple blind, sham-controlled study. Brain Stimul. 2020, 13, 329–336. [Google Scholar] [CrossRef]
- Valiengo, L.C.; Goulart, A.C.; de Oliveira, J.F.; Benseñor, I.M.; Lotufo, P.A.; Brunoni, A.R. Transcranial direct current stimulation for the treatment of post-stroke depression: Results from a randomised, sham-controlled, double-blinded trial. J. Neurol. Neurosurg. Psychiatry 2017, 88, 170–175. [Google Scholar] [CrossRef]
- Shen, X.; Liu, M.; Cheng, Y.; Jia, C.; Pan, X.; Gou, Q.; Liu, X.; Cao, H.; Zhang, L. Repetitive transcranial magnetic stimulation for the treatment of post-stroke depression: A systematic review and meta-analysis of randomized controlled clinical trials. J. Affect. Disord. 2017, 211, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Hordacre, B.; Comacchio, K.; Williams, L.; Hillier, S. Repetitive transcranial magnetic stimulation for post-stroke depression: A randomised trial with neurophysiological insight. J. Neurol. 2021, 268, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Zhao, Z.; Zhang, Y.; Zhou, X.; Zhao, L.; Dong, M.; Xu, F.; Xiang, Y.; Luo, H. Efficacy of repetitive transcranial magnetic stimulation for post-stroke depression: A systematic review and meta-analysis of randomized clinical trials. Braz. J. Med. Biol. Res. 2021, 54, e10010. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, J.M.; LeReun, C.; Diamantopoulos, A.; Mitchell, S.; Gaynes, B.N. Vagus nerve stimulation (VNS) therapy in patients with treatment resistant depression: A systematic review and meta-analysis. Compr. Psychiatry 2019, 98, 152156. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.L.; Robinson, R.G.; Raphael, B.; Bishop, D. The relationship between the perception of social support and post-stroke depression in hospitalized patients. Psychiatry 1991, 54, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Woranush, W.; Moskopp, M.L.; Sedghi, A.; Stuckart, I.; Noll, T.; Barlinn, K.; Siepmann, T. Preventive Approaches for Post-Stroke Depression: Where Do We Stand? A Systematic Review. Neuropsychiatr. Dis. Treat. 2021, 17, 3359–3377. [Google Scholar] [CrossRef]
- Wang, S.M.; Han, C.; Bahk, W.M.; Lee, S.J.; Patkar, A.A.; Masand, P.S.; Pae, C.U. Addressing the Side Effects of Contemporary Antidepressant Drugs: A Comprehensive Review. Chonnam Med. J. 2018, 54, 101–112. [Google Scholar] [CrossRef]





| Antidepressants | Advantages | Disadvantages |
|---|---|---|
| SSRIs | GAD, fibromyalgia | Sexual dysfunction, nausea, vomiting, insomnia, serotonin syndrome, HTN |
| SNRIs | GAD, fibromyalgia | Sexual side effects, insomnia, nausea, vomiting, HTN, serotonin syndrome |
| NDRIs | Alcohol, smoking cessation, no sexual dysfunction, no weight gain | Anxiety, suicidal ideation, seizures, general side effects |
| MAOIs | Effectiveness for atypical depression, PTSD | Low sex drive, weight gain, high or low blood pressure, keep off during alcoholism, kidney and heart disease, food restriction |
| TCA | Proven efficacy, low cost | Urinary retention, xerostomia, tachycardia, VF, SCD |
| Ketamine | Severe, treatment-resistant depression, increasing libido | Numbness, tingling, dizziness, transient cognitive deficits, and increasing blood pressure |
| Memantine | Moderate and severe form depression, highly tolerated by many patients, improving cognition and general quality of life | Constipation, nausea, weight gain, diarrhea, confusion, sleeplessness, shortness of breath, and hallucination |
| D-cycloserine | Well tolerated, do not produce psychotomimetic effects | Dizziness, hyperexcitability, anxiety, memory loss, and gastrointestinal problems |
| Magnesium | Anxiety, anticonvulsive effect | Muscle weakness, lethargy, nausea, diarrhea, and a fall in blood pressure |
| Riluzole | Anticonvulsive effect | Fatigue, nausea, and weight loss |
| Mirtazapine | Can be used as a preventive treatment for PSD | Increased appetite, weight gain, headache, nausea or vomiting, diarrhea, and constipation |
| Agomelatine | Short-term and long-term efficacy in treating PSD | Hepatic impairment, anxiety, nausea and vomiting, stomach pain, insomnia, dizziness, fatigue, and an increase in weight |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, D.; Gruenbaum, B.F.; Zlotnik, A.; Semyonov, M.; Frenkel, A.; Boyko, M. Pathophysiology and Current Drug Treatments for Post-Stroke Depression: A Review. Int. J. Mol. Sci. 2022, 23, 15114. https://doi.org/10.3390/ijms232315114
Frank D, Gruenbaum BF, Zlotnik A, Semyonov M, Frenkel A, Boyko M. Pathophysiology and Current Drug Treatments for Post-Stroke Depression: A Review. International Journal of Molecular Sciences. 2022; 23(23):15114. https://doi.org/10.3390/ijms232315114
Chicago/Turabian StyleFrank, Dmitry, Benjamin F. Gruenbaum, Alexander Zlotnik, Michael Semyonov, Amit Frenkel, and Matthew Boyko. 2022. "Pathophysiology and Current Drug Treatments for Post-Stroke Depression: A Review" International Journal of Molecular Sciences 23, no. 23: 15114. https://doi.org/10.3390/ijms232315114
APA StyleFrank, D., Gruenbaum, B. F., Zlotnik, A., Semyonov, M., Frenkel, A., & Boyko, M. (2022). Pathophysiology and Current Drug Treatments for Post-Stroke Depression: A Review. International Journal of Molecular Sciences, 23(23), 15114. https://doi.org/10.3390/ijms232315114