

Similarity-Based Virtual Screening to Find Antituberculosis Agents Based on Novel Scaffolds: Design, Syntheses and Pharmacological Assays


 , , , and
, , , and
Abstract
:1. Introduction
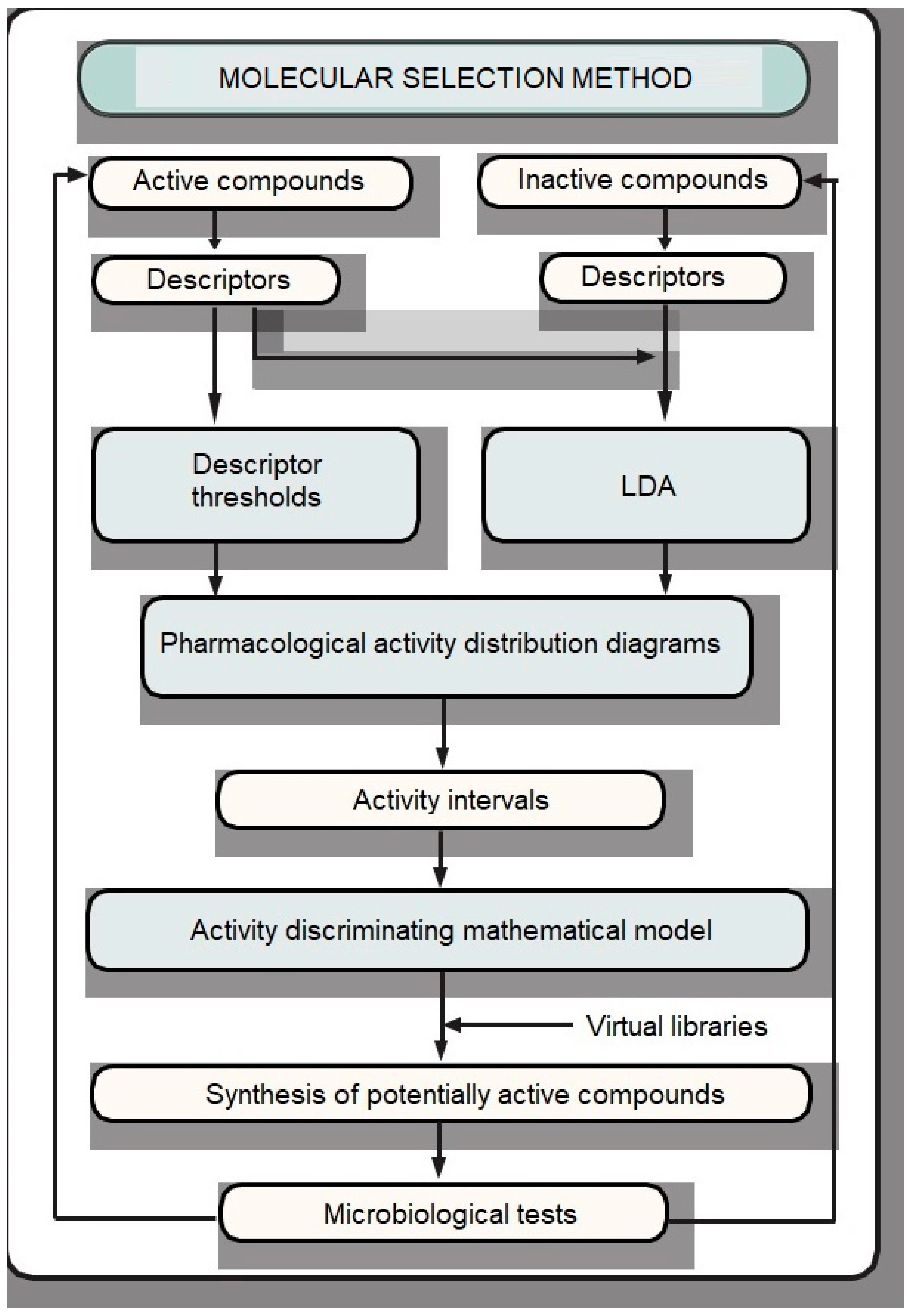
2. Results and Discussion
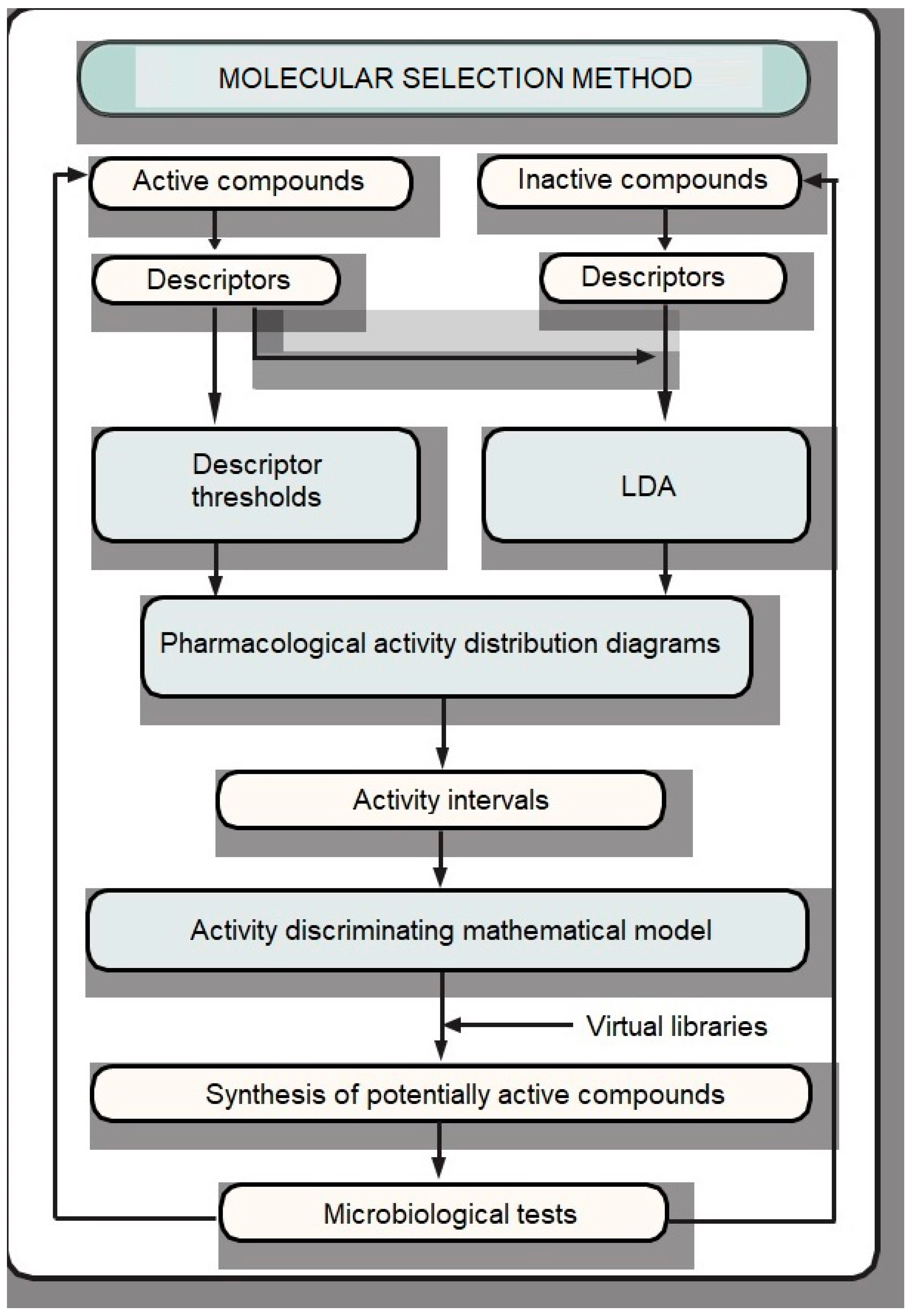
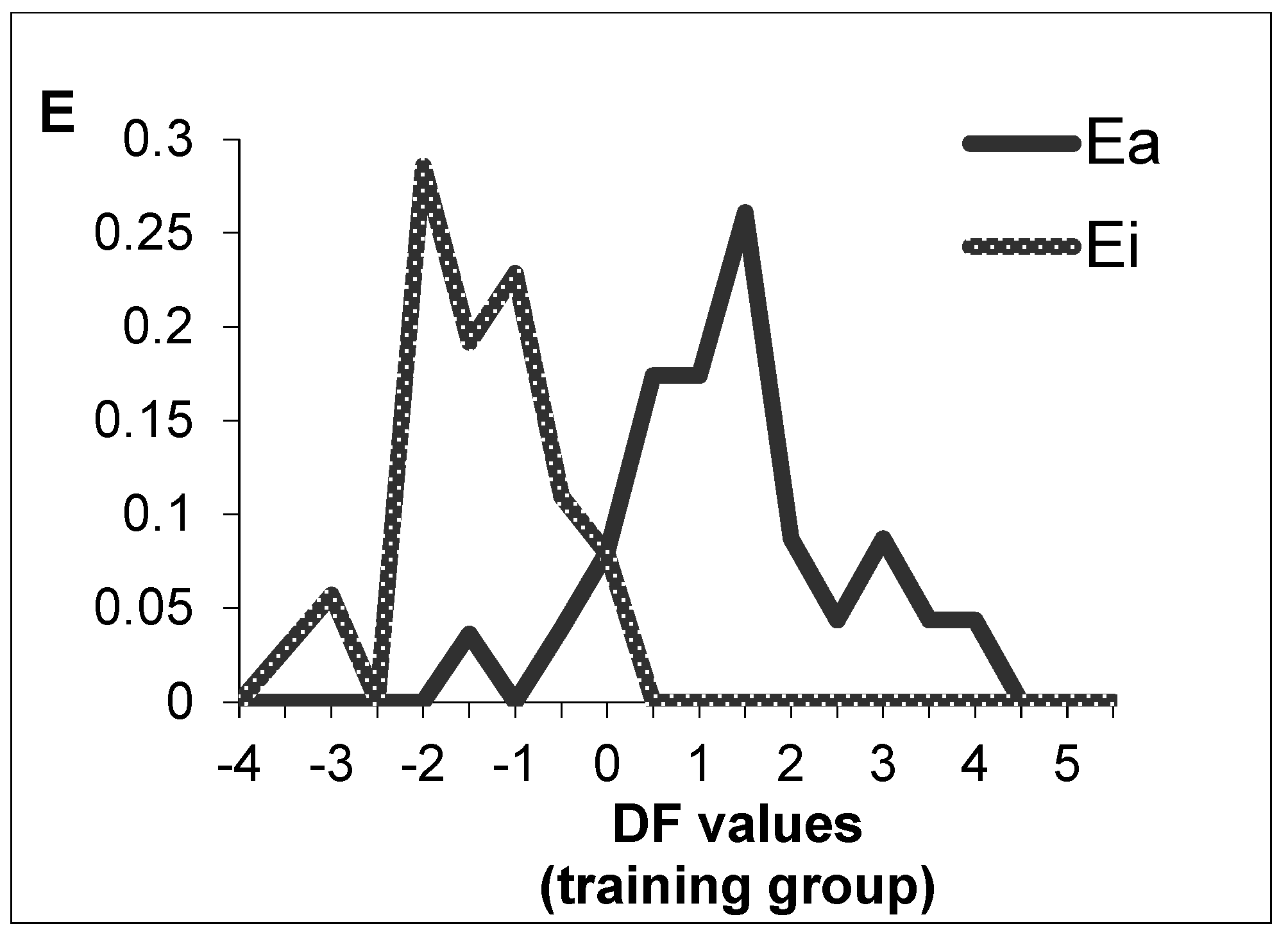
2.1. Antituberculosis Activity Modelling
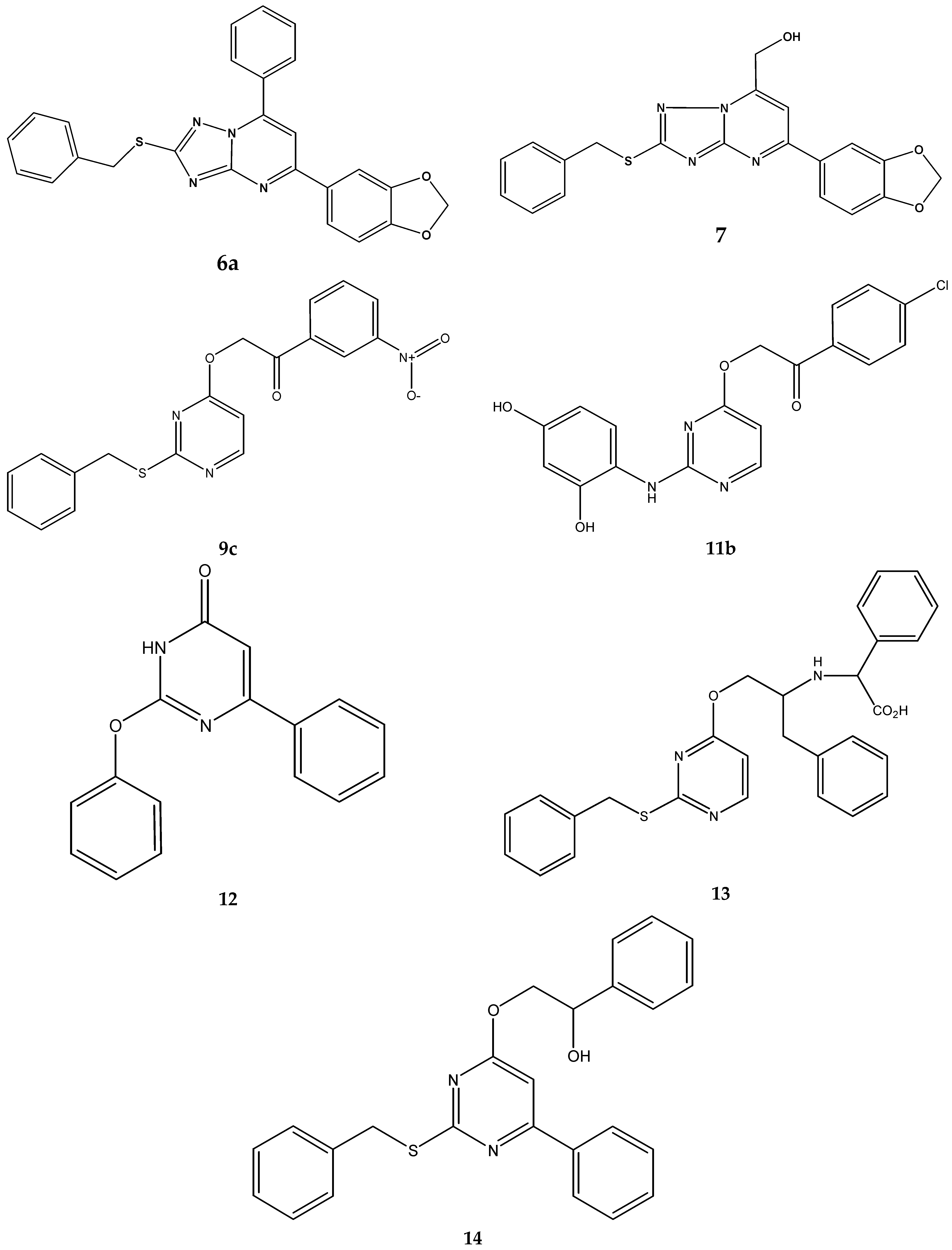
2.2. Similarity-Based Virtual Screening
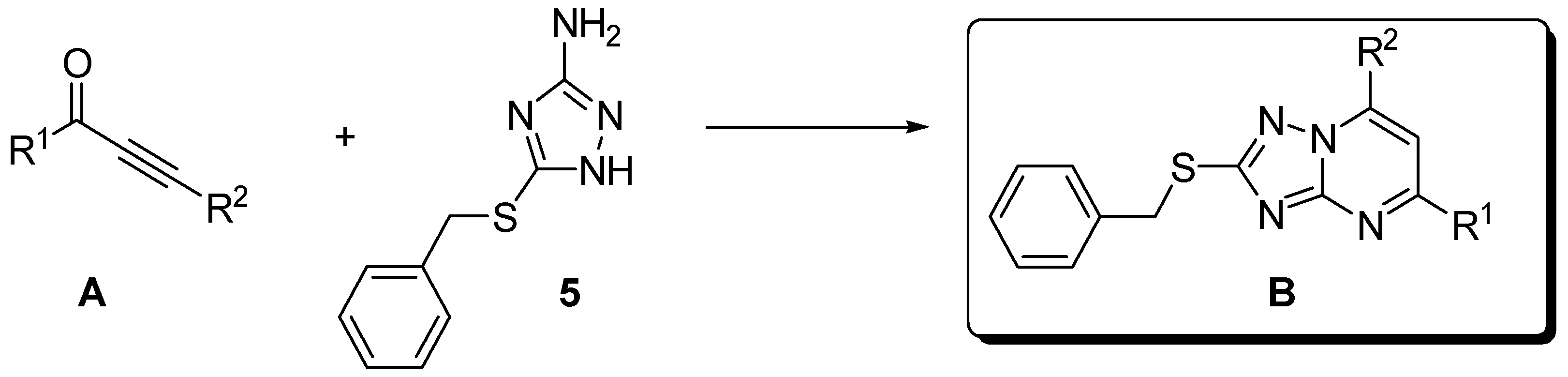
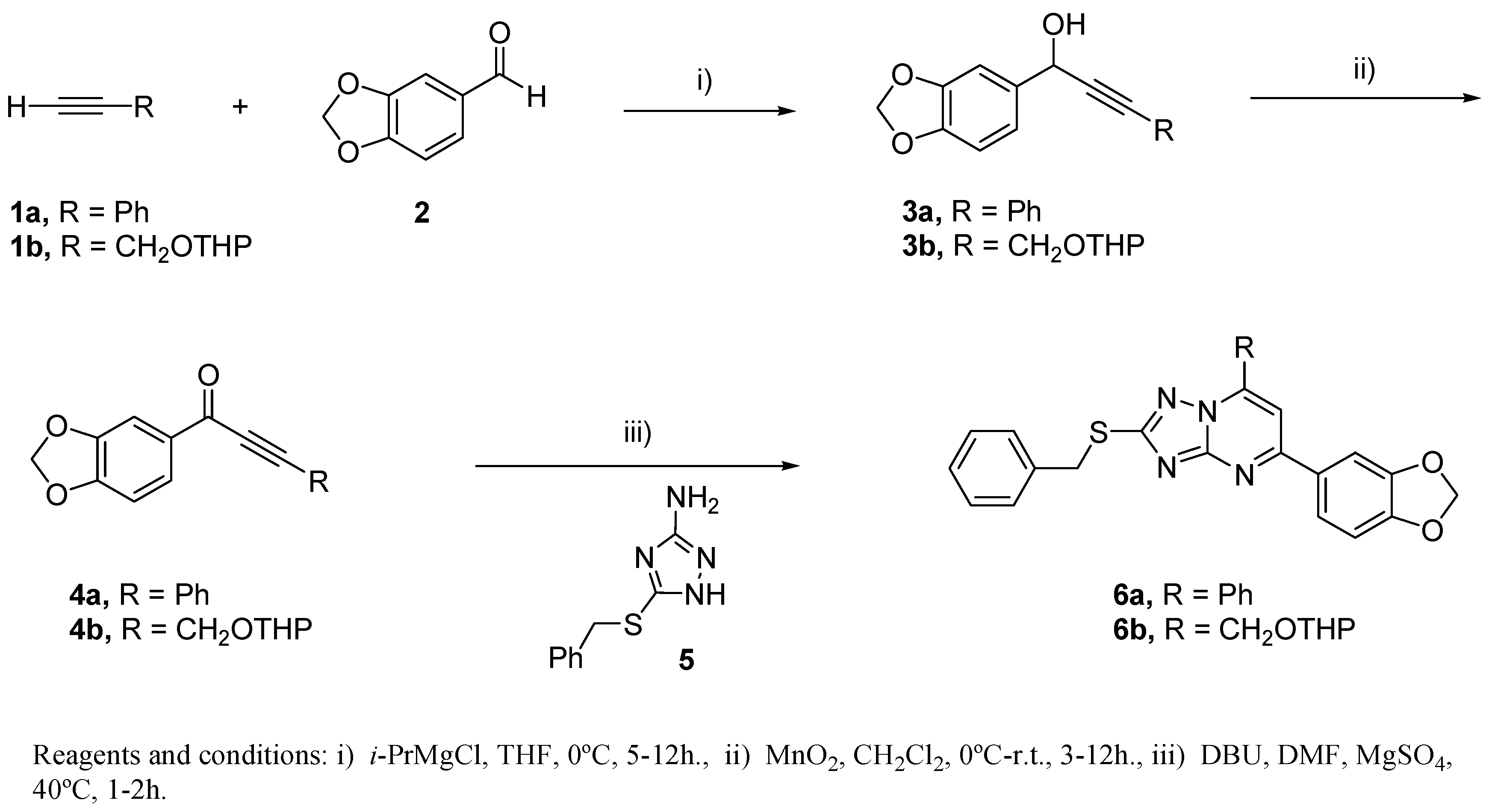
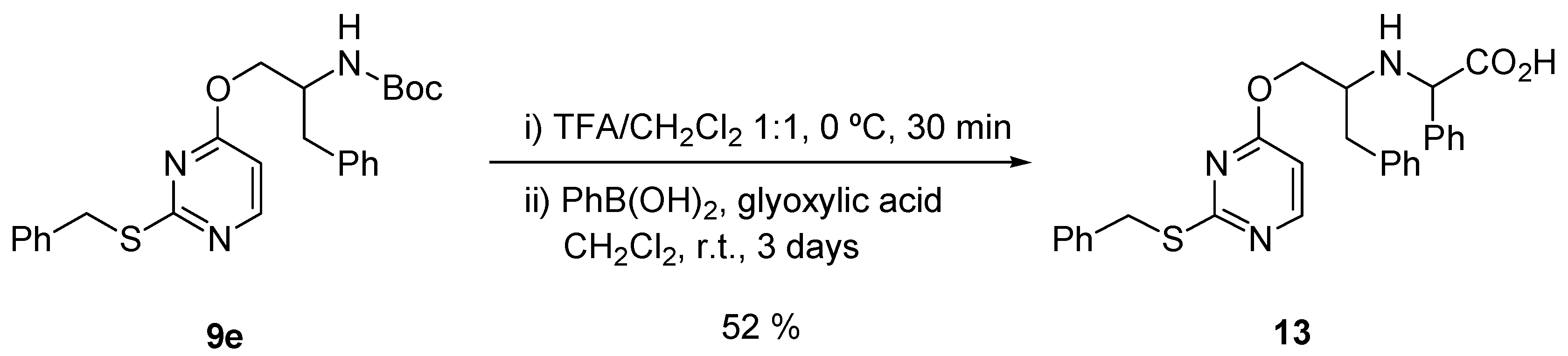
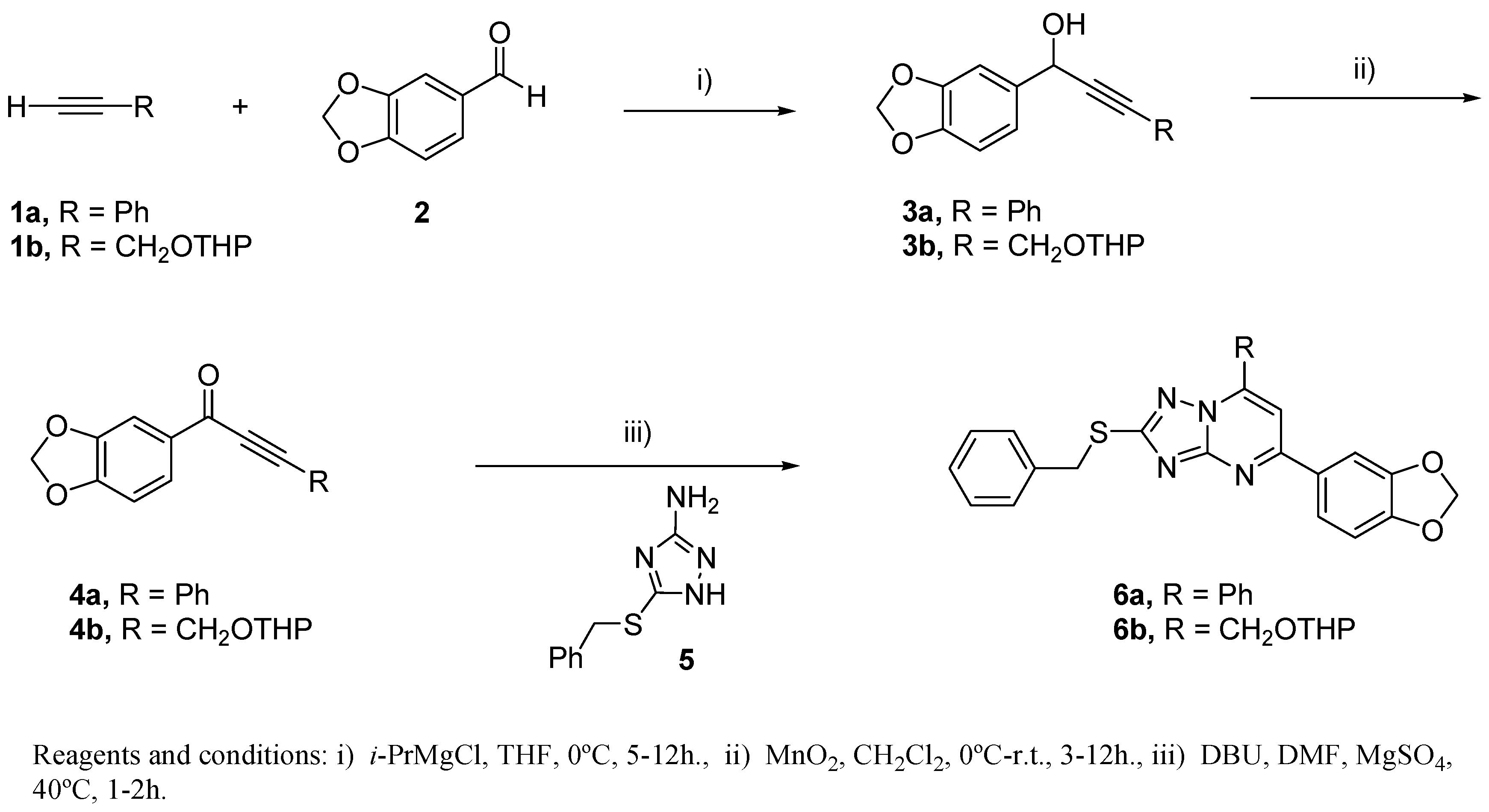
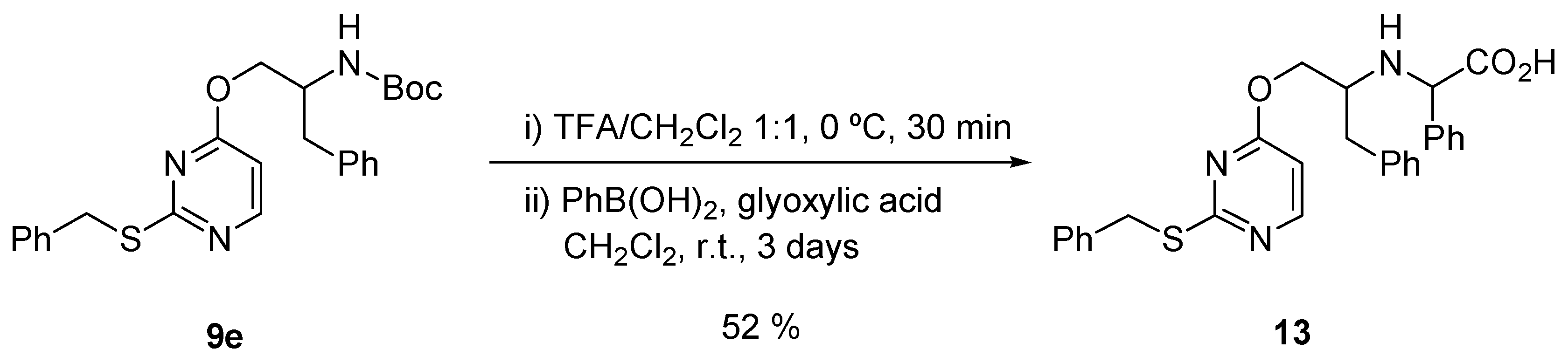
2.3. Chemistry
3. Materials and Methods
3.1. Antituberculosis Activity Modelling
3.2. Similarity-Based Virtual Screening
3.3. Chemical Methods
3.4. Microbiological Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tuberculosis. Available online: https://www.who.int/westernpacific/health-topics/tuberculosis (accessed on 25 June 2022).
- Dhote, A.; Kawishwar, V. Utility of Concentration Bleach Method and Fluorescent Auramine Rhodamine Staining in Diagnosis of Mycobacterium tuberculosis on Formalin Fixed Tissue. New Horiz. Med. Med. Res. 2022, 4, 67–70. [Google Scholar]
- Besalú, E.; Ponec, R.; de Julián-Ortiz, J.V. Virtual generation of agents against Mycobacterium tuberculosis. A QSAR study. Mol. Divers. 2003, 6, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.; Acharya, A.; Gautam, S.; Ghimire, S.P.; Mishra, G.; Parajuli, N.; Sapkota, B. Advances in Diagnosis of Tuberculosis: An Update into Molecular Diagnosis of Mycobacterium Tuberculosis. Molec. Biol. Rep. 2020, 47, 4065–4075. [Google Scholar] [CrossRef] [PubMed]
- Hoffner, S.E.; Gezelius, L.; Olsson-Liljequist, B. In vitro activity of fluorinated quinolones and macrolides against drug-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 1997, 40, 885–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomioka, H. Prospects for development of new antituberculous drug. Kekkaku 2002, 77, 573–584. [Google Scholar]
- Koketsu, M.; Tanaka, K.; Takenaka, Y.; Kwong, C.D.; Ishihara, H. Synthesis of 1,3-thiazine derivates and their evaluation as potential antimicobacterial agents. Eur. J. Pharm. Sci. 2002, 15, 307–310. [Google Scholar] [CrossRef]
- Fernandes, G.F.D.S.; Jornada, D.H.; de Souza, P.C.; Chin, C.M.; Pavan, F.R.; Santos, J.L.D. Current Advances in Antitubercular Drug Discovery: Potent Prototypes and New Targets. Curr. Med. Chem. 2015, 22, 3133–3161. [Google Scholar] [CrossRef]
- Fernandes, G.F.; Thompson, A.M.; Castagnolo, D.; Denny, W.A.; Dos Santos, J.L. Tuberculosis Drug Discovery: Challenges and New Horizons. J. Med. Chem. 2022, 65, 7489–7531. [Google Scholar] [CrossRef]
- Obrecht, D.; Weiss, B. A new method for the preparation of (E)-3-acylprop-2-enoics acids. Helv. Chim. Acta 1989, 72, 117–122. [Google Scholar] [CrossRef]
- Utimoto, K.; Miwa, H.; Nozaki, H. Palladium-catalyzed synthesis of pyrroles. Tetrahedron Lett. 1981, 22, 4277–4278. [Google Scholar] [CrossRef]
- Obrecht, D. Acid-catalyzed cyclization reactions of substituted acetylenic ketones: A new approach for the synthesis of 3-halofurans, flavones and styrylchromones. Helv. Chim. Acta 1989, 72, 447–456. [Google Scholar] [CrossRef]
- Masquelin, T.; Obrecht, D. A facile preparation of 2- and 5-substituted 3-bromothiophenes. Tetrahedron Lett. 1994, 35, 9387–9390. [Google Scholar] [CrossRef]
- Garvey, D.S.; Wasicak, J.T.; Elliot, R.L.; Lebold, S.A.; Hettinger, A.-M.; Carrera, G.M.; Lin, N.-H.; He, Y.; Holladay, M.W.; Anderson, D.J.; et al. Ligands for brain cholinergic channel receptors: Synthesis and in vitro characterization of novel isoxazoles and isothiazoles as bioisosteric replacements for the pyridine ring in nicotine. J. Med. Chem. 1994, 37, 4455–4463. [Google Scholar] [CrossRef] [PubMed]
- Masquelin, T.; Obrecht, D. A new approach to the synthesis of N-protected 2- and 5-substituted 3-halopyrroles. Synthesis 1995, 1995, 276–384. [Google Scholar] [CrossRef]
- Degl’Inocenti, A.; Scafato, P.; Capperucci, A.; Bartoletti, L.; Mordini, A.; Reginato, G. Azide cyclocondensation with acetylenic silyl ketone: A general access to funtionalized-1,2,3-triazolylacylsilanes and –aldehydes. Tetrahedron Lett. 1995, 36, 9031–9034. [Google Scholar]
- Masquelin, T.; Obrecht, D. A novel access to 2,4-substituted quinolines from acetylenic ketones. Tetrahedron 1997, 53, 641–646. [Google Scholar] [CrossRef]
- Falorni, M.; Giacomelli, G.; Spanedda, A.M. Synthesis of chiral pyrazoles and isoxazoles as constrained amino acids. Tetrahedron Asymmetry 1998, 9, 3039–3046. [Google Scholar] [CrossRef]
- Cabarrocas, G.; Rafel, S.; Ventura, M.; Villalgordo, J.M. A new approach toward the stereoselective synthesis of novel quinolyl glycines: Synthesis of the enantiomerically pure quinolyl-β-amino alcohol precursors. Synlett 2000, 5, 595–598. [Google Scholar] [CrossRef]
- Cabarrocas, G.; Ventura, M.; Maestro, M.; Mahı́a, J.; Villalgordo, J.M. Synthesis of Novel Optically Pure Quinolyl-β-Amino Alcohol Derivatives from 2-Amino Thiophenol and Chiral α-Acetylenic Ketones and Their IBX-Mediated Oxidative Cleavage to N-BOC Quinolyl Carboxamides. Tetrahedron Asymmetry 2001, 12, 1851–1863. [Google Scholar] [CrossRef]
- Satheesha Rai, N.; Kalluraya, B.; Lingappa, B.; Shenoy, S.; Puranic, V.G. Convenient Access to 1,3,4-Trisubstituted Pyrazoles Carrying 5-Nitrothiophene Moiety via 1,3-Dipolar Cycloaddition of Sydnones with Acetylenic Ketones and Their Antimicrobial Evaluation. Eur. J. Med. Chem. 2008, 43, 1715–1720. [Google Scholar] [CrossRef]
- Harigae, R.; Moriyama, K.; Togo, H. Preparation of 3,5-Disubstituted Pyrazoles and Isoxazoles from Terminal Alkynes, Aldehydes, Hydrazines, and Hydroxylamine. J. Org. Chem. 2014, 79, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Adlington, R.M.; Baldwin, J.E.; Catterick, D.; Pritchard, G.J. The Synthesis of Pyrimidin-4-Yl Substituted α-Amino Acids. A Versatile Approach from Alkynyl Ketones. J. Chem. Soc. Perkin Trans. 1 1999, 8, 855–866. [Google Scholar] [CrossRef]
- Nájera, C.; Sydnes, L.K.; Yus, M. Conjugated Ynones in Organic Synthesis. Chem. Rev. 2019, 119, 11110–11244. [Google Scholar] [CrossRef] [PubMed]
- Chucholowski, A.; Masquelin, T.; Obrecht, D.; Stadlwieser, J.; Villalgordo, J.M. Cheminform Abstract: Novel Solution- and Solid-Phase Strategies for the Parallel and Combinatorial Synthesis of Low-Molecular-Weight Compound Libraries. CHIMIA Int. J. Chem. 2010, 28, 525–530. [Google Scholar] [CrossRef]
- Obrecht, D.; Abrecht, C.; Grieder, A.; Villalgordo, J.M. A novel and efficient approach for the combinatorial synthesis of structurally diverse pyrimidines on solid support. Helv. Chim. Acta 1997, 80, 65–72. [Google Scholar] [CrossRef]
- Aparna, E.P.; Devaky, K.S. Advances in the Solid-Phase Synthesis of Pyrimidine Derivatives. ACS Comb. Sci. 2019, 21, 35–68. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. Convenient synthesis of acetylenes. Catalytic substitutions of acetylenic hydrogen with bromo alkenes, iodo arenes and bromopyridines. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar] [CrossRef]
- Tohda, Y.; Sonogashira, K.; Hagihara, N. A convenient synthesis of 1-alkynyl ketones and 2-alkynamides. Synthesis 1977, 777–778. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Bolin, G.; Iqbal, J.; Moreau, J.J.E.; Vernhet, C. Aminosilanes in organic synthesis. Addition of organocopper reagents on γ-bis(trimethylsilyl)amino-α-acetylenic amides, esters and ketones. Stereochemistry and some synthetic uses. Tetrahedron 1993, 49, 4603–4618. [Google Scholar] [CrossRef]
- Verkruijsse, H.D.; Heus-Kloos, Y.A.; Brandsma, L. Efficient methods for the preparation of acetylenic ketones. J. Organomet. Chem. 1988, 338, 289–294. [Google Scholar] [CrossRef]
- Larson, D.P.; Heathcock, C.H. Total synthesis of tricolorin A. J. Org. Chem. 1997, 62, 8406–8418. [Google Scholar] [CrossRef] [PubMed]
- Palombi, L.; Arista, L.; Lattanzi, A.; Bonadies, F.; Scettri, Q. Zeolite-catalized oxidation of benzylic and acetylenic alcohols with t-butyl hidroperoxide. Tetrahedron Lett. 1996, 37, 7849–7850. [Google Scholar] [CrossRef]
- Serrat, X.; Cabarrocas, G.; Rafel, S.; Ventura, M.; Linden, A.; Villalgordo, J.M. A highly efficient and straightforward stereoselective synthesis of novel chiral α-acetylenic ketones. Tetrahedron Asymmetry 1999, 10, 3417–3430. [Google Scholar] [CrossRef]
- Frigerio, M.; Santagostino, M.; Sputore, S.; Palmisano, G. Oxidation of alcohols with o-iodoxybenzoic acid in DMSO: A new insight into an old hypervalent iodine reagent. J. Org. Chem. 1995, 60, 7272–7276. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; Rashed, N. 1,2,4-triazolo- and tetrazolo[x,y-z]pyrimidines. Adv. Heterocycl. Chem. 1999, 72, 127–171. [Google Scholar]
- El Ashry, E.S.H.; El Kilany, Y.; Rashed, N. Assafir, H.R. Dimroth rearrangement: Translocation of heteroatoms in heterocyclic rings and its role in ring transformations of heterocycles. Adv. Heterocycl. Chem. 1999, 75, 79–167. [Google Scholar]
- Pugmire, R.J.; Smith, J.C.; Grant, D.M.; Stanovnik, B.; Tisler, M.; Vercek, B. Correlation of ring nitrogen substituents with carbon-13 nuclear magnetic resonance data in azoloazines. J. Heterocycl. Chem. 1987, 24, 805–809. [Google Scholar] [CrossRef]
- Salas, J.M.; Romero, M.A.; Sánchez, M.P.; Quirós, M. Metal complexes of 1,2,4-triazolo[1,5-a]pyrimidine derivatives. Coord. Chem. Rev. 1999, 193, 1119–1142. [Google Scholar] [CrossRef]
- Sato, Y.; Shimoji, Y.; Fujita, H.; Nishino, H.; Mizuno, H.; Kobayashi, S.; Kamakura, S. Studies on cardiovascular agents. 6. Synthesis and coronary vasodilating and antihypertensive activities of 1,2,4-triazolo[1,5-a]pyrimidines fused to heterocyclic systems. J. Med. Chem. 1980, 23, 927–937. [Google Scholar] [CrossRef]
- Tenor, E.; Ludwig, R. Pharmaceutical-chemical research on s-triazolo[1,5-a]pyrimidines. Pharmazie 1971, 26, 534–539. [Google Scholar]
- Navarro, J.A.R.; Salas, J.M.; Romero, M.A.; Faure, R. Influence of anions and crystallization conditions on the solid-state structure of some binuclear silver (I) complexes supported by triazolopyrimidine bridges. J. Chem. Soc. Dalton Trans. 1998, 6, 901–904. [Google Scholar] [CrossRef]
- Velders, A.H.; Pazderski, L.; Ugozzoli, F.; Biagini-Cingi, M.; Manotti-Lanfredi, A.M.; Haasnot, J.G.; Reedijk, J. Synthesis, characterization and crystal structure of trans-aquatrichlorobis(5,7-dimethyl-1,2,4-triazolo[1,5-a]pyrimidine-N3)ruthenium (III) monohydrate. Inorg. Chim. Acta 1998, 273, 259–265. [Google Scholar] [CrossRef]
- Navarro, J.A.R.; Romero, M.A.; Salas, J.M.; Molina, J.; Tiekink, E.R.T. Ternary copper (II) complexos with the versatile 4,7-dihydro-5-methyl-7-oxo-1,2,4-triazolo[1,5-a]pyrimidine ligand. Inorg. Chim. Acta 1998, 274, 53–63. [Google Scholar] [CrossRef]
- Davies, G.E. Anthibronchoconstrictor activity of two new phosphodiesterase inhibitors, a triazolopyrazine (ICI 58301) and a triazolopyrimidine (ICI 63197). J. Pharm. Pharmacol. 1973, 25, 681–689. [Google Scholar] [CrossRef]
- Fischer, G. 1,2,4-Triazolo[1,5-a]pyrimidines. Adv. Heterocycl. Chem. 1993, 57, 81–138. [Google Scholar]
- Gupton, J.T.; Petrich, S.A.; Hicks, F.A.; Wilkinson, D.R.; Vargas, M.; Hosein, K.N.; Sikorski, J.A. The preparation of heterocyclic appended vinylogous iminium salts and their application to the regioselective preparation of biheterocyclic systems. Heterocycles 1998, 47, 689–702. [Google Scholar] [CrossRef]
- Tominaga, Y.; Sakai, S.; Kohra, S.; Tsuka, J.; Matsuda, Y.; Kobayashi, G. Pyrimidine and fused pyrimidine derivatives. III. Synthesis of s-triazolo[1,5-a]pyrimidines derivatives by using ketene dithioacetals. Chem. Pharm. Bull. 1985, 33, 962–970. [Google Scholar] [CrossRef]
- Oukoloff, K.; Lucero, B.; Francisco, K.R.; Brunden, K.R.; Ballatore, C. 1,2,4-Triazolo[1,5-a]Pyrimidines in Drug Design. Eur. J. Med. Chem. 2019, 165, 332–346. [Google Scholar] [CrossRef]
- Namgoong, S.K.; Lee, H.J.; Kim, Y.S.; Shin, J.-H.; Che, J.-K.; Jang, D.Y.; Kim, G.S.; Yoo, J.K.; Kang, M.-K.; Kil, M.-W.; et al. Synthesis of the quinoline-linked triazolopyrimidine analogues and their interections with the recombinant tobacco acetolactate synthase. Biochem. Biophys. Res. Commun. 1999, 258, 797–801. [Google Scholar] [CrossRef]
- Johnson, T.C.; Mann, R.K.; Schmitzer, P.R.; Gast, R.E. Acetohydroxyacid Synthase Inhibiting Triazolopyrimidine Herbicides. In Bioactive Heterocyclic Compound Classes. Agrochemicals, 1st ed.; Wiley-VCH Verlag & Co.: Weinheim, Germany, 2012; pp. 51–60. [Google Scholar]
- Monte, W.T.; Kleschick, W.A.; Meikle, R.W.; Snider, S.W.; Bordner, J. Methods for controlling the regioselection in the reaction of 3-amino-5-(benzylthiol-1,2,4-triazole with acetylacetaldehyde dimethyl acetal. J. Heterocycl. Chem. 1989, 26, 1393–1396. [Google Scholar] [CrossRef]
- Kleschick, W.A.; Bordner, J. Regioselection in the reaction of 3-amino-5-benzylthio-1,2,4-triazole with unsymmetrical 1,3-diketones. J. Heterocycl. Chem. 1989, 26, 1489–1493. [Google Scholar] [CrossRef]
- Monte, W.T.; Kleschick, W.A.; Bordner, J. Controlling substitution patterns on the 1,2,4-triazolo[1,5-a]pyrimidine ring. Selective removal of chlorine at the 7-position. J. Heterocycl. Chem. 1999, 36, 183–188. [Google Scholar] [CrossRef]
- Shankar, R.B.; Pews, R.G. Synthesis of 1,2,4-triazolo[1,5-a]pyrimidines-2-sulfonamides. J. Heterocycl. Chem. 1993, 30, 169–172. [Google Scholar] [CrossRef]
- Okabe, T.; Bhooshan, B.; Novinson, T.; Hillyard, I.W.; Garner, G.E.; Robins, R.K. Dialkyl bicyclic heterocycles with a bridgehead nitrogen as purine analogs possessing significant cardiac inotropic activity. J. Heterocycl. Chem. 1983, 20, 735–751. [Google Scholar] [CrossRef]
- Williams, L.A. Structure of certain polyazaindenes. VI. Structure of some products obtained from 3-amino-1,2,4-triazoles with acetylacetone and ethyl acetoacetate. J. Chem. Soc. 1960, 1960, 1829–1832. [Google Scholar] [CrossRef]
- Novinson, T.; Springer, R.H.; O’Brien, D.E.; Scholten, M.B.; Miller, J.P.; Robins, R.K. 2-(alkylthiol-1,2,4-triazolo[1,5-a]pyrimidines as adenosine 3′,5′-monophosphate phosphodiesterase inhibitors with potential as new cardiovascular agents. J. Med. Chem. 1982, 25, 420–426. [Google Scholar] [CrossRef]
- Kidwai, M.; Chauhan, R. Nafion-H® Catalyzed Efficient One-Pot Synthesis of Triazolo[5,1-b]Quinazolinones and Triazolo[1,5-a]Pyrimidines: A Green Strategy. J. Mol. Catal. A Chem. 2013, 377, 1–6. [Google Scholar] [CrossRef]
- Petrick, S.A.; Qian, Z.; Santiago, L.M.; Grupton, J.T. The application of unsymmetrical vinylogous iminium salts and related synthons to the preparation of monosubstituted triazolo[1,5-a]pyrimidines. Tetrahedron 1994, 50, 12113–12124. [Google Scholar] [CrossRef]
- Petrick, S.A.; Qian, Z.; Santiago, L.M.; Gupton, J.T. The application of symmetrical vinamidinium salts to the preparation of monosubstituted triazolo[1,5-a]pyrimidines. Heterocycles 1995, 40, 729–742. [Google Scholar]
- Tominaga, Y.; Kohra, S.; Honkawa, H.; Hosomi, A. Synthesis of pyrimidine derivatives and their related compounds using ketene dithioacetals. Heterocycles 1989, 29, 1409–1429. [Google Scholar] [CrossRef]
- Gami, S.P.; Vilapara, K.V.; Khunt, H.R.; Babariya, J.S.; Naliapara, Y.T. Synthesis and Antimicrobal Activities of Some Novel Triazolo[1,5-a]Pyrimidine Derivatives. Int. Lett. Chem., Phys. Astron. 2014, 30, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Gasse, C.; Douguet, D.; Huteau, V.; Marchal, G.; Munier-Lehmann, H.; Pochet, S. Substituted Benzyl-Pyrimidines Targeting Thymidine Monophosphate Kinase of Mycobacterium Tuberculosis: Synthesis and in Vitro Anti-Mycobacterial Activity. Bioorg. Med. Chem. 2008, 16, 6075–6085. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.B.; Fatema, S.; Bhagat, S.S.; Farooqui, M. Thiazolo[3,2- a] Pyrimidones as a Novel Anti-TB Agents. J. Heterocycl. Chem. 2018, 55, 2893–2900. [Google Scholar] [CrossRef]
- Bhatt, J.D.; Chudasama, C.J.; Patel, K.D. Pyrazole Clubbed Triazolo[1,5-a]Pyrimidine Hybrids as an Anti-Tubercular Agents: Synthesis, in Vitro Screening and Molecular Docking Study. Bioorg. Med. Chem. 2015, 23, 7711–7716. [Google Scholar] [CrossRef]
- Anand, P.; Akhter, Y. A Review on Enzyme Complexes of Electron Transport Chain from Mycobacterium tuberculosis as Promising Drug Targets. Int. J. Biol. Macromol. 2022, 212, 474–494. [Google Scholar] [CrossRef]
- Gálvez, J.; García-Domenech, R.; de Julián-Ortiz, J.V.; Soler, R. Topological approach to drug design. J. Chem. Inf. Comp. Sci. 1995, 35, 272–284, Erratum in J. Chem. Inf. Comp. Sci. 1995, 35, 938. [Google Scholar] [CrossRef]
- García-March, F.J.; García-Domenech, R.; Gálvez, J.; Antón-Fos, G.M.; de Julián-Ortiz, J.V.; Giner-Pons, R.; Recio-Iglesias, M.C. Pharmacological Studies of 1-(p-Chlorophenyl)Propanol and 2-(1-Hydroxy-3-Butenyl)Phenol: Two New Non-Narcotic Analgesics Designed by Molecular Connectivity. J. Pharm. Pharmacol. 1997, 49, 10–15. [Google Scholar] [CrossRef] [PubMed]
- García-Domenech, R.; de Julián-Ortiz, J.V. Antimicrobial activity characterization in a heterogeneous group of compounds. J. Chem. Inf. Comput. Sci. 1998, 38, 445–449. [Google Scholar] [CrossRef]
- De Julián-Ortiz, J.V.; Gálvez, J.; Muñoz-Collado, C.; García-Domenech, R.; Gimeno-Cardona, C. Virtual Combinatorial Syntheses and Computational Screening of New Potential Anti-Herpes Compounds. J. Med. Chem. 1999, 42, 3308–3314. [Google Scholar] [CrossRef]
- Bruno-Blanch, L.; Galvez, J.; Garcia-Domenech, R. Topological virtual screening: A way to find new anticonvulsant drugs from chemical diversity. Bioorg. Med. Chem. Lett. 2003, 13, 2749–2754. [Google Scholar] [CrossRef]
- De Julián-Ortiz, J.V.; Besalú, E. Internal Test Sets Studies in a Group of Antimalarials. Int. J. Molec. Sci. 2006, 7, 456–468. [Google Scholar] [CrossRef]
- García-Domenech, R.; Gálvez, J.; de Julián-Ortiz, J.V.; Pogliani, L. Some New Trends in Chemical Graph Theory. Chem. Rev. 2008, 108, 1127–1169. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Domenech, R.; Zanni, R.; Galvez-Llompart, M.; de Julian-Ortiz, J.V. Modeling Anti-Allergic Natural Compounds by Molecular Topology. Comb. Chem. High Throughput Screening 2013, 16, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Zanni, R.; Galvez-Llompart, M.; Morell, C.; Rodríguez-Henche, N.; Díaz-Laviada, I.; Recio-Iglesias, M.C.; Garcia-Domenech, R.; Galvez, J. Novel Cancer Chemotherapy Hits by Molecular Topology: Dual Akt and Beta-Catenin Inhibitors. PLoS ONE 2015, 10, e0124244. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Garit, J.A.; Flores-Balmaseda, N.; Álvarez, O.; Pham-The, H.; Pérez-Doñate, V.; Torrens, F.; Pérez-Giménez, F. Computational Identification of Chemical Compounds with Potential Activity against Leishmania Amazonensis Using Nonlinear Machine Learning Techniques. Curr. Top. Med. Chem. 2019, 18, 2347–2354. [Google Scholar] [CrossRef] [PubMed]
- García-Domenech, R.; de Julián-Ortiz, J.V.; Duart, M.J.; García-Torrecillas, J.M.; Antón-Fos, G.M.; Ríos-Santamarina, I.; de Gregorio Alapont, C.; Gálvez, J. Search of a topological pattern to evaluate toxicity of heterogeneous compounds. SAR QSAR Environ. Res. 2001, 12, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Lavado, G.J.; Baderna, D.; Carnesecchi, E.; Toropova, A.P.; Toropov, A.A.; Dorne, J.L.; Benfenati, E. QSAR Models for Soil Ecotoxicity: Development and Validation of Models to Predict Reproductive Toxicity of Organic Chemicals in the Collembola Folsomia Candida. J. Hazard. Mater. 2022, 423, 127236. [Google Scholar] [CrossRef]
- Gálvez, J.; de Julián-Ortiz, J.V.; García-Domenech, R. General topological patterns of known drugs. J. Mol. Graphics Modell. 2001, 20, 84–94. [Google Scholar] [CrossRef]
- Kier, L.B.; Murray, W.J.; Randić, M.; Hall, L.H. Molecular Connectivity V: Connectivity series concept applied to density. J. Pharm. Sci. 1976, 65, 1226–1230. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. Molecular Connectivity VII: Specific treatment of heteroatoms. J. Pharm. Sci. 1976, 65, 1806–1809. [Google Scholar] [CrossRef]
- Randić, M. The connectivity index 25 years after. J. Mol. Graphics Modell. 2001, 20, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E.; Uriarte, E. Recent advances on the role of topological indices in drug discovery research. Curr. Med. Chem. 2001, 8, 1573–1588. [Google Scholar] [CrossRef] [PubMed]
- Selassie, C.D.; Mekapati, S.B.; Verma, R.P. QSAR: Then and now. Curr. Top. Med. Chem. 2002, 2, 1357–1379. [Google Scholar] [CrossRef]
- Ivanciuc, O. Cheminform Abstract: Chemical Graphs, Molecular Matrices and Topological Indices in Chemoinformatics and Quantitative Structure-Activity Relationships. ChemInform 2013, 44, 153–163. [Google Scholar] [CrossRef]
- Havare, Ö.Ç. Quantitative Structure Analysis of Some Molecules in Drugs Used in the Treatment of COVID-19 with Topological Indices. Polycyclic Aromat. Compd. 2021, 42, 1–12. [Google Scholar] [CrossRef]
- Balaban, A.T. Highly discriminating distance based topological index. Chem. Phys. Lett. 1982, 89, 399–404. [Google Scholar] [CrossRef]
- Bonchev, D. Overall connectivity: A next generation molecular connectivity. J. Mol. Graphics Modell. 2001, 20, 65–75. [Google Scholar] [CrossRef]
- Estrada, E.; Molina, E. Novel local (fragment-based) topological molecular descriptors for QSpr/QSAR and molecular design. J. Mol. Graphics Modell. 2001, 20, 54–64. [Google Scholar] [CrossRef]
- Tropsha, A.; Zheng, W. Identification of the descriptor pharmacophores using variable selection QSAR: Applications to database mining. Curr. Pharm. Des. 2001, 7, 599–612. [Google Scholar] [CrossRef]
- Gozalbes, R.; Doucet, J.P.; Derouin, F. Application of topological descriptors in QSAR and drug design: History and new trends. Curr. Drug. Targets Infect. Disord. 2002, 2, 93–102. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Ling, L.H.; Tham, S.Y.; Alany, R.G. Molecular descriptors that influence the amount of drugs transfer into human breast milk. J. Pharm. Biomed. Anal. 2002, 29, 103–119. [Google Scholar] [CrossRef]
- Basak, S.C.; Mills, D.R.; Balaban, A.T.; Gute, B.D. Cheminform Abstract: Prediction of Mutagenicity of Aromatic and Heteroaromatic Amines from Structure: A Hierarchical QSAR Approach. ChemInform 2010, 32. [Google Scholar] [CrossRef]
- Orosz, Á.; Héberger, K.; Rácz, A. Comparison of Descriptor- and Fingerprint Sets in Machine Learning Models for ADME-Tox Targets. Front. Chem. 2022, 10, 852893. [Google Scholar] [CrossRef]
- Jacobs, M.R. Activity of Quinolones against Mycobacteria. Drugs 1995, 49 (Suppl. 2), 67–75. [Google Scholar] [CrossRef]
- García-Sánchez, J.E.; López, R.; Prieto, J. Antimicrobianos en Medicina; Prous Science: Barcelona, Spain, 2006. [Google Scholar]
- O’Neil, M.J. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- García-Domenech, R. DesMol. In Unidad de Investigación de Diseño de Fármacos y Conectividad Molecular, Dep; Química-Física, Facultad de Farmacia, Universitat de València: Burjassot, Valencia, Spain, 2013. [Google Scholar]
- Wiener, H. Structural Determination of Paraffin Boiling Points. J. Am. Chem. Soc. 1947, 69, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, J.; García-Domenech, R.; Salabert, M.T.; Soler, R. Charge indices. New topological descriptors. J. Chem. Inf. Comp. Sci. 1994, 34, 520–525. [Google Scholar] [CrossRef]
- De Julián-Ortiz, J.V.; Besalú, E.; García-Domenech, R. True prediction by consensus for small sets of cyclooxigenase-2 inhibitors. Indian J. Chem. 2003, 42, 1392–1404. [Google Scholar]
- Font, D.; Heras, M.; Villalgordo, J.M. Development of an efficient and straightforward methodology toward the synthesis of molecularly diverse 2,6-disubstituted 3,4-dihydropyrimidin-4(3h)-ones. Synthesis 2002, 2002, 1833–1842. [Google Scholar] [CrossRef]
- Font, D.; Heras, M.; Villalgordo, J.M. Solution- and solid-phase parallel synthesis of 4-alkoxy-substituted pyrimidines with high molecular diversity. J. Comb. Chem. 2003, 5, 311–321. [Google Scholar] [CrossRef]
- Petasis, N.A.; Goodman, A.; Zavialov, I.A. A new synthesis of α-arylglycines from aryl boronic acids. Tetrahedron 1997, 53, 16463–16470. [Google Scholar] [CrossRef]
- Petasis, N.A.; Zavialov, I.A. A new and practical synthesis of α-amino acids from alkenyl boronic acids. J. Am. Chem. Soc. 1997, 119, 445–446. [Google Scholar] [CrossRef]
- BMDP New System 2.0; Statistical Solutions Ltd.: Cork, Ireland, 2004.
- Gálvez, J.; García-Domenech, R.; de Gregorio Alapont, C.; de Julián-Ortiz, J.V.; Popa, L. Pharmacological distribution diagrams: A tool for the new drug design. J. Mol. Graphics 1996, 14, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Shinnick, T.M.; Good, R.C. Mycobacterial taxonomy. Eur. J. Clin. Microbiol. Infect. Dis. 1994, 13, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Franzblau, S.G.; Witzig, R.S.; McLaughinhlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid low technology MIC determination with clinical Mycobacterium isolates by using the microplate alamar blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [CrossRef] [PubMed]
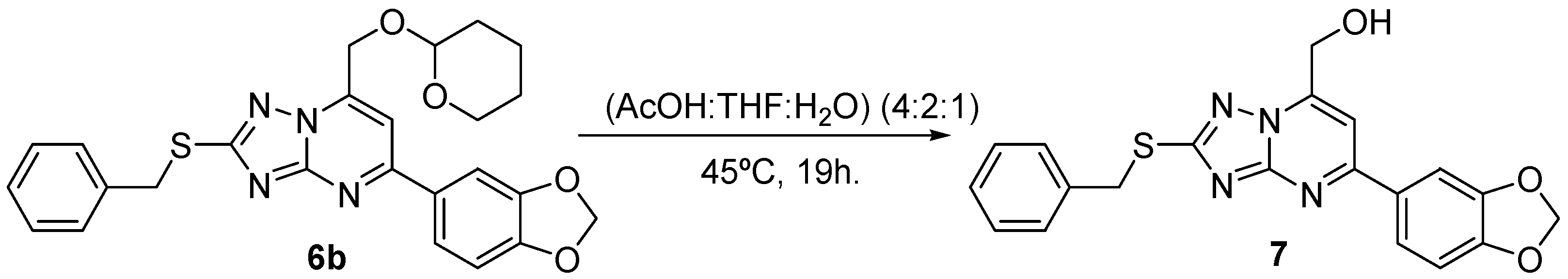

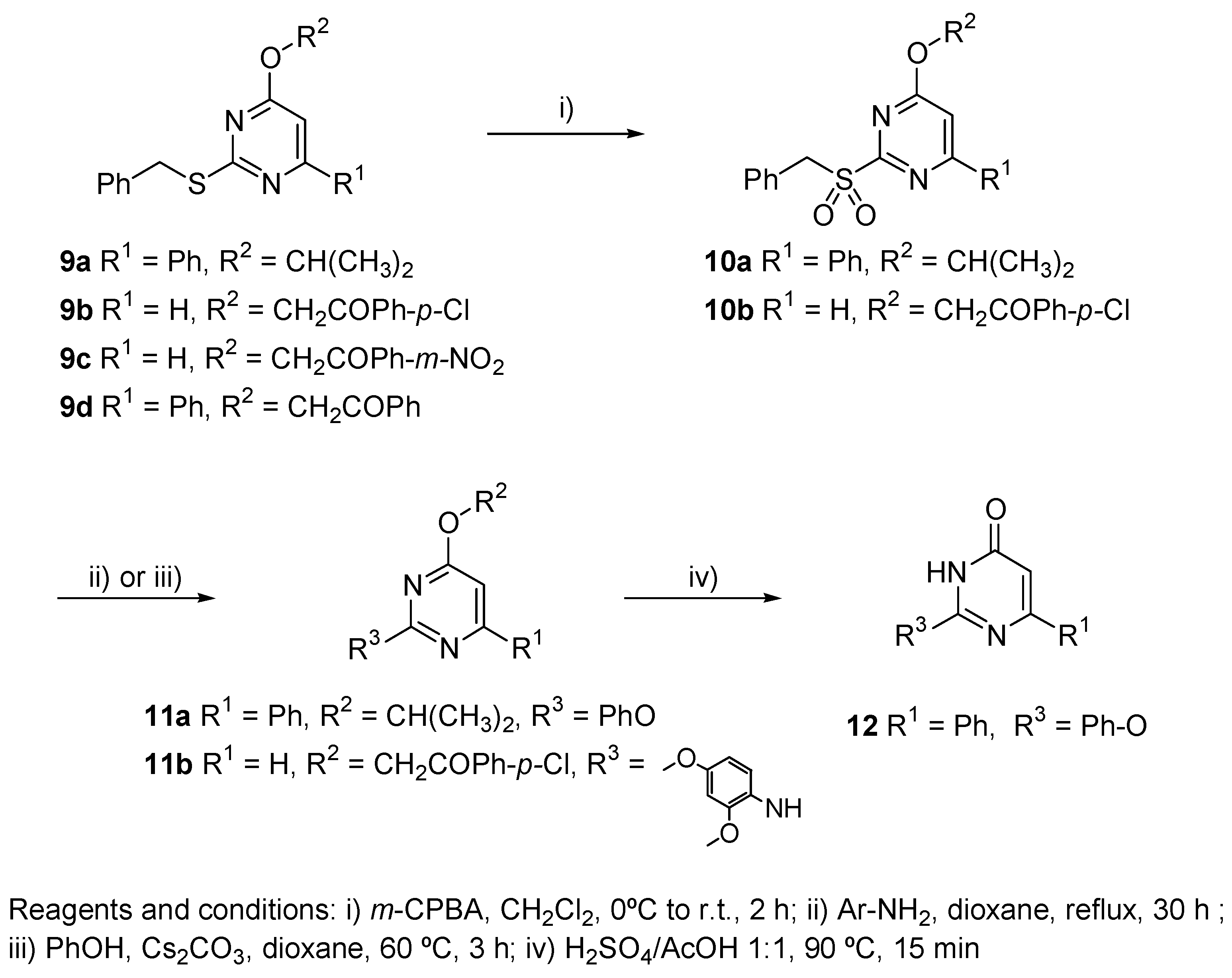


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
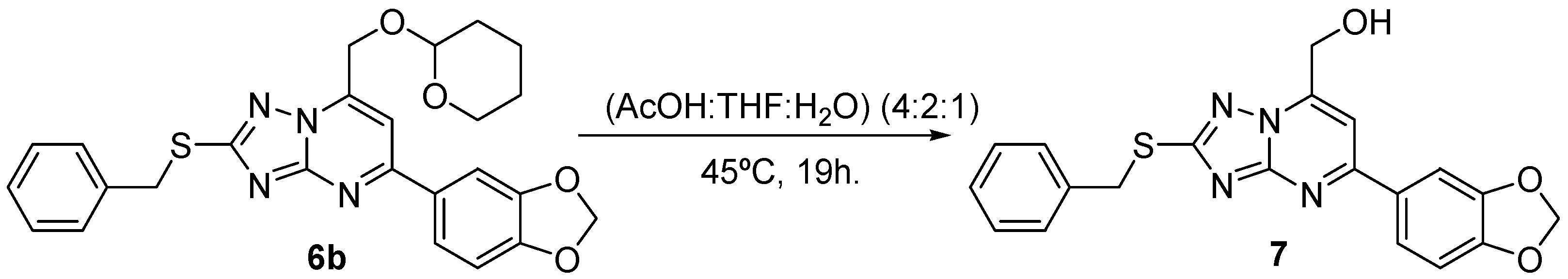{kind=link}
{kind=link}
{kind=link}
{kind=link}
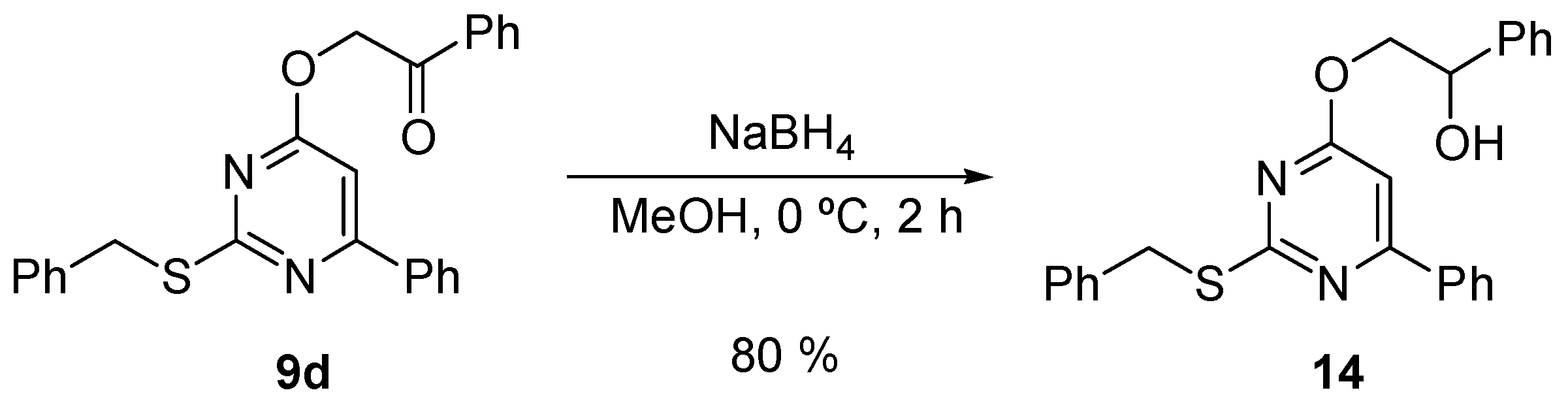{kind=link}
| Symbol | Name | Definition | Reference |
|---|---|---|---|
| N | Molecular size | Number of non-hydrogen atoms. | [68] |
| Vk k = 3, 4 | Vertices of degree k | Number of atoms having k bonds, σ or π, to non-hydrogen atoms. | [68] |
| R | Ramification | Number of single structural branches. | [68] |
| W | Wiener path number | Sum of the distances between any two atoms in terms of bonds. | [100] |
| L | Length | Maximal distance between atoms in terms of bonds. | [68] |
| PRk k = 0–3 | Pairs of ramifications at distance k | Number of pairs of single branches at distance k in terms of bonds. | [68] |
| kχt k = 0–4 t = p, c, pc | Randić-like indices of order k and type path (p), cluster (c) and path-cluster (pc) | δi, number of bonds, σ or π, of the atom i to non-hydrogen atoms. Sj, jth sub-structure of order k and type t. | [71,81,82] |
| kχtv k = 0–4 t = p, c, pc | Kier-Hall indices of order k and type path (p), cluster (c) and path-cluster (pc) | δiv, Kier-Hall valence of the atom i. Sj,jth sub-structure of order k and type t. | [71,81,82] |
| Gk k = 1–5 | Topological charge indices of order k | , product of the adjacency and inverse squared distance matrices for the hydrogen-depleted molecular graph. D, distance matrix. δ, Kronecker delta. | [68,101] |
| Gkv k = 1–5 | Valence topological charge indices of order k | product of the electronegativity-modified adjacency and inverse squared distance matrices for the hydrogen-depleted molecular graph. D, distance matrix. δ, Kronecker delta. | [68,101] |
| Jk k = 1–5 | Pondered topological charge indices of order k | [68,101] | |
| Jkv k = 1–5 | Pondered valence topological charge indices of order k | [68,101] | |
| kDt k = 0–4 t = p, c, pc | Connectivity differences of order k and type path (p), cluster (c) and path-cluster (pc) | [68] | |
| Ek k = 1–5 | Topological charge differences of order k | [102] | |
| Fk k = 1–5 | Pondered topological charge differences of order k | [102] | |
| kCt k = 0–4 t = p, c, pc | Connectivity quotients of order k and type path (p), cluster (c) and path-cluster (pc) | [68] | |
| kQt k = 0–4 t = p, c, pc | Inverse connectivity quotients of order k and type path (p), cluster (c) and path-cluster (pc) | [102] | |
| CGk k = 1–5 | Topological charge quotients of order k | [102] | |
| QGk k = 1–5 | Inverse topological charge quotients of order k | [102] |
| Active Group | Inactive Group | ||||||
|---|---|---|---|---|---|---|---|
| Compound | DF | Prob. a Active | Predicted Activity b | Compound | DF | Prob. a Inactive | Predicted Activity b |
| Streptonicozid | 3.14 | 1.000 | + | Butibufen | −3.14 | 1.000 | − |
| Tobramycin | 3.49 | 1.000 | + | Aldicarb | −2.74 | 1.000 | − |
| Kanamycin | 3.88 | 1.000 | + | Antrafenine | −2.55 | 1.000 | − |
| Amikacin | 4.14 | 1.000 | + | Carprofen | −1.89 | 0.997 | − |
| Dihydrostreptomycin | 2.80 | 0.999 | + | Beclobrate | −1.84 | 0.997 | − |
| Streptomycin | 2.32 | 0.997 | + | Benzoctamine | −1.77 | 0.996 | − |
| Ethambutol | 2.23 | 0.996 | + | Carmofur | −1.77 | 0.996 | − |
| Pyrazinamide | 1.97 | 0.992 | + | Aminothiazole | −1.73 | 0.996 | − |
| Enviomycin | 1.92 | 0.991 | + | Acifran | −1.72 | 0.996 | − |
| Ofloxacin | 1.82 | 0.988 | + | Brilliant Blue | −1.72 | 0.996 | − |
| Moxifloxacin | 1.66 | 0.982 | + | Amitraz | −1.68 | 0.995 | − |
| Gatifloxacin | 1.66 | 0.981 | + | Clofibrate | −1.62 | 0.994 | − |
| Rifampin | 1.60 | 0.978 | + | Paraoxon | −1.52 | 0.992 | − |
| Azithromycin | 1.49 | 0.971 | + | Piroxicam | −1.39 | 0.989 | − |
| Verazide | 1.44 | 0.967 | + | Carmustine | −1.11 | 0.977 | − |
| Isoniazid | 1.20 | 0.938 | + | Ornithine | −1.11 | 0.977 | − |
| Capreomycin | 1.16 | 0.930 | + | Alpidem | −1.10 | 0.976 | − |
| Sparfloxacin | 0.98 | 0.889 | + | Alprazolam | −1.04 | 0.972 | − |
| Clarithromycin | 0.93 | 0.874 | + | Bixin | −1.04 | 0.972 | − |
| Salinazid | 0.92 | 0.871 | + | Benzoic Acid | −1.03 | 0.971 | − |
| Tuberin | 0.91 | 0.869 | + | Amsacrine | −0.98 | 0.967 | − |
| Clofazimine | 0.13 | 0.432 | NC | Azaserine | −0.96 | 0.965 | − |
| Imipenem | 0.09 | 0.404 | NC | Theofibrate | −0.88 | 0.957 | − |
| Ph−Aminosalicylate | −0.200 | 0.230 | − | Azacosterol | −0.83 | 0.951 | − |
| PAS | −1.000 | 0.031 | − | Allicin | −0.77 | 0.943 | − |
| Prazepam | −0.77 | 0.943 | − | ||||
| Camazepam | −0.71 | 0.933 | − | ||||
| Aminopromazine | −0.57 | 0.904 | − | ||||
| Bromazepam | −0.45 | 0.870 | − | ||||
| Carnitine | −0.32 | 0.824 | − | ||||
| Acipimox | −0.28 | 0.809 | − | ||||
| Buspirone | −0.03 | 0.677 | − | ||||
| Azapicyl | 0.03 | 0.640 | − | ||||
| Acronine | 0.11 | 0.588 | NC | ||||
| Captodiamine | 0.28 | 0.470 | NC | ||||
| Run No. | λ | Training Group | Test Group | ||
|---|---|---|---|---|---|
| (+) | (−) | (+) | (−) | ||
| 1 | 0.28 | 90% (18/2) | 100% (0/26) | 60% (3/2) | 100% (0/9) |
| 2 | 0.31 | 87% (20/3) | 96% (1/27) | 50% (1/1) | 100% (0/7) |
| 3 | 0.35 | 86% (19/3) | 89% (3/24) | 100% (3/0) | 75% (2/6) |
| 4 | 0.33 | 82% (14/3) | 100% (0/27) | 88% (7/1) | 100% (0/8) |
| 5 | 0.33 | 91% (20/2) | 96% (1/27) | 100% (3/0) | 86% (1/6) |
| Average | - | 87% | 96% | 80% | 92% |
| DF | 0.34 | 84% (21/4) | 97% (1/34) | No | No |
| Active Group | Inactive Group | ||||||
|---|---|---|---|---|---|---|---|
| Compound | DF | Prob. a Active | Predicted Activity b | Compound | DF | Prob. a Inactive | Predicted Activity b |
| Morphazinamide | 4.32 | 1.000 | + | Canthaxanthin | −3.46 | 1.000 | − |
| Neomycin | 5.02 | 1.000 | + | Genite | −2.57 | 1.000 | − |
| Tubercidin | 2.85 | 0.999 | + | Altretamine | −1.91 | 0.997 | − |
| Ciprofloxacin | 2.15 | 0.995 | + | Etifoxin | −1.86 | 0.997 | − |
| Viomycin | 1.75 | 0.986 | + | Dichlone | −1.78 | 0.996 | − |
| Rifabutin | 0.73 | 0.799 | + | Feprazone | −1.47 | 0.991 | − |
| Ethionamide | −1.30 | 0.014 | − | Antipyrine | −0.44 | 0.868 | − |
| Benorylate | −0.25 | 0.792 | − | ||||
| Chloropal | 0.19 | 0.526 | NC | ||||
| Glucosamine | 1.77 | 0.014 | + | ||||
| Chemical Scaffolds | R1 | R2 |
|---|---|---|
 |  | 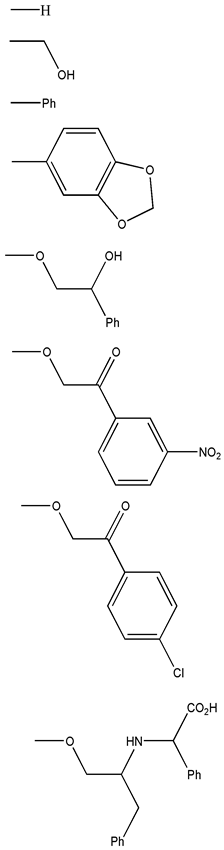 |
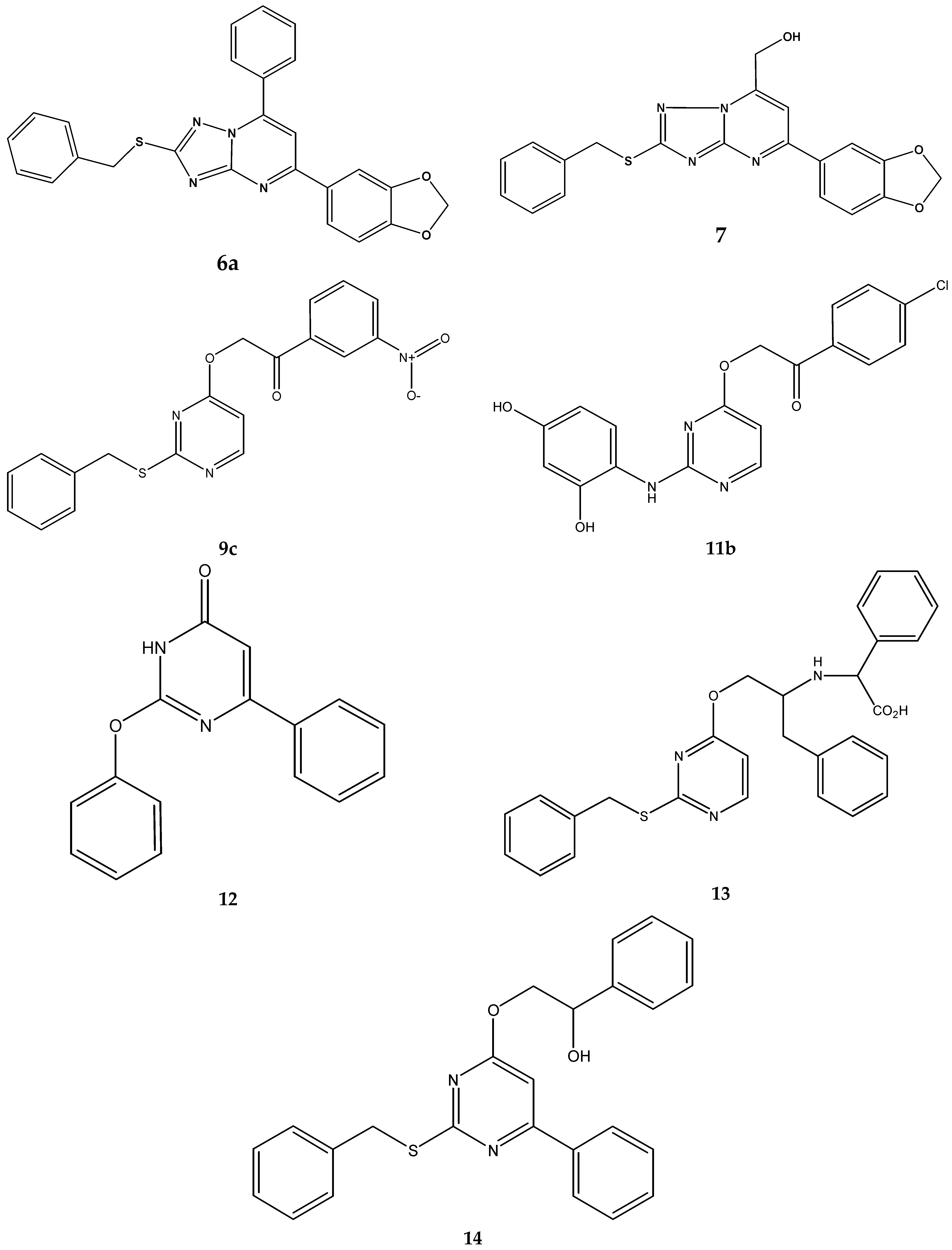
| Compound | MIC Range/mg/L | MIC50/mg/L | MIC90/mg/L | Molecular Mass/Da | MIC50/μM | MIC90/μM |
|---|---|---|---|---|---|---|
| Ethambutol | 1–8 | 4.0 | 4.0 | 204.3 | 19.6 | 19.6 |
| Isoniazid | 0.05–0.2 | 0.05 | 0.05 | 137.1 | 0.4 | 0.4 |
| Rifampin | 0.125–1 | 0.125 | 0.5 | 823.0 | 0.2 | 0.6 |
| Streptomycin | 0.125–0.5 | 0.25 | 0.5 | 581.6 | 0.4 | 0.9 |
| Compound | MIC Range/mg/L | MIC50/mg/L | MIC90/mg/L | Molecular Mass/Da | MIC50/μM | MIC90/μM |
|---|---|---|---|---|---|---|
| 6a | 64 | 64 | 64 | 438.5 | 146.0 | 146.0 |
| 7 | 32 | 32 | 32 | 392.4 | 81.5 | 81.5 |
| 9c | 64 | 64 | 64 | 381.4 | 167.8 | 167.8 |
| 11b | 16 - 64 | 32 | 64 | 399.8 | 80.0 | 160.1 |
| 12 | 32 | 32 | 32 | 264.3 | 121.1 | 121.1 |
| 13 | 32 - 64 | 64 | 64 | 485.6 | 131.8 | 131.8 |
| 14 | >128 | >128 | >128 | 414.5 | >308.8 | >308.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-García, Á.; Julián-Ortiz, J.V.d.; Gálvez, J.; Font, D.; Ayats, C.; Guna Serrano, M.d.R.; Muñoz-Collado, C.; Borrás, R.; Villalgordo, J.M. Similarity-Based Virtual Screening to Find Antituberculosis Agents Based on Novel Scaffolds: Design, Syntheses and Pharmacological Assays. Int. J. Mol. Sci. 2022, 23, 15057. https://doi.org/10.3390/ijms232315057
García-García Á, Julián-Ortiz JVd, Gálvez J, Font D, Ayats C, Guna Serrano MdR, Muñoz-Collado C, Borrás R, Villalgordo JM. Similarity-Based Virtual Screening to Find Antituberculosis Agents Based on Novel Scaffolds: Design, Syntheses and Pharmacological Assays. International Journal of Molecular Sciences. 2022; 23(23):15057. https://doi.org/10.3390/ijms232315057
Chicago/Turabian StyleGarcía-García, Ángela, Jesus Vicente de Julián-Ortiz, Jorge Gálvez, David Font, Carles Ayats, María del Remedio Guna Serrano, Carlos Muñoz-Collado, Rafael Borrás, and José Manuel Villalgordo. 2022. "Similarity-Based Virtual Screening to Find Antituberculosis Agents Based on Novel Scaffolds: Design, Syntheses and Pharmacological Assays" International Journal of Molecular Sciences 23, no. 23: 15057. https://doi.org/10.3390/ijms232315057
APA StyleGarcía-García, Á., Julián-Ortiz, J. V. d., Gálvez, J., Font, D., Ayats, C., Guna Serrano, M. d. R., Muñoz-Collado, C., Borrás, R., & Villalgordo, J. M. (2022). Similarity-Based Virtual Screening to Find Antituberculosis Agents Based on Novel Scaffolds: Design, Syntheses and Pharmacological Assays. International Journal of Molecular Sciences, 23(23), 15057. https://doi.org/10.3390/ijms232315057