
The Role of Aldose Reductase in Beta-Amyloid-Induced Microglia Activation
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
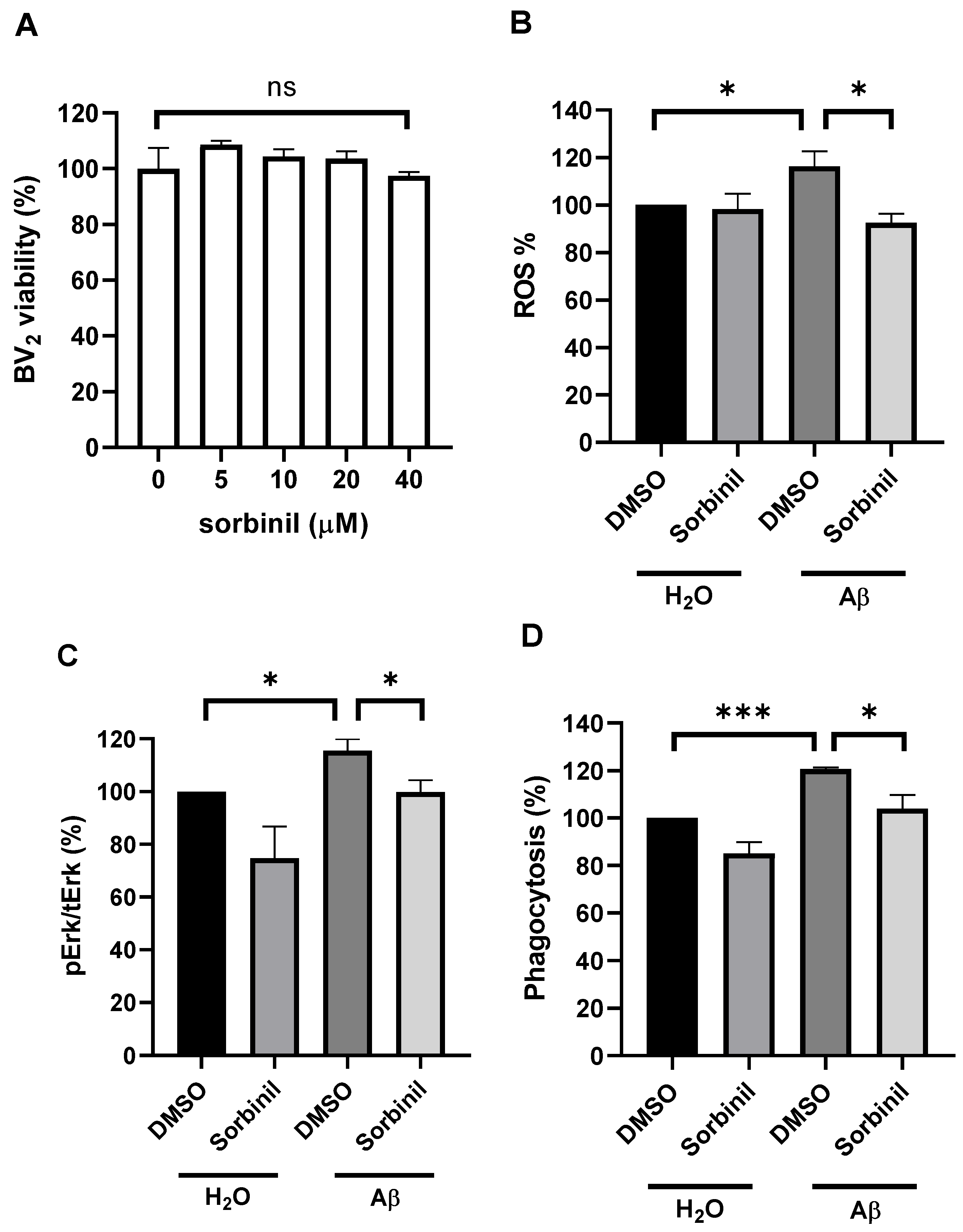
2.1. AR Inhibition Alleviates β-Amyloid-Induced TNF-α Secretion in Microglia
2.2. AR Inhibition Decreases β-Amyloid-Induced ROS Production
2.3. AR Inhibition Attenuates β-Amyloid-Induced BV2 Cell Migration
2.4. AR Inhibition in BV2 Cells Protects Neurons from Death under β-Amyloid Exposure
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Aβ 1-42 Preparation
4.3. Co-Culture Assay
4.4. ROS Assay
4.5. ELISA Assay
4.6. Real-Time qPCR (RT-qPCR)
4.7. Western Blot
4.8. Migration Assay
4.9. Phagocytosis Assay
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- 2022 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2022, 18, 700–789. [CrossRef] [PubMed]
- Pfundstein, G.; Nikonenko, A.G.; Sytnyk, V. Amyloid precursor protein (APP) and amyloid beta (Abeta) interact with cell adhesion molecules: Implications in Alzheimer’s disease and normal physiology. Front. Cell Dev. Biol. 2022, 10, 969547. [Google Scholar] [CrossRef]
- Song, M. The Asparaginyl Endopeptidase Legumain: An Emerging Therapeutic Target and Potential Biomarker for Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 223. [Google Scholar] [CrossRef] [PubMed]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Chiba, K. Role of microglial m1/m2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 2014, 7, 1028–1048. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Petrash, J.M. All in the family: Aldose reductase and closely related aldo-keto reductases. Cell Mol. Life Sci. 2004, 61, 737–749. [Google Scholar] [CrossRef]
- Chang, K.C.; Petrash, J.M. Aldo-Keto Reductases: Multifunctional Proteins as Therapeutic Targets in Diabetes and Inflammatory Disease. Adv. Exp. Med. Biol. 2018, 1032, 173–202. [Google Scholar] [CrossRef]
- Fichtner, J.E.; Patnaik, J.; Christopher, K.L.; Petrash, J.M. Cataract inhibitors: Present needs and future challenges. Chem. Biol. Interact. 2021, 349, 109679. [Google Scholar] [CrossRef]
- Hupy, M.L.; Pedler, M.G.; Shieh, B.; Wang, D.; Wang, X.J.; Petrash, J.M. Suppression of epithelial to mesenchymal transition markers in mouse lens by a Smad7-based recombinant protein. Chem. Biol. Interact. 2021, 344, 109495. [Google Scholar] [CrossRef]
- Chang, K.C.; Shieh, B.; Petrash, J.M. Influence of aldose reductase on epithelial-to-mesenchymal transition signaling in lens epithelial cells. Chem. Biol. Interact. 2017, 276, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Petrash, J.M. Aldose Reductase Mediates Transforming Growth Factor beta2 (TGF-beta2)-Induced Migration and Epithelial-To-Mesenchymal Transition of Lens-Derived Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4198–4210. [Google Scholar] [CrossRef] [PubMed]
- Jannapureddy, S.; Sharma, M.; Yepuri, G.; Schmidt, A.M.; Ramasamy, R. Aldose Reductase: An Emerging Target for Development of Interventions for Diabetic Cardiovascular Complications. Front. Endocrinol. 2021, 12, 636267. [Google Scholar] [CrossRef]
- Samuels, I.S.; Lee, C.A.; Petrash, J.M.; Peachey, N.S.; Kern, T.S. Exclusion of aldose reductase as a mediator of ERG deficits in a mouse model of diabetic eye disease. Vis. Neurosci. 2012, 29, 267–274. [Google Scholar] [CrossRef]
- Yeung, C.M.; Lo, A.C.; Cheung, A.K.; Chung, S.S.; Wong, D.; Chung, S.K. More severe type 2 diabetes-associated ischemic stroke injury is alleviated in aldose reductase-deficient mice. J. Neurosci. Res. 2010, 88, 2026–2034. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Shieh, B.; Petrash, J.M. Aldose reductase mediates retinal microglia activation. Biochem. Biophys. Res. Commun. 2016, 473, 565–571. [Google Scholar] [CrossRef]
- Petrash, J.M.; Shieh, B.; Ammar, D.A.; Pedler, M.G.; Orlicky, D.J. Diabetes-Independent Retinal Phenotypes in an Aldose Reductase Transgenic Mouse Model. Metabolites 2021, 11, 450. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Rodriguez, J.; Giambartolomei, G.H. Microglia at the Crossroads of Pathogen-Induced Neuroinflammation. ASN Neuro. 2022, 14, 17590914221104566. [Google Scholar] [CrossRef]
- Tanimura, S.; Takeda, K. ERK signalling as a regulator of cell motility. J. Biochem. 2017, 162, 145–154. [Google Scholar] [CrossRef]
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1704–1721. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Laffin, B.; Ponder, J.; Enzsoly, A.; Nemeth, J.; LaBarbera, D.V.; Petrash, J.M. Beta-glucogallin reduces the expression of lipopolysaccharide-induced inflammatory markers by inhibition of aldose reductase in murine macrophages and ocular tissues. Chem. Biol. Interact. 2013, 202, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Grewal, A.S.; Bhardwaj, S.; Pandita, D.; Lather, V.; Sekhon, B.S. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-diabetic Diseases. Mini. Rev. Med. Chem. 2016, 16, 120–162. [Google Scholar] [CrossRef] [PubMed]
- Song, X.M.; Yu, Q.; Dong, X.; Yang, H.O.; Zeng, K.W.; Li, J.; Tu, P.F. Aldose reductase inhibitors attenuate beta-amyloid-induced TNF-alpha production in microlgia via ROS-PKC-mediated NF-kappaB and MAPK pathways. Int. Immunopharmacol. 2017, 50, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Srivastava, S.K. Aldose reductase: A novel therapeutic target for inflammatory pathologies. Int. J. Biochem. Cell. Biol. 2010, 42, 17–20. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Yadav, U.C.S.; Reddy, A.B.M.; Saxena, A.; Tammali, R.; Shoeb, M.; Ansari, N.H.; Bhatnagar, A.; Petrash, M.J.; Srivastava, S.; et al. Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chemico-Biological. Interact. 2011, 191, 330–338. [Google Scholar] [CrossRef]
- Srivastava, S.; Dixit, B.L.; Cai, J.; Sharma, S.; Hurst, H.E.; Bhatnagar, A.; Srivastava, S.K. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: Role of aldose reductase. Free Radic. Biol. Med. 2000, 29, 642–651. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- de Souza, A.P.; Vale, V.L.; Silva Mda, C.; Araujo, I.B.; Trindade, S.C.; de Moura-Costa, L.F.; Rodrigues, G.C.; Sales, T.S.; dos Santos, H.A.; de Carvalho-Filho, P.C.; et al. MAPK involvement in cytokine production in response to Corynebacterium pseudotuberculosis infection. BMC Microbiol. 2014, 14, 230. [Google Scholar] [CrossRef]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, Z.; Wang, D.; Wang, Z.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15. [Google Scholar] [CrossRef]
- Chang, K.C.; Ponder, J.; Labarbera, D.V.; Petrash, J.M. Aldose reductase inhibition prevents endotoxin-induced inflammatory responses in retinal microglia. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2853–2861. [Google Scholar] [CrossRef]
- Ramana, K.V.; Reddy, A.B.M.; Tammali, R.; Srivastava, S.K. Aldose reductase mediates endotoxin-induced production of nitric oxide and cytotoxicity in murine macrophages. Free Radic. Biol. Med. 2007, 42, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Elmazoglu, Z.; Prnova, M.S.; Stefek, M.; Ceylan, A.F.; Aschner, M.; Rangel-Lopez, E.; Santamaria, A.; Karasu, C. Protective Effects of Novel Substituted Triazinoindole Inhibitors of Aldose Reductase and Epalrestat in Neuron-like PC12 Cells and BV2 Rodent Microglial Cells Exposed to Toxic Models of Oxidative Stress: Comparison with the Pyridoindole Antioxidant Stobadine. Neurotox. Res. 2021, 39, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Han, F.X.; Zhang, R.; Yang, X.X.; Ma, S.B.; Hu, S.J.; Li, B. Neuroprotective effect of aldose reductase knockout in a mouse model of spinal cord injury involves NF-kappaB pathway. Exp. Brain Res. 2022, 240, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.W.; Li, J.; Dong, X.; Wang, Y.H.; Ma, Z.Z.; Jiang, Y.; Jin, H.W.; Tu, P.F. Anti-neuroinflammatory efficacy of the aldose reductase inhibitor FMHM via phospholipase C/protein kinase C-dependent NF-kappaB and MAPK pathways. Toxicol. Appl. Pharmacol. 2013, 273, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, D.; Li, X.; Jiang, Y.; Wang, C.; Zhang, Y.; Kong, Q.; Tian, C.; Dai, Y.; Zhao, W.; et al. Excess salt intake promotes M1 microglia polarization via a p38/MAPK/AR-dependent pathway after cerebral ischemia in mice. Int. Immunopharmacol. 2020, 81, 106176. [Google Scholar] [CrossRef]
- Chang, K.C.; Snow, A.; LaBarbera, D.V.; Petrash, J.M. Aldose reductase inhibition alleviates hyperglycemic effects on human retinal pigment epithelial cells. Chem. Biol. Interact. 2015, 234, 254–260. [Google Scholar] [CrossRef]
- Zhang, K.; Lu, W.C.; Zhang, M.; Zhang, Q.; Xian, P.P.; Liu, F.F.; Chen, Z.Y.; Kim, C.S.; Wu, S.X.; Tao, H.R.; et al. Reducing host aldose reductase activity promotes neuronal differentiation of transplanted neural stem cells at spinal cord injury sites and facilitates locomotion recovery. Neural. Regen. Res. 2022, 17, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bian, G.; Chen, P.; Liu, L.; Yu, C.; Liu, F.; Xue, Q.; Chung, S.K.; Song, B.; Ju, G.; et al. Aldose Reductase Regulates Microglia/Macrophages Polarization Through the cAMP Response Element-Binding Protein After Spinal Cord Injury in Mice. Mol. Neurobiol. 2016, 53, 662–676. [Google Scholar] [CrossRef]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezo, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia contribute to the propagation of Abeta into unaffected brain tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Shieh, B.; Petrash, J.M. Role of aldose reductase in diabetes-induced retinal microglia activation. Chem. Biol. Interact. 2019, 302, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yuan, S.; Li, Y.; Zhang, Z.; Xiao, W.; Tang, D.; Ye, K.; Liu, Z.; Wang, C.; Zheng, Y.; et al. Regulation on SIRT1-PGC-1alpha/Nrf2 pathway together with selective inhibition of aldose reductase makes compound hr5F a potential agent for the treatment of diabetic complications. Biochem. Pharmacol. 2018, 150, 54–63. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Brown, G.C. Primary phagocytosis of neurons by inflamed microglia: Potential roles in neurodegeneration. Front. Pharmacol. 2012, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef]
- Mori, K.; Uchida, T.; Yoshie, T.; Mizote, Y.; Ishikawa, F.; Katsuyama, M.; Shibanuma, M. A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS-activated cancer cells. FEBS J. 2019, 286, 459–478. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Reis, S.A.; Adams, E.T.; Fass, D.M.; Angus, S.P.; Stuhlmiller, T.J.; Richardson, J.; Olafson, H.; Wang, E.T.; Patnaik, D.; et al. High-content image-based analysis and proteomic profiling identifies Tau phosphorylation inhibitors in a human iPSC-derived glutamatergic neuronal model of tauopathy. Sci. Rep. 2021, 11, 17029. [Google Scholar] [CrossRef] [PubMed]
- Bolos, M.; Hu, Y.; Young, K.M.; Foa, L.; Small, D.H. Neurogenin 2 mediates amyloid-beta precursor protein-stimulated neurogenesis. J. Biol. Chem. 2014, 289, 31253–31261. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Nabet, A.M.; Wernig, M.; Sudhof, T.C. Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer’s Disease Risk. J. Neurosci. 2019, 39, 7408–7427. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Gomez, M.A.; Llorens-Alvarez, E.; Alom, J.; Del Ser, T.; Avila, J.; Saez-Valero, J.; Garcia-Ayllon, M.S. Tau phosphorylation by glycogen synthase kinase 3beta modulates enzyme acetylcholinesterase expression. J. Neurochem. 2021, 157, 2091–2105. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Suzen, S.; Buyukbingol, E. Recent studies of aldose reductase enzyme inhibition for diabetic complications. Curr. Med. Chem. 2003, 10, 1329–1352. [Google Scholar] [CrossRef]
- Chalk, C.; Benstead, T.J.; Moore, F. Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. Cochrane Database Syst. Rev. 2007, 2007, CD004572. [Google Scholar] [CrossRef]
- Gabbay, K.H. Aldose reductase inhibition in the treatment of diabetic neuropathy: Where are we in 2004? Curr. Diab. Rep. 2004, 4, 405–408. [Google Scholar] [CrossRef]
- Prnova, M.S.; Rackova, L.; Kovacikova, L.; Ballekova, J.; Viskupicova, J.; Michalikova, S.; Taskoparan, B.; Elmazoglu, Z.; Rizner, T.L.; Karasu, C.; et al. General toxicity assessment of the novel aldose reductase inhibitor cemtirestat. Interdiscip. Toxicol. 2019, 12, 120–128. [Google Scholar] [CrossRef]
- Alford, M.A.; Tian, Z.; Menard, F.; Klegeris, A. Characterization of novel kainic acid analogs as inhibitors of select microglial functions. Eur. J. Pharmacol. 2019, 851, 25–35. [Google Scholar] [CrossRef]
- Ramana, K.V.; Friedrich, B.; Tammali, R.; West, M.B.; Bhatnagar, A.; Srivastava, S.K. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes 2005, 54, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, K.; Ueda, H.; Moriyasu, M. Aldose reductase inhibitors from the nature. Curr. Med. Chem. 2003, 10, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A Cell Surface Receptor Complex for Fibrillar β-Amyloid Mediates Microglial Activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-K.; Liu, C.-C.; Wang, S.; Cheng, H.-C.; Meadows, C.; Chang, K.-C. The Role of Aldose Reductase in Beta-Amyloid-Induced Microglia Activation. Int. J. Mol. Sci. 2022, 23, 15088. https://doi.org/10.3390/ijms232315088
Huang Y-K, Liu C-C, Wang S, Cheng H-C, Meadows C, Chang K-C. The Role of Aldose Reductase in Beta-Amyloid-Induced Microglia Activation. International Journal of Molecular Sciences. 2022; 23(23):15088. https://doi.org/10.3390/ijms232315088
Chicago/Turabian StyleHuang, Yu-Kai, Chia-Chun Liu, Shining Wang, Hui-Chun Cheng, Chandler Meadows, and Kun-Che Chang. 2022. "The Role of Aldose Reductase in Beta-Amyloid-Induced Microglia Activation" International Journal of Molecular Sciences 23, no. 23: 15088. https://doi.org/10.3390/ijms232315088
APA StyleHuang, Y.-K., Liu, C.-C., Wang, S., Cheng, H.-C., Meadows, C., & Chang, K.-C. (2022). The Role of Aldose Reductase in Beta-Amyloid-Induced Microglia Activation. International Journal of Molecular Sciences, 23(23), 15088. https://doi.org/10.3390/ijms232315088