
Validation of Promoters and Codon Optimization on CRISPR/Cas9-Engineered Jurkat Cells Stably Expressing αRep4E3 for Interfering with HIV-1 Replication
 and
and 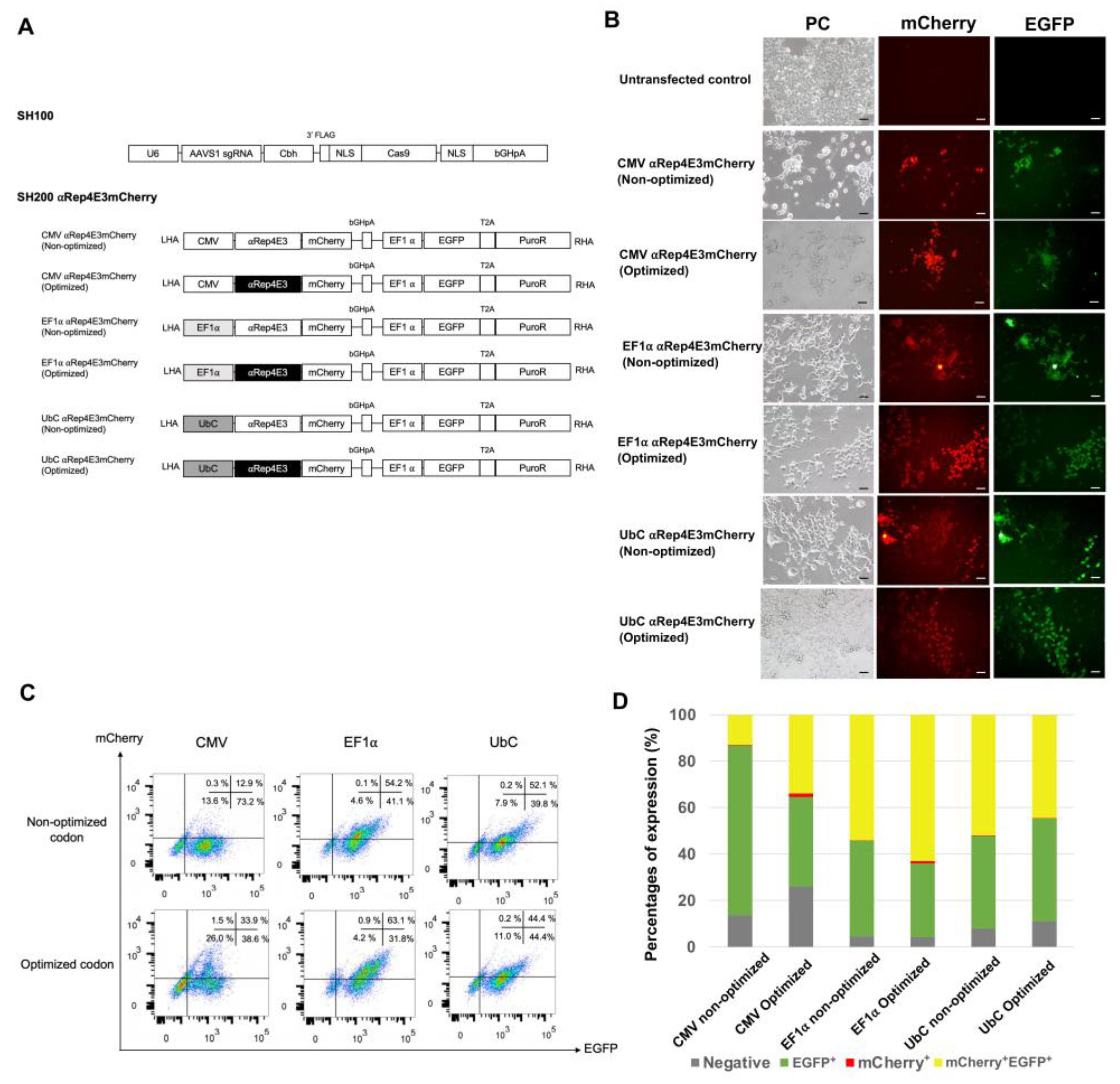{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Expression of the αRep4E3mCherry Protein in HEK293T Cells under Various Constitutive Promoters
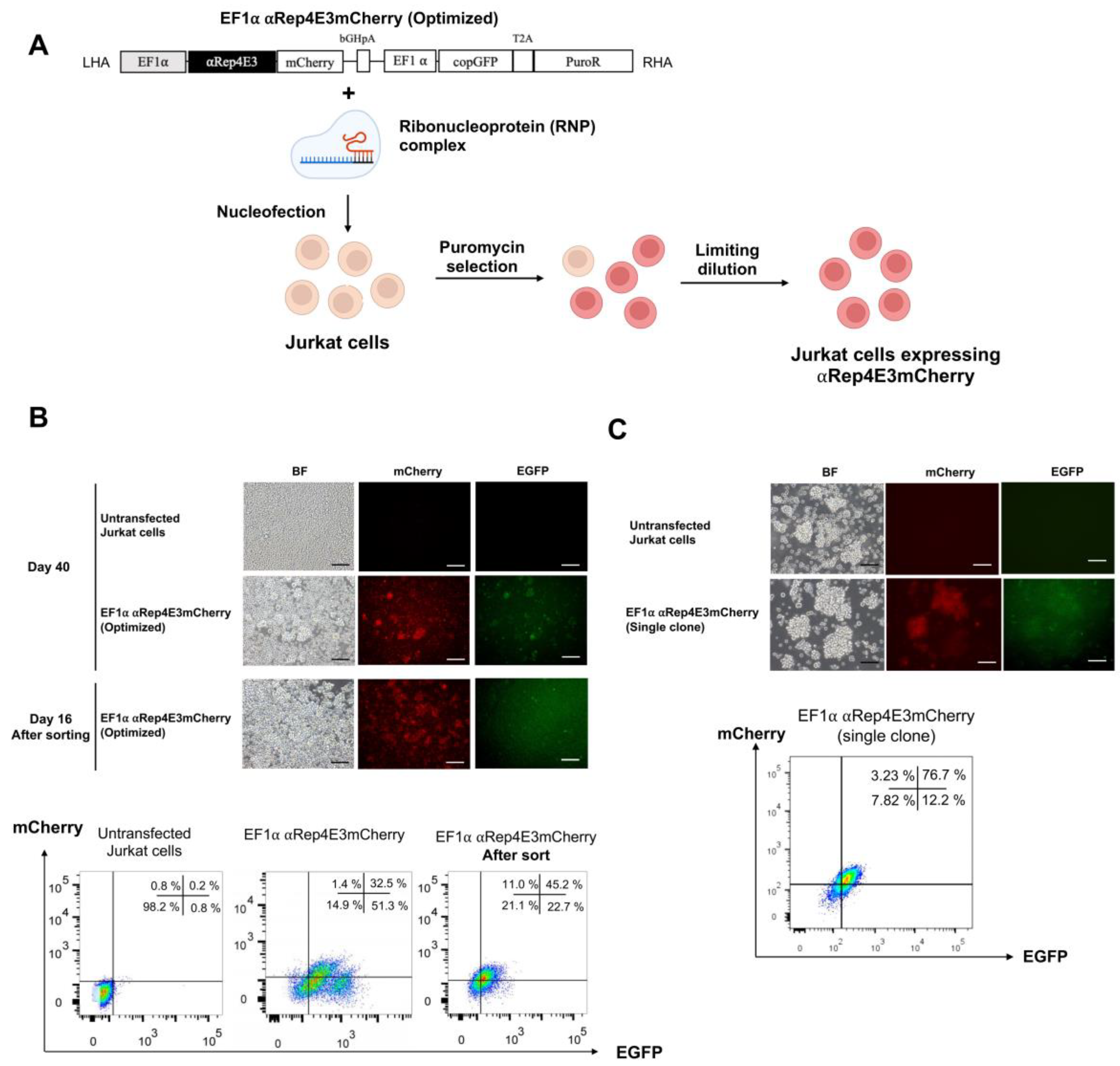
2.2. CRISPR/Cas9-Mediated αRep4E3mCherry Gene Knock-in in the Jurkat Cell Line
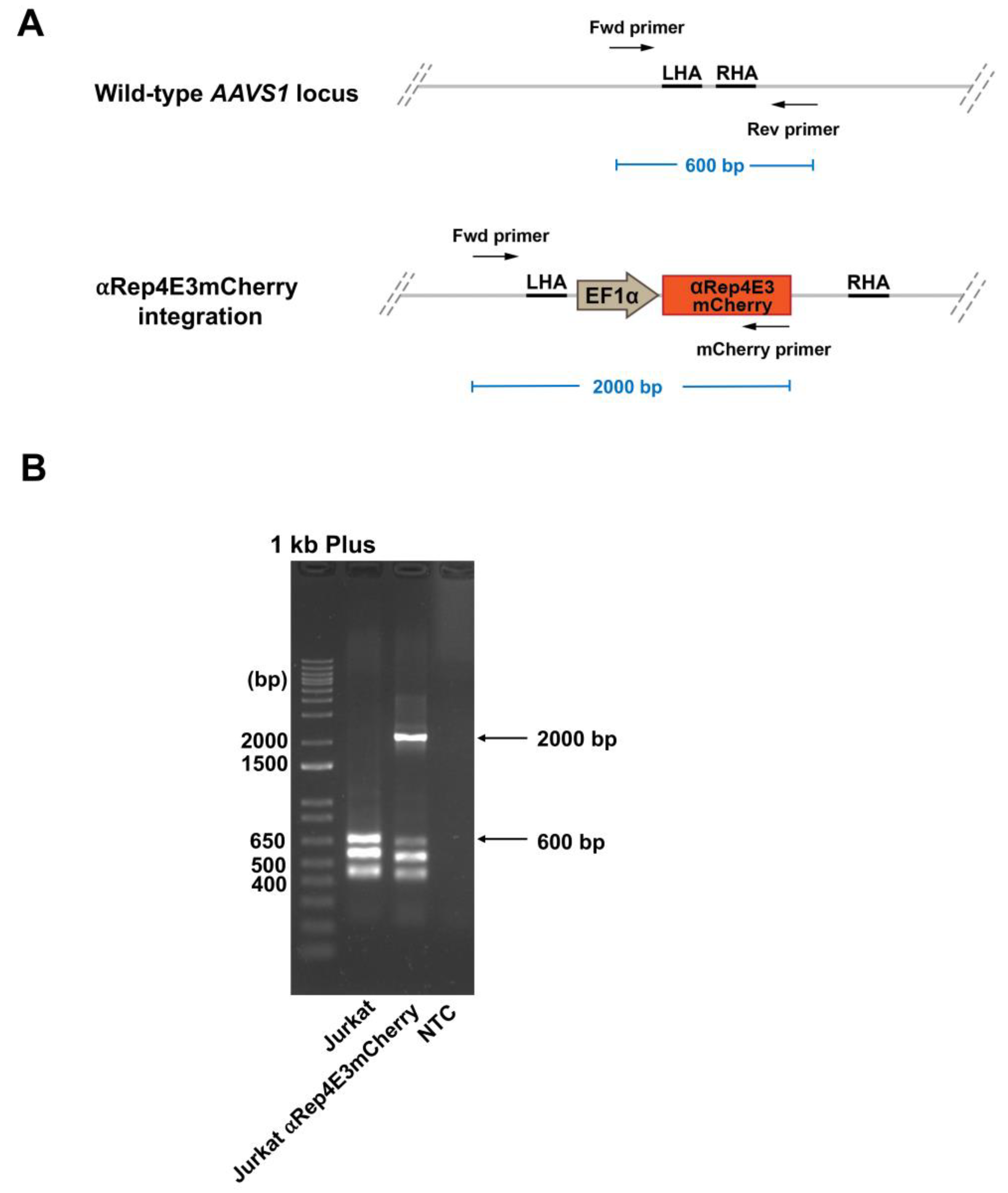
2.3. Analysis of the αRep4E3mcherry Transgene Integration in the Jurkat Clone by Multiplex PCR
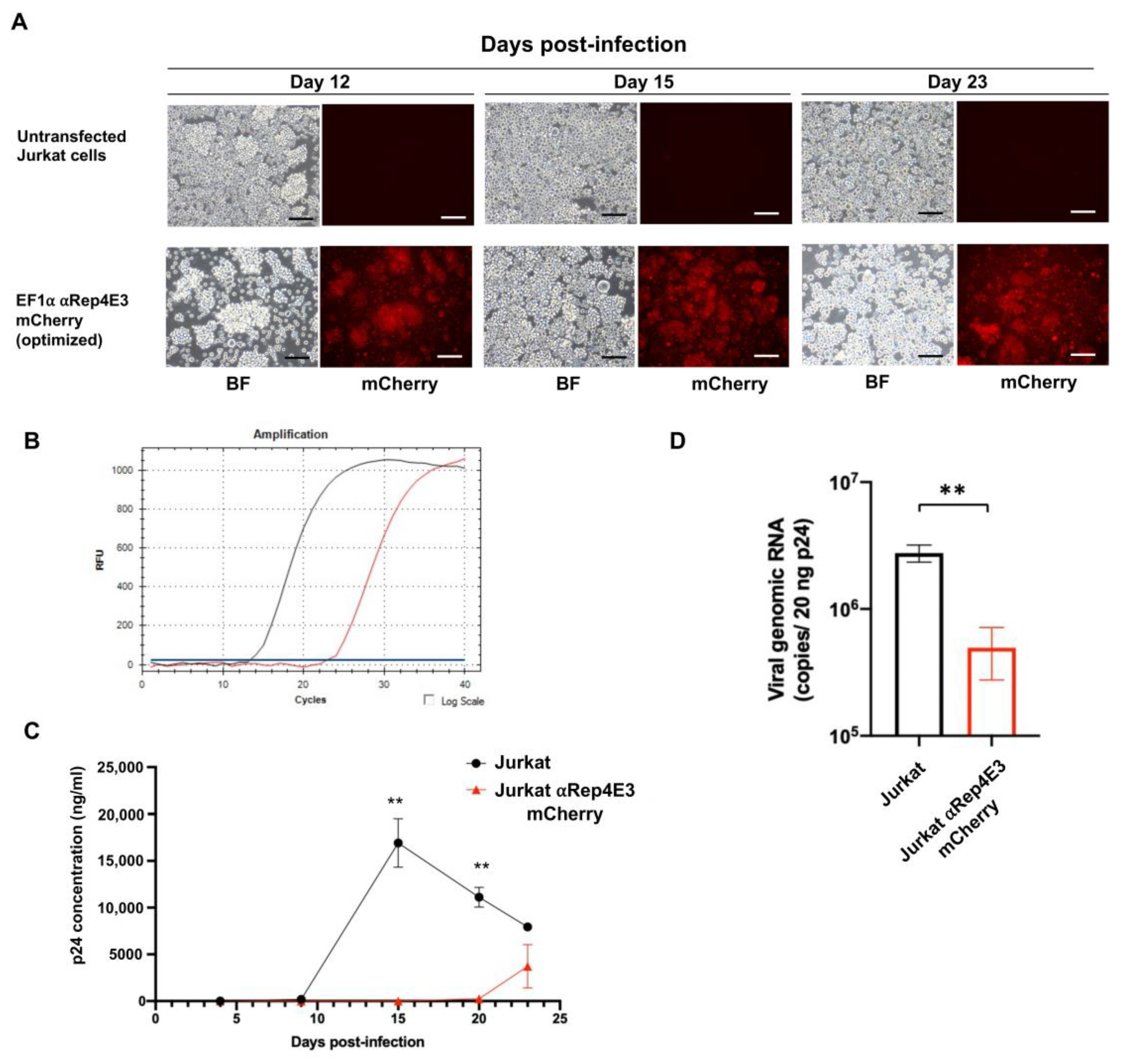
2.4. The αRep4E3mCherry Jurkat Clone Conferred Resistance to HIV-1 Infection
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Optimization of Codon Usages
4.3. Construction of the Donor Plasmid Vector Harboring αRep4E3mCherry Gene
4.4. CRISPR/Cas9 αRep4E3mCherry Gene Delivery and Expression in Cell Lines
4.5. Fluorescence-Activated Cell Sorting (FACS)
4.6. Clonal Isolation by Limiting Dilution
4.7. Determination of the αRep4E3mCherry Integration Site in the Knock-in Cell Lines
4.8. Viral Stocks
4.9. HIV-1 Infection
4.10. HIV-1 Integration Assay
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Maguire, C.A.; Ramirez, S.H.; Merkel, S.F.; Sena-Esteves, M.; Breakefield, X.O. Gene therapy for the nervous system: Challenges and new strategies. Neurotherapeutics 2014, 11, 817–839. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.; Derrick-Roberts, A.L.; Byers, S.; Anson, D.S. Correction of murine mucopolysaccharidosis type IIIA central nervous system pathology by intracerebroventricular lentiviral-mediated gene delivery. J. Gene Med. 2014, 16, 374-87. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Li, H.P.; Hung, Y.H.; Leu, Y.W.; Wu, W.H.; Wang, F.S.; Lee, K.D.; Chang, P.J.; Wu, C.S.; Lu, Y.J.; et al. Targeted methylation of CMV and E1A viral promoters. Biochem. Biophys. Res. Commun. 2010, 402, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Osterlehner, A.; Simmeth, S.; Gopfert, U. Promoter methylation and transgene copy numbers predict unstable protein production in recombinant Chinese hamster ovary cell lines. Biotechnol. Bioeng. 2011, 108, 2670-81. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Z.; Tian, Z.; Zhang, X.; Xu, D.; Li, Q.; Zhang, J.; Wang, T. The EF-1alpha promoter maintains high-level transgene expression from episomal vectors in transfected CHO-K1 cells. J. Cell. Mol. Med. 2017, 21, 3044–3054. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Uetsuki, T.; Kaziro, Y.; Yamaguchi, N.; Sugano, S. Use of the human elongation factor 1 alpha promoter as a versatile and efficient expression system. Gene 1990, 91, 217–223. [Google Scholar] [CrossRef]
- Liew, C.G.; Draper, J.S.; Walsh, J.; Moore, H.; Andrews, P.W. Transient and stable transgene expression in human embryonic stem cells. Stem Cells 2007, 25, 1521–1528. [Google Scholar] [CrossRef]
- Norrman, K.; Fischer, Y.; Bonnamy, B.; Wolfhagen Sand, F.; Ravassard, P.; Semb, H. Quantitative comparison of constitutive promoters in human ES cells. PLoS ONE 2010, 5, e12413. [Google Scholar] [CrossRef]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.; Claassens, N.J.; Soll, D.; van der Oost, J. Codon Bias as a Means to Fine-Tune Gene Expression. Mol. Cell. 2015, 59, 149-61. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. A code within the genetic code: Codon usage regulates co-translational protein folding. Cell Commun. Signal. 2020, 18, 145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.H.; Fu, J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 2016, 113, E6117-E25. [Google Scholar] [CrossRef] [PubMed]
- Jeacock, L.; Faria, J.; Horn, D. Codon usage bias controls mRNA and protein abundance in trypanosomatids. eLife 2018, 7, e32496. [Google Scholar] [CrossRef]
- Coghlan, A.; Wolfe, K.H. Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae. Yeast 2000, 16, 1131-45. [Google Scholar] [CrossRef]
- dos Reis, M.; Wernisch, L.; Savva, R. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res. 2003, 31, 6976–6985. [Google Scholar] [CrossRef]
- Huo, X.; Liu, S.; Li, Y.; Wei, H.; Gao, J.; Yan, Y.; Zhang, G.; Liu, M. Analysis of synonymous codon usage of transcriptome database in Rheum palmatum. PeerJ 2021, 9, e10450. [Google Scholar] [CrossRef]
- Hadpech, S.; Nangola, S.; Chupradit, K.; Fanhchaksai, K.; Furnon, W.; Urvoas, A.; Valerio-Lepiniec, M.; Minard, P.; Boulanger, P.; Hong, S.S.; et al. Alpha-helicoidal HEAT-like Repeat Proteins (alphaRep) Selected as Interactors of HIV-1 Nucleocapsid Negatively Interfere with Viral Genome Packaging and Virus Maturation. Sci. Rep. 2017, 7, 16335. [Google Scholar] [CrossRef]
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated With Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 2016, 58, 1159–1166. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281-308. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Maguire, S.; Davis, L.A.; Alexander, M.; Yang, F.; Chandran, S.; Ffrench-Constant, C.; Pedersen, R.A. Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells 2008, 26, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Inderbitzin, A.; Loosli, T.; Kouyos, R.D.; Metzner, K.J. Quantification of transgene expression in GSH AAVS1 with a novel CRISPR/Cas9-based approach reveals high transcriptional variation. Mol. Ther. Methods Clin. Dev. 2022, 26, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Schambach, A. Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 2016, 24, 678-84. [Google Scholar] [CrossRef]
- Chupradit, K.; Thongsin, N.; Tayapiwatana, C.; Wattanapanitch, M. A precise gene delivery approach for human induced pluripotent stem cells using Cas9 RNP complex and recombinant AAV6 donor vectors. PLoS ONE 2022, 17, e0270963. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; de Saint Basile, G.; Cavazzana-Calvo, M. Gene therapy of X-linked severe combined immunodeficiency. Methods Mol. Biol. 2003, 215, 247–259. [Google Scholar]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef]
- Chen, K.; Gao, C. Targeted genome modification technologies and their applications in crop improvements. Plant Cell Rep. 2014, 33, 575–583. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.; Jin, X.; Wang, Q.; Yang, K.; Li, C.; Xiao, Q.; Hou, P.; Liu, S.; Wu, S.; et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci. 2017, 7, 47. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Nangola, S.; Urvoas, A.; Valerio-Lepiniec, M.; Khamaikawin, W.; Sakkhachornphop, S.; Hong, S.S.; Boulanger, P.; Minard, P.; Tayapiwatana, C. Antiviral activity of recombinant ankyrin targeted to the capsid domain of HIV-1 Gag polyprotein. Retrovirology 2012, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Sakkhachornphop, S.; Barbas, C.F., 3rd; Keawvichit, R.; Wongworapat, K.; Tayapiwatana, C. Zinc finger protein designed to target 2-long terminal repeat junctions interferes with human immunodeficiency virus integration. Hum. Gene. Ther. 2012, 23, 932–942. [Google Scholar] [CrossRef]
- Darlix, J.L.; Lapadat-Tapolsky, M.; de Rocquigny, H.; Roques, B.P. First glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Mol. Biol. 1995, 254, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.G.; Guo, J.; Rouzina, I.; Musier-Forsyth, K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: Critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 2005, 80, 217–286. [Google Scholar]
- Muriaux, D.; Darlix, J.L. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 2010, 7, 744–753. [Google Scholar] [CrossRef]
- Mougel, M.; Cimarelli, A.; Darlix, J.L. Implications of the nucleocapsid and the microenvironment in retroviral reverse transcription. Viruses 2010, 2, 939–960. [Google Scholar] [CrossRef]
- Shin, S.; Kim, S.H.; Shin, S.W.; Grav, L.M.; Pedersen, L.E.; Lee, J.S.; Lee, G.M. Comprehensive Analysis of Genomic Safe Harbors as Target Sites for Stable Expression of the Heterologous Gene in HEK293 Cells. ACS Synth. Biol. 2020, 9, 1263–1269. [Google Scholar] [CrossRef]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef]
- Leppek, K.; Byeon, G.W.; Kladwang, W.; Wayment-Steele, H.K.; Kerr, C.H.; Xu, A.F.; Kim, D.S.; Topkar, V.V.; Choe, C.; Rothschild, D.; et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 2022, 13, 1356. [Google Scholar] [CrossRef]
- Agashe, D.; Martinez-Gomez, N.C.; Drummond, D.A.; Marx, C.J. Good codons, bad transcript: Large reductions in gene expression and fitness arising from synonymous mutations in a key enzyme. Mol. Biol. Evol. 2013, 30, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Sauna, Z.E.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M.; Nussinov, R. Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J. Mol. Biol. 2008, 383, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P.; Oliveira, J.L.; Frommlet, J.; Santos, M.A.; Moura, G. EuGene: Maximizing synthetic gene design for heterologous expression. Bioinformatics 2012, 28, 2683–2684. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Guillemet, E.; Reiss, C.; Cullin, C. Enhanced expression of the yeast Ure2 protein in Escherichia coli: The effect of synonymous codon substitutions at a selected place in the gene. Biol. Chem. 1998, 379, 1295–1300. [Google Scholar]
- Inouye, S.; Sahara-Miura, Y.; Sato, J.; Suzuki, T. Codon optimization of genes for efficient protein expression in mammalian cells by selection of only preferred human codons. Protein Expr. Purif. 2015, 109, 47–54. [Google Scholar] [CrossRef]
- Selik, R.M.; Linley, L. Viral Loads Within 6 Weeks After Diagnosis of HIV Infection in Early and Later Stages: Observational Study Using National Surveillance Data. JMIR Public Health Surveill. 2018, 4, e10770. [Google Scholar] [CrossRef]
- O’Doherty, U.; Swiggard, W.J.; Jeyakumar, D.; McGain, D.; Malim, M.H. A sensitive, quantitative assay for human immunodeficiency virus type 1 integration. J. Virol. 2002, 76, 10942–10950. [Google Scholar] [CrossRef]
- Agosto, L.M.; Yu, J.J.; Dai, J.; Kaletsky, R.; Monie, D.; O’Doherty, U. HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology 2007, 368, 60–72. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chupradit, K.; Sornsuwan, K.; Saiprayong, K.; Wattanapanitch, M.; Tayapiwatana, C. Validation of Promoters and Codon Optimization on CRISPR/Cas9-Engineered Jurkat Cells Stably Expressing αRep4E3 for Interfering with HIV-1 Replication. Int. J. Mol. Sci. 2022, 23, 15049. https://doi.org/10.3390/ijms232315049
Chupradit K, Sornsuwan K, Saiprayong K, Wattanapanitch M, Tayapiwatana C. Validation of Promoters and Codon Optimization on CRISPR/Cas9-Engineered Jurkat Cells Stably Expressing αRep4E3 for Interfering with HIV-1 Replication. International Journal of Molecular Sciences. 2022; 23(23):15049. https://doi.org/10.3390/ijms232315049
Chicago/Turabian StyleChupradit, Koollawat, Kanokporn Sornsuwan, Kritayaporn Saiprayong, Methichit Wattanapanitch, and Chatchai Tayapiwatana. 2022. "Validation of Promoters and Codon Optimization on CRISPR/Cas9-Engineered Jurkat Cells Stably Expressing αRep4E3 for Interfering with HIV-1 Replication" International Journal of Molecular Sciences 23, no. 23: 15049. https://doi.org/10.3390/ijms232315049
APA StyleChupradit, K., Sornsuwan, K., Saiprayong, K., Wattanapanitch, M., & Tayapiwatana, C. (2022). Validation of Promoters and Codon Optimization on CRISPR/Cas9-Engineered Jurkat Cells Stably Expressing αRep4E3 for Interfering with HIV-1 Replication. International Journal of Molecular Sciences, 23(23), 15049. https://doi.org/10.3390/ijms232315049