
Cis-eQTL Analysis and Functional Validation of Candidate Genes for Carcass Yield Traits in Beef Cattle

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Summary Statistics of Sequencing Dataset
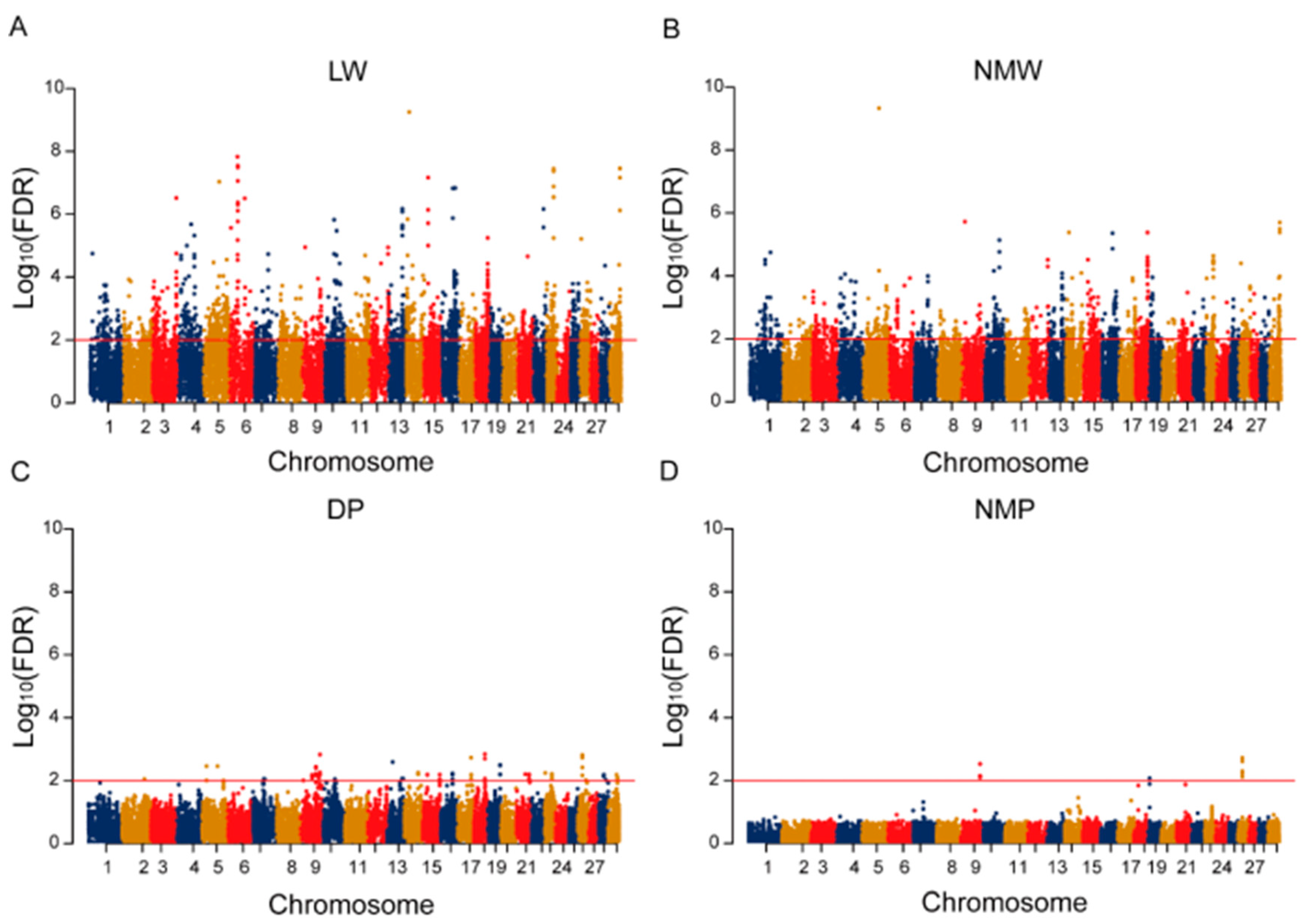
2.2. Gene-Based GWAS
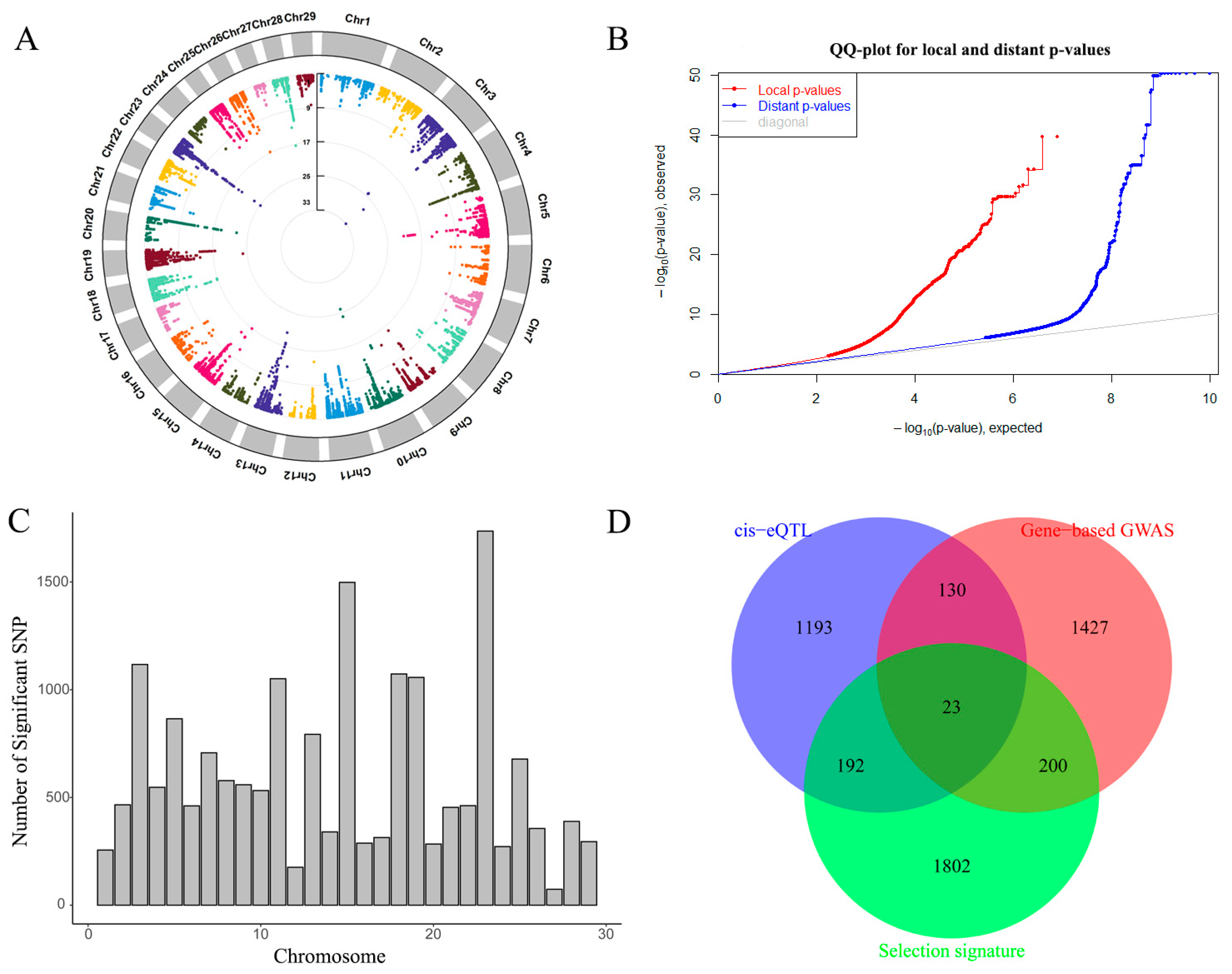
2.3. Cis-eQTL
2.4. Gene Overlap between Gene-Based GWAS and Cis-eQTLs
2.5. SNPs Associated with PON3 and PRIM2
2.6. Gene Expression Patterns in Myogenic Differentiation
2.7. Overexpression of PON3 and PRIM2 in BSCs
3. Discussion
4. Materials and Methods
4.1. Animal Sampling and Phenotype
4.2. Genotyping and Data Quality Control
4.3. RNA Preparation and Sequencing
4.4. Quantification of Molecular Phenotypes
4.5. Cis eQTL Detection
4.6. Gene-Based GWAS
4.7. Candidate Genes’ Annotation and Enrichment
4.8. Enrichment of Cis-eQTLs in GWAS Hit Regions
4.9. Cell Isolation and Culture
4.10. Plasmid Construction and Transfection
4.11. RNA Extraction, RT-PCR and qRT-PCR
4.12. Cell Counting Kit-8 (CCK8) Assay
4.13. 5-Ethynyl-2′-Deoxyuridine (EdU) Assay
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turk, J. Meeting projected food demands by 2050: Understanding and enhancing the role of grazing ruminants. J. Anim. Sci. 2016, 94, 53–62. [Google Scholar] [CrossRef]
- Igoshin, A.V.; Yudin, N.S.; Belonogova, N.M.; Larkin, D.M. Genome-wide association study for body weight in cattle populations from Siberia. Anim. Genet. 2019, 50, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Zalewska, M.; Puppel, K.; Sakowski, T. Associations between gene polymorphisms and selected meat traits in cattle—A review. Anim. Biosci. 2021, 34, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Silva-Vignato, B.; Coutinho, L.L.; Cesar, A.S.M.; Poleti, M.D.; Regitano, L.C.A.; Balieiro, J.C.C. Comparative muscle transcriptome associated with carcass traits of Nellore cattle. BMC Genom. 2017, 18, 506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Zhang, F.; Mukiibi, R.; Chen, L.H.; Vinsky, M.; Plastow, G.; Basarab, J.; Stothard, P.; Li, C.X. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: II: Carcass merit traits. BMC Genom. 2020, 21, 38. [Google Scholar] [CrossRef]
- Begum, F.; Ghosh, D.; Tseng, G.C.; Feingold, E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 2012, 40, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E.; Hayes, B.J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 2009, 10, 381–391. [Google Scholar] [CrossRef]
- MacLeod, I.M.; Bowman, P.J.; Vander Jagt, C.J.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef]
- Goddard, M.E.; Kemper, K.E.; MacLeod, I.M.; Chamberlain, A.J.; Hayes, B.J. Genetics of complex traits: Prediction of phenotype, identification of causal polymorphisms and genetic architecture. Proc. R. Soc. B Boil. Sci. 2016, 283, 20160569. [Google Scholar] [CrossRef]
- Falker-Gieske, C.; Blaj, I.; Preuss, S.; Bennewitz, J.; Thaller, G.; Tetens, J. GWAS for Meat and Carcass Traits Using Imputed Sequence Level Genotypes in Pooled F2-Designs in Pigs. G3-Genes Genom. Genet. 2019, 9, 2823–2834. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; He, Y.T.; Ye, S.P.; Wang, J.Y.; Yuan, X.L.; Zhang, H.; Li, J.Q.; Zhang, X.Q.; Zhang, Z. Genome-wide association study on chicken carcass traits using sequence data imputed from SNP array. J. Appl. Genet. 2018, 59, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Kizilaslan, M.; Arzik, Y.; Behrem, S.; Ata, N.; Karaca, O.; Elmaci, C.; Cemal, I. Genome-wide association studies of preweaning growth and in vivo carcass composition traits in Esme sheep. J. Anim. Breed. Genet. 2022, 139, 26–39. [Google Scholar] [CrossRef]
- Kemper, K.E.; Goddard, M.E. Understanding and predicting complex traits: Knowledge from cattle. Hum. Mol. Genet. 2012, 21, R45–R51. [Google Scholar] [CrossRef] [PubMed]
- Pasaniuc, B.; Price, A.L. Dissecting the genetics of complex traits using summary association statistics. Nat. Rev. Genet. 2017, 18, 117–127. [Google Scholar] [CrossRef]
- Xu, L.Y.; Bickhart, D.M.; Cole, J.B.; Schroeder, S.G.; Song, J.Z.; Van Tassell, C.P.; Sonstegard, T.S.; Liu, G.E. Genomic Signatures Reveal New Evidences for Selection of Important Traits in Domestic Cattle. Mol. Biol. Evol. 2015, 32, 711–725. [Google Scholar] [CrossRef]
- Fan, H.; Wu, Y.; Zhou, X.; Xia, J.; Zhang, W.; Song, Y.; Liu, F.; Chen, Y.; Zhang, L.; Gao, X.; et al. Pathway-Based Genome-Wide Association Studies for Two Meat Production Traits in Simmental Cattle. Sci. Rep. 2015, 5, 18389. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Zhang, T.; Xu, L.; Wang, T.; Wang, Z.; Zhu, B.; Zhang, L.; Gao, H.; Song, J.; Li, J.; et al. Integration of selection signatures and multi-trait GWAS reveals polygenic genetic architecture of carcass traits in beef cattle. Genomics 2021, 113, 3325–3336. [Google Scholar] [CrossRef]
- Naserkheil, M.; Mehrban, H.; Lee, D.; Park, M.N. Genome-wide Association Study for Carcass Primal Cut Yields Using Single-step Bayesian Approach in Hanwoo Cattle. Front. Genet. 2021, 12, 752424. [Google Scholar] [CrossRef] [PubMed]
- Rafter, P.; Gormley, I.C.; Purfield, D.; Parnell, A.C.; Naderi, S.; Berry, D.P. Genome-wide association analyses of carcass traits using copy number variants and raw intensity values of single nucleotide polymorphisms in cattle. BMC Genom. 2021, 22, 757. [Google Scholar] [CrossRef]
- Li, L.; Kabesch, M.; Bouzigon, E.; Demenais, F.; Farrall, M.; Moffatt, M.F.; Lin, X.; Liang, L. Using eQTL weights to improve power for genome-wide association studies: A genetic study of childhood asthma. Front. Genet. 2013, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.G.; Fitzsimons, C.; McClure, M.C.; McKenna, C.; Conroy, S.; Kenny, D.A.; McGee, M.; Waters, S.M.; Morris, D.W. GWAS and eQTL analysis identifies a SNP associated with both residual feed intake and GFRA2 expression in beef cattle. Sci. Rep. 2018, 8, 14301. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.H.; Zhang, F.T.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; van de Bunt, M.; Segre, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Broadaway, K.A.; Raulerson, C.K.; Scott, L.J.; Pan, C.; Ko, A.; He, A.Q.; Tilford, C.; Fuchsberger, C.; Locke, A.E.; et al. Colocalization of GWAS and eQTL signals at loci with multiple signals identifies additional candidate genes for body fat distribution. Hum. Mol. Genet. 2019, 28, 4162–4173. [Google Scholar] [CrossRef] [PubMed]
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120362. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.; Berg, I.V.D.; MacLeod, I.M.; Hayes, B.J.; Prowse-Wilkins, C.P.; Wang, M.; Bolormaa, S.; Liu, Z.; Rochfort, S.J.; Reich, C.M.; et al. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc. Natl. Acad. Sci. USA 2019, 116, 19398–19408. [Google Scholar] [CrossRef]
- Leal-Gutierrez, J.D.; Elzo, M.A.; Mateescu, R.G. Identification of eQTLs and sQTLs associated with meat quality in beef. BMC Genom. 2020, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- de las Heras-Saldana, S.; Clark, S.A.; Duijvesteijn, N.; Gondro, C.; van der Werf, J.H.J.; Chen, Y.Z. Combining information from genome-wide association and multi-tissue gene expression studies to elucidate factors underlying genetic variation for residual feed intake in Australian Angus cattle. BMC Genom. 2019, 20, 939. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Smemo, S.; Tena, J.J.; Kim, K.H.; Gamazon, E.R.; Sakabe, N.J.; Gomez-Marin, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 507, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.Z.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.B.; Dermitzakis, E.T. From expression QTLs to personalized transcriptomics. Nat. Rev. Genet. 2011, 12, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Khansefid, M.; Pryce, J.E.; Bolormaa, S.; Chen, Y.; Millen, C.A.; Chamberlain, A.J.; Vander Jagt, C.J.; Goddard, M.E. Comparing allele specific expression and local expression quantitative trait loci and the influence of gene expression on complex trait variation in cattle. BMC Genom. 2018, 19, 793. [Google Scholar] [CrossRef]
- Ding, R.; Zhuang, Z.; Qiu, Y.; Wang, X.; Wu, J.; Zhou, S.; Ruan, D.; Xu, C.; Hong, L.; Gu, T.; et al. A composite strategy of genome-wide association study and copy number variation analysis for carcass traits in a Duroc pig population. BMC Genom. 2022, 23, 590. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Tokunaga, T.; Ishida, T.; Kobayashi, I.; Katahama, Y.; Yano, A.; Erickson, L.; Kawahara, S. Changes in expression of the autophagy-related genes microtubule-associated protein 1 light chain 3beta and autophagy related 7 in skeletal muscle of fattening Japanese Black cattle: A pilot study. Asian-Australas. J. Anim. Sci. 2019, 32, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, C. Proteomics discovery of protein biomarkers linked to yak meat tenderness as determined by label-free mass spectrometry. Anim. Sci. J. 2021, 92, e13669. [Google Scholar] [CrossRef] [PubMed]
- Muniz, M.M.M.; Fonseca, L.F.S.; Scalez, D.C.B.; Vega, A.S.; Silva, D.B.D.; Ferro, J.A.; Chardulo, A.L.; Baldi, F.; Canovas, A.; de Albuquerque, L.G. Characterization of novel lncRNA muscle expression profiles associated with meat quality in beef cattle. Evol. Appl. 2022, 15, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chu, S.F.; Xu, X.; Jiang, J.Y.; Wang, W.Q.; Shen, H.L.; Li, M.X.; Zhang, H.M.; Mao, Y.J.; Yang, Z.P. Analysis of longissimus muscle quality characteristics and associations with DNA methylation status in cattle. Genes Genom. 2019, 41, 1147–1163. [Google Scholar] [CrossRef]
- Hamill, R.M.; McBryan, J.; McGee, C.; Mullen, A.M.; Sweeney, T.; Talbot, A.; Cairns, M.T.; Davey, G.C. Functional analysis of muscle gene expression profiles associated with tenderness and intramuscular fat content in pork. Meat Sci. 2012, 92, 440–450. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, I.; Hayes, B.J.; Chamberlain, A.J.; Goddard, M.E. Overlap between eQTL and QTL associated with production traits and fertility in dairy cattle. BMC Genom. 2019, 20, 291. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, B.; Beaudry, D.; Mayhue, M.; Halle, C.; Bordignon, V.; Murphy, B.D.; Palin, M.F. Molecular characterization and expression analysis of the porcine paraoxonase 3 (PON3) gene. Gene 2009, 443, 110–120. [Google Scholar] [CrossRef]
- An, B.X.; Xu, L.; Xia, J.W.; Wang, X.Q.; Miao, J.; Chang, T.P.; Song, M.H.; Ni, J.Q.; Xu, L.Y.; Zhang, L.P.; et al. Multiple association analysis of loci and candidate genes that regulate body size at three growth stages in Simmental beef cattle. BMC Genet. 2020, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Kempster, S.L.; Belteki, G.; Licence, D.; Charnock-Jones, D.S.; Smith, G.C.S. Disruption of paraoxonase 3 impairs proliferation and antioxidant defenses in human A549 cells and causes embryonic lethality in mice. Am. J. Physiol. Endoc. M 2012, 302, E103–E107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.L.; Shen, Y.Y.; Sun, W. Paraoxonase 3 promotes cell proliferation and metastasis by PI3K/Akt in oral squamous cell carcinoma. Biomed. Pharmacother. 2017, 85, 712–717. [Google Scholar] [CrossRef]
- Shiratori, A.; Okumura, K.; Nogami, M.; Taguchi, H.; Onozaki, T.; Inoue, T.; Ando, T.; Shibata, T.; Izumi, M.; Miyazawa, H.; et al. Assignment of the 49-Kda (Prim1) and 58-Kda (Prim2a and Prim2b) Subunit Genes of the Human DNA Primase to Chromosome Bands 1q44 and 6p11.1-P12. Genomics 1995, 28, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Yatsula, B.; Galvao, C.; McCrann, M.; Perkins, A.S. Assessment of F-MuLV-induced tumorigenesis reveals new candidate tumor genes including Pecam1, St7, and Prim2. Leukemia 2006, 20, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Liao, F.; Shi, Z.Z.; Ren, Y.; Deng, X.L.; Yang, T.T.; Li, D.Y.; Li, R.F.; Pu, D.D.; Wang, Y.J.; et al. Dihydroartemisinin Inhibits the Proliferation, Colony Formation and Induces Ferroptosis of Lung Cancer Cells by Inhibiting PRIM2/SLC7A11 Axis. OncoTargets Ther. 2020, 13, 10829–10840. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Guo, H.; Zong, Y.; Zhao, X.F. Curcumin restrains hepatocellular carcinoma progression depending on the regulation of the circ_0078710/miR-378b/PRIM2 axis. J. Recept. Sig. Transd. 2022, 42, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Tang, T.S.; Jiang, Y.G.; He, T.; Qi, L.Y.; Chang, H.K.; Qiao, Y.Y.; Sun, M.M.; Shan, C.L.; Zhu, X.Y.; et al. PRIM2 Promotes Cell Cycle and Tumor Progression in p53-Mutant Lung Cancer. Cancers 2022, 14, 3370. [Google Scholar] [CrossRef] [PubMed]
- Suravajhala, P.; Kogelman, L.J.A.; Kadarmideen, H.N. Multi-omic data integration and analysis using systems genomics approaches: Methods and applications in animal production, health and welfare. Genet. Sel. Evol. 2016, 48, 38. [Google Scholar] [CrossRef]
- National Academies of Sciences Engineering and Medicine (U.S.). Committee on Nutrient Requirements of Beef Cattle. In Nutrient Requirements of Beef Cattle, 8th ed.; National Academies Press: Washington, DC, USA, 2016. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Yuan, Z.H.; Sunduimijid, B.; Xiang, R.D.; Behrendt, R.; Knight, M.I.; Mason, B.A.; Reich, C.M.; Prowse-Wilkins, C.; Vander Jagt, C.J.; Chamberlain, A.J.; et al. Expression quantitative trait loci in sheep liver and muscle contribute to variations in meat traits. Genet. Sel. Evol. 2021, 53, 8. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, Y.M.; Zhang, W.R.; Liu, X.F.; Li, X.; Ding, X.B.; Guo, H. The role of microRNA-1 and microRNA-206 in the proliferation and differentiation of bovine skeletal muscle satellite cells. Vitr. Cell. Dev. Biol. Anim. 2016, 52, 27–34. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Niu, Q.; Zhang, T.; Zheng, X.; Li, H.; Gao, X.; Chen, Y.; Gao, H.; Zhang, L.; Liu, G.E.; et al. Cis-eQTL Analysis and Functional Validation of Candidate Genes for Carcass Yield Traits in Beef Cattle. Int. J. Mol. Sci. 2022, 23, 15055. https://doi.org/10.3390/ijms232315055
Wang T, Niu Q, Zhang T, Zheng X, Li H, Gao X, Chen Y, Gao H, Zhang L, Liu GE, et al. Cis-eQTL Analysis and Functional Validation of Candidate Genes for Carcass Yield Traits in Beef Cattle. International Journal of Molecular Sciences. 2022; 23(23):15055. https://doi.org/10.3390/ijms232315055
Chicago/Turabian StyleWang, Tianzhen, Qunhao Niu, Tianliu Zhang, Xu Zheng, Haipeng Li, Xue Gao, Yan Chen, Huijiang Gao, Lupei Zhang, George E. Liu, and et al. 2022. "Cis-eQTL Analysis and Functional Validation of Candidate Genes for Carcass Yield Traits in Beef Cattle" International Journal of Molecular Sciences 23, no. 23: 15055. https://doi.org/10.3390/ijms232315055
APA StyleWang, T., Niu, Q., Zhang, T., Zheng, X., Li, H., Gao, X., Chen, Y., Gao, H., Zhang, L., Liu, G. E., Li, J., & Xu, L. (2022). Cis-eQTL Analysis and Functional Validation of Candidate Genes for Carcass Yield Traits in Beef Cattle. International Journal of Molecular Sciences, 23(23), 15055. https://doi.org/10.3390/ijms232315055