

Revisiting AGAMOUS-LIKE15, a Key Somatic Embryogenesis Regulator, Using Next Generation Sequencing Analysis in Arabidopsis


Abstract
:1. Introduction
2. Results
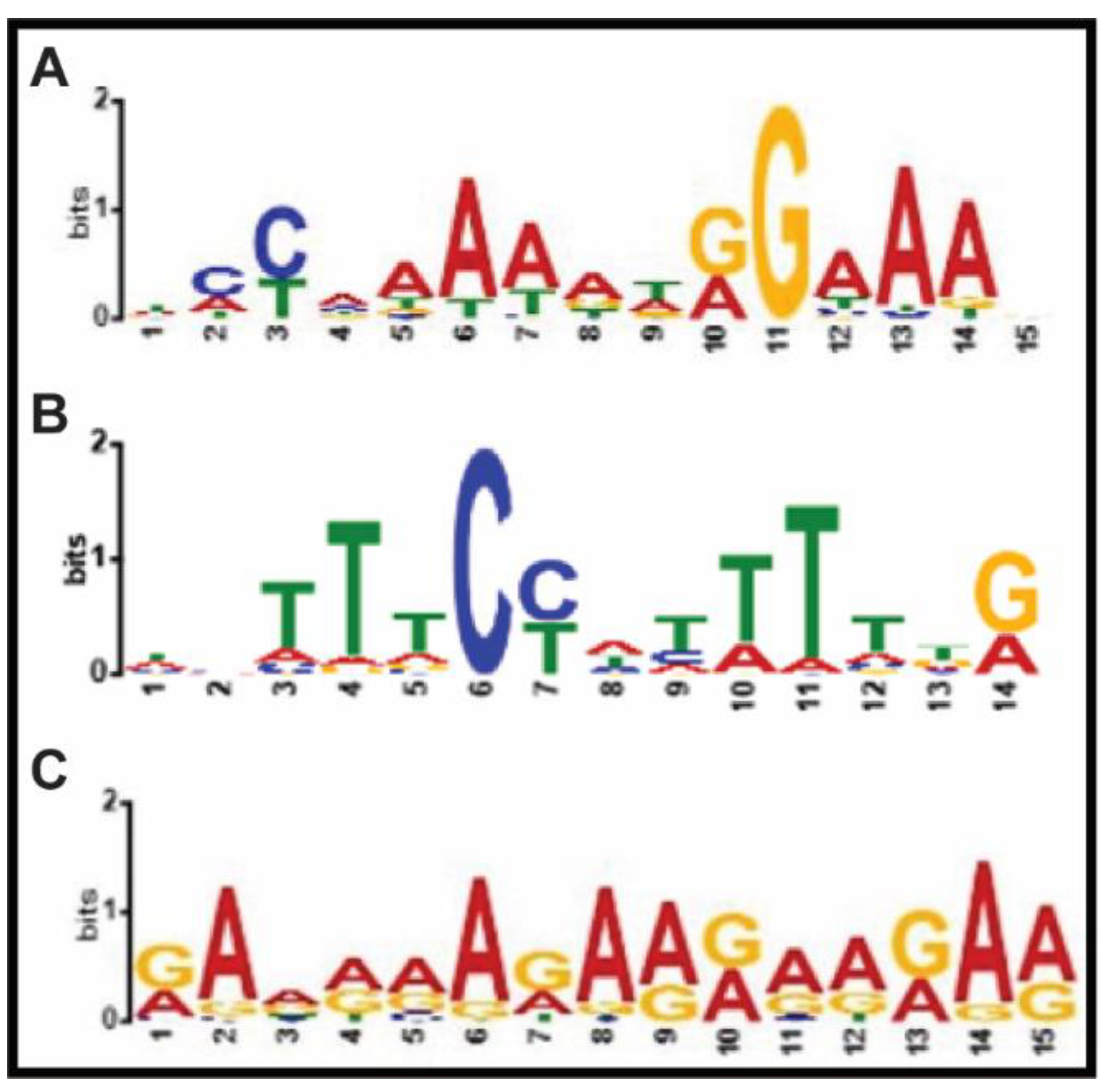
2.1. Genome-Wide Mapping of Regions bound by AGL15 Using Chip-Seq
2.2. Gene Expression Changes in Response to AGL15 Accumulation
2.3. Identification of Putative Direct and Indirect Targets of AGL15 from NGS Data
3. Discussion
3.1. Comparison of ChIP-Chip and ChIP-Seq
3.2. Newly Discovered AGL15 Direct Targets Using NGS
3.3. AGL15 and Hormones
4. Materials and Methods
4.1. Plant Material
4.2. ChIP-Seq, Data Analysis, and qPCR
4.3. RNA-Isolation, RNA-Seq, and qRT-PCR
4.4. Data Accessions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tian, R.; Wang, F.; Zheng, Q.; Niza, V.M.A.G.E.; Downie, A.B.; Perry, S.E. Direct and indirect targets of the Arabidopsis seed transcription factor ABSCISIC ACID INSENSITIVE3. Plant J. 2020, 103, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Harding, E.W.; Tang, W.; Nichols, K.W.; Fernandez, D.E.; Perry, S.E. Expression and maintenance of embryogenic potential is enhanced through constitutive expression of AGAMOUS-Like 15. Plant Physiol. 2003, 133, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakare, D.; Tang, W.; Hill, K.; Perry, S.E. The MADS-domain transcriptional regulator AGAMOUS-LIKE15 promotes somatic embryo development in Arabidopsis and soybean. Plant Physiol. 2008, 146, 1663–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Li, C.; Wang, Y.; Zhang, C.; Wu, Z.; Zhang, X.; Liu, C.; Li, F. GhAGL15s, preferentially expressed during somatic embryogenesis, promote embryogenic callus formation in cotton (Gossypium hirsutum L.). Mol. Gen. Genom. 2014, 289, 873–883. [Google Scholar] [CrossRef]
- Zheng, Y.; Ren, N.; Wang, H.; Stromberg, A.J.; Perry, S.E. Global identification of targets of the Arabidopsis MADS domain protein AGAMOUS-Like15. Plant Cell 2009, 21, 2563–2577. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Perry, S.E. Identification of direct targets of FUSCA3, a key regulator of Arabidopsis seed development. Plant Physiol. 2013, 161, 1251–1264. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.; Joshi, S.; Tian, R.; Diogo Junior, R.; Chakrabarti, M.; Perry, S.E. The MADS-domain factor AGAMOUS-Like18 promotes somatic embryogenesis. Plant Physiol. 2021, 188, 1617–1631. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, F.; Dyer, N.P.; Wong, W.H. CisGenome Browser: A flexible tool for genomic data visualization. Bioinformatics 2010, 26, 1781–1782. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nuc. Acids Res. 2020, 49, D394–D403. [Google Scholar] [CrossRef]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2011, 27, 1696–1697. [Google Scholar] [CrossRef]
- Tang, W.; Perry, S.E. Binding site selection for the plant MADS domain protein AGL15: An in vitro and in vivo study. J. Biol. Chem. 2003, 278, 28154–28159. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tang, W.; Zhu, C.; Perry, S. A chromatin immunoprecipitation (ChIP) approach to isolate genes regulated by AGL15, a MADS-domain protein that preferentially accumulates in embryos. Plant J. 2002, 32, 831–843. [Google Scholar] [CrossRef]
- Ruan, J.; Chen, H.; Zhu, T.; Yu, Y.; Lei, Y.; Yuan, L.; Liu, J.; Wang, Z.-Y.; Kuang, J.-F.; Lu, W.-J. Brassinosteroids repress the seed maturation program during the seed-to-seedling transition. Plant Physiol. 2021, 186, 534–548. [Google Scholar] [CrossRef]
- Kaufmann, K.; Muino, J.M.; Jauregui, R.; Airoldi, C.A.; Smaczniak, C.; Krajewski, P.; Angenent, G.C. Target genes of the MADS transcription factor SEPALLATA3: Integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol. 2009, 7, e1000090. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.; Muiño, J.M.; Østerås, M.; Farinelli, L.; Krajewski, P.; Angenent, G.C. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat. Protoc. 2010, 5, 457–472. [Google Scholar] [CrossRef]
- Ho, J.W.K.; Bishop, E.; Karchenko, P.V.; Nègre, N.; White, K.P.; Park, P.J. ChIP-chip versus ChIP-seq: Lessons for experimental design and data analysis. BMC Genom. 2011, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- Park, P.J. ChIP–seq: Advantages and challenges of a maturing technology. Nat. Rev. Gen. 2009, 10, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Grafi, G.; Chalifa-Caspi, V.; Nagar, T.; Plaschkes, I.; Barak, S.; Ransbotyn, V. Plant response to stress meets dedifferentiation. Planta 2011, 233, 433–438. [Google Scholar] [CrossRef]
- Hill, K.; Wang, H.; Perry, S.E. A transcriptional repression motif in the MADS factor AGL15 is involved in recruitment of histone deacetylase complex components. Plant J. 2008, 53, 172–185. [Google Scholar] [CrossRef]
- Joshi, S.; Keller, C.; Perry, S.E. The EAR Motif in the Arabidopsis MADS Transcription Factor AGAMOUS-Like 15 Is not necessary to promote somatic embryogenesis. Plants 2021, 10, 758. [Google Scholar] [CrossRef]
- Lee, J.; He, K.; Stolc, V.; Lee, H.; Figueroa, P.; Gao, Y.; Tongprasit, W.; Zhao, H.Y.; Lee, I.; Deng, X. Analysis of transcription factor HY5 genomic binding sites revealed its hierarchical role in light regulation of development. Plant Cell 2007, 19, 731–749. [Google Scholar] [CrossRef] [Green Version]
- Wyrick, J.J.; Young, R.A. Deciphering gene expression regulatory networks. Curr. Opin. Gen. Develop. 2002, 12, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.A.; Bailey-Serres, J. Integrative analysis from the epigenome to translatome uncovers patterns of dominant nuclear regulation during transient stress. Plant Cell 2019, 31, 2573–2595. [Google Scholar] [CrossRef] [Green Version]
- Salleh, F.M.; Evans, K.; Goodall, B.; Machin, H.; Mowla, S.B.; Mur, L.A.; Runions, J.; Theodoulou, F.L.; Foyer, C.H.; Rogers, H.J. A novel function for a redox-related LEA protein (SAG21/AtLEA5) in root development and biotic stress responses. Plant Cell Env. 2012, 35, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Gong, W.; Boscá, S.; Tucker, M.; Vaucheret, H.; Laux, T. Dose-dependent AGO1-mediated inhibition of the miRNA165/166 pathway modulates stem cell maintenance in arabidopsis shoot apical meristem. Plant Comm. 2020, 1, 100002. [Google Scholar] [CrossRef]
- Xue, T.; Dai, X.; Wang, R.; Wang, J.; Liu, Z.; Xiang, F. ARGONAUTE10 inhibits in vitro shoot regeneration via repression of miR165/166 in Arabidopsis thaliana. Plant Cell Physiol. 2017, 58, 1789–1800. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Paul, P.; Hartman, J.M.; Perry, S.E. AGL15 Promotion of somatic embryogenesis: Role and molecular mechanism. Front. Plant Sci. 2022, 13, 861556. [Google Scholar] [CrossRef]
- Zheng, Q.; Zheng, Y.; Perry, S.E. AGAMOUS-Like15 promotes somatic embryogenesis in Arabidopsis and Soybean in part by the control of ethylene biosynthesis and response. Plant Physiol. 2013, 161, 2113–2127. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Zheng, Y.; Perry, S.E. Decreased GmAGL15 expression and reduced ethylene synthesis may contribute to reduced somatic embryogenesis in a poorly embryogenic cultivar of Glycine max. Plant Signal. Behavoir 2013, 8, e25422. [Google Scholar] [CrossRef] [Green Version]
- Ceserani, T.; Trofka, A.; Gandotra, N.; Nelson, T. VH1/BRL2 receptor-like kinase interacts with vascular-specific adaptor proteins VIT and VIK to influence leaf venation. Plant J. 2009, 57, 1000–1014. [Google Scholar] [CrossRef]
- Gou, X.P.; Yin, H.J.; He, K.; Du, J.B.; Yi, J.; Xu, S.B.; Lin, H.H.; Clouse, S.D.; Li, J. Genetic evidence for an indispensable role of somatic embryogenesis receptor kinases in brassinosteroid signaling. PLoS Genet. 2012, 8, e1002452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassel, G.W.; Fung, P.; Chow, T.F.F.; Foong, J.A.; Provart, N.J.; Cutler, S.R. Elucidating the germination transcriptional program using small molecules. Plant Physiol. 2008, 147, 143–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An “electronic fluorescent pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef] [PubMed]
- Klepikova, A.V.; Kasianov, A.S.; Gerasimov, E.S.; Logacheva, M.D.; Penin, A.A. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 2016, 88, 1058–1070. [Google Scholar] [CrossRef]
- Schmid, M.; Davison, T.S.; Henz, S.R.; Pape, U.J.; Demar, M.; Vingron, M.; Scholkopf, B.; Weigel, D.; Lohmann, J.U. A gene expression map of Arabidopsis thaliana development. Nat. Gen. 2005, 37, 501–506. [Google Scholar] [CrossRef]
- Belmonte, M.F.; Kirkbride, R.C.; Stone, S.L.; Pelletier, J.M.; Bui, A.Q.; Yeung, E.C.; Hashimoto, M.; Fei, J.; Harada, M.; Munoz, M.D.; et al. Comprehensive developmental profiles of gene activity in regions and subregions of the Arabidopsis seed. Proc. Natl. Acad. Sci. USA 2013, 110, E435–E444. [Google Scholar] [CrossRef] [Green Version]
- Le, B.H.; Cheng, C.; Bui, A.Q.; Wagmaister, J.A.; Henry, K.F.; Pelletier, J.; Kwong, L.; Belmonte, M.; Kirkbride, R.; Horvath, S.; et al. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 8063–8070. [Google Scholar] [CrossRef] [Green Version]
- Heck, G.R.; Perry, S.E.; Nichols, K.W.; Fernandez, D.E. AGL15, a MADS domain protein expressed in developing embryos. Plant Cell 1995, 7, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.E.; Nichols, K.W.; Fernandez, D.E. The MADS domain protein AGL15 localizes to the nucleus during early stages of seed development. Plant Cell 1996, 8, 1977–1989. [Google Scholar] [CrossRef]
- Rounsley, S.D.; Ditta, G.S.; Yanofsky, M.F. Diverse roles for MADS box genes in Arabidopsis development. Plant Cell 1995, 7, 1259–1269. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Categories | Fold Enrichment | FDR |
|---|---|---|
| Regulation of seed maturation (GO:2000034) | 4.09 | 2.74 × 10−3 |
| Regulation of abscisic acid biosynthetic process (GO:0010115) | 3.77 | 4.58 × 10−2 |
| Regulation of response to salt stress (GO:1901000) | 3.66 | 9.51 × 10−4 |
| Regulation of seed development (GO:0080050) | 3.63 | 6.15 × 10−3 |
| Very long-chain fatty acid biosynthetic process (GO:0042761) | 3.4 | 4.59 × 10−2 |
| Positive regulation of seed germination (GO:0010030) | 3.28 | 3.65 × 10−2 |
| Response to gibberellin (GO:0009739) | 3.11 | 1.45 × 10−7 |
| Regulation of response to water deprivation (GO:2000070) | 3.06 | 9.09 × 10−3 |
| Regulation of seed germination (GO:0010029) | 2.86 | 5.50 × 10−5 |
| Regulation of hormone levels (GO:0010817) | 2.63 | 8.56 × 10−12 |
| Response to hormone (GO:0009725) | 2.5 | 4.51 × 10−50 |
| Regulation of post-embryonic development (GO:0048580) | 2.39 | 3.46 × 10−12 |
| Seed development (GO:0048316) | 1.67 | 5.08 × 10−7 |
| GO Categories for Induced Genes Enriched for: | Fold Enrichment | FDR |
|---|---|---|
| Regulation of cytokinin-activated signaling pathway (0080036) | 6.94 | 4.44 × 10−2 |
| Cellular response to oxygen levels (0071453) | 5.62 | 2.12 × 10−22 |
| Water transport (0006833) | 5.55 | 1.09 × 10−2 |
| Fatty acid beta-oxidation (0006635) | 4.5 | 2.78 × 10−2 |
| Positive regulation of abscisic acid signaling (0009789) | 4.32 | 6.52 × 10−3 |
| Response to gibberellin (0009739) | 3.25 | 3.37 × 10−3 |
| Positive regulation of response to stimulus (0048584) | 2.29 | 6.60 × 10−3 |
| GO categories for repressed genes enriched for: | Fold Enrichment | FDR |
| Embryonic meristem initiation (0090421) Ethylene biosynthetic process (0009693) | 7.40 5.14 | 2.16 × 10−4 1.85 × 10−2 |
| Auxin polar transport (0009926) | 3.43 | 2.19 × 10−3 |
| Response to fatty acid (0070542) | 2.84 | 1.91 × 10−5 |
| Response to lipid (0033993) | 2.42 | 1.02 × 10−15 |
| Response to gibberellin (0009739) | 2.33 | 4.75 × 10−2 |
| Response to abscisic acid (0009737) | 2.33 | 3.63 × 10−8 |
| Response to water deprivation (0009414) | 2.14 | 6.35 × 10−5 |
| Response to salt stress (0009651) | 2.14 | 5.35 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, S.; Awan, H.; Paul, P.; Tian, R.; Perry, S.E. Revisiting AGAMOUS-LIKE15, a Key Somatic Embryogenesis Regulator, Using Next Generation Sequencing Analysis in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 15082. https://doi.org/10.3390/ijms232315082
Joshi S, Awan H, Paul P, Tian R, Perry SE. Revisiting AGAMOUS-LIKE15, a Key Somatic Embryogenesis Regulator, Using Next Generation Sequencing Analysis in Arabidopsis. International Journal of Molecular Sciences. 2022; 23(23):15082. https://doi.org/10.3390/ijms232315082
Chicago/Turabian StyleJoshi, Sanjay, Hadia Awan, Priyanka Paul, Ran Tian, and Sharyn E. Perry. 2022. "Revisiting AGAMOUS-LIKE15, a Key Somatic Embryogenesis Regulator, Using Next Generation Sequencing Analysis in Arabidopsis" International Journal of Molecular Sciences 23, no. 23: 15082. https://doi.org/10.3390/ijms232315082
APA StyleJoshi, S., Awan, H., Paul, P., Tian, R., & Perry, S. E. (2022). Revisiting AGAMOUS-LIKE15, a Key Somatic Embryogenesis Regulator, Using Next Generation Sequencing Analysis in Arabidopsis. International Journal of Molecular Sciences, 23(23), 15082. https://doi.org/10.3390/ijms232315082