
Pt-Based Nanostructures for Electrochemical Oxidation of CO: Unveiling the Effect of Shapes and Electrolytes

 ,
,  ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
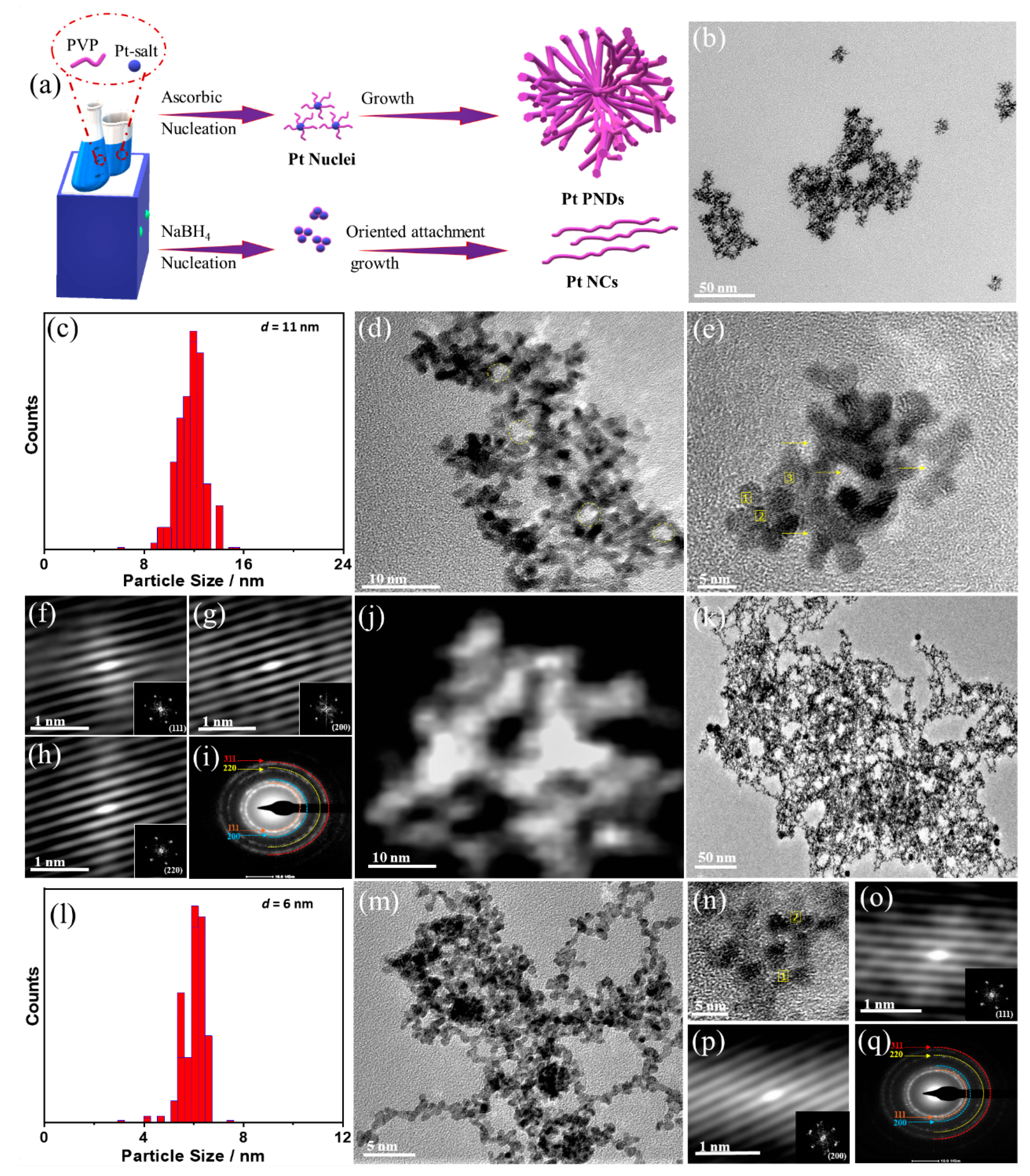
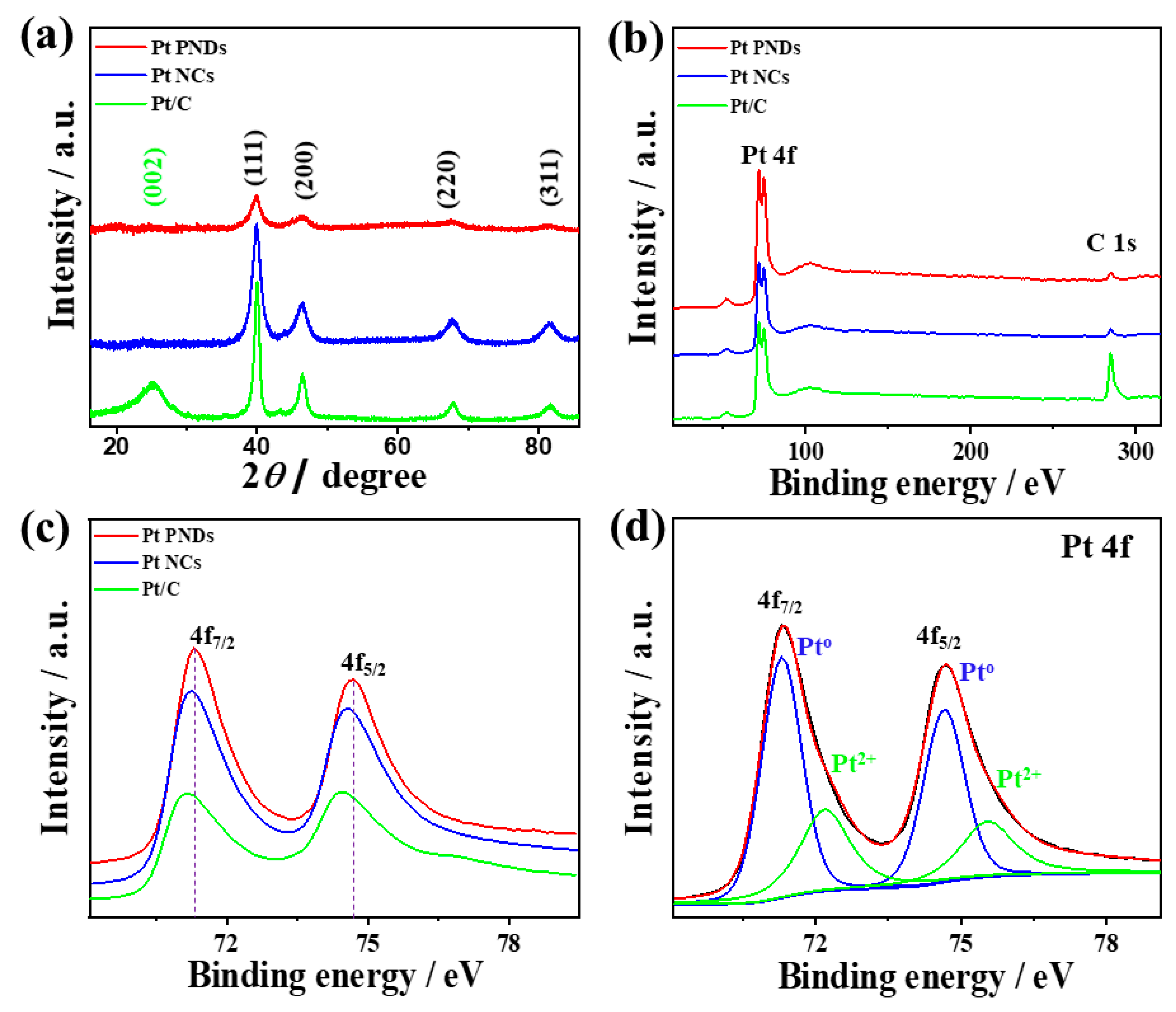
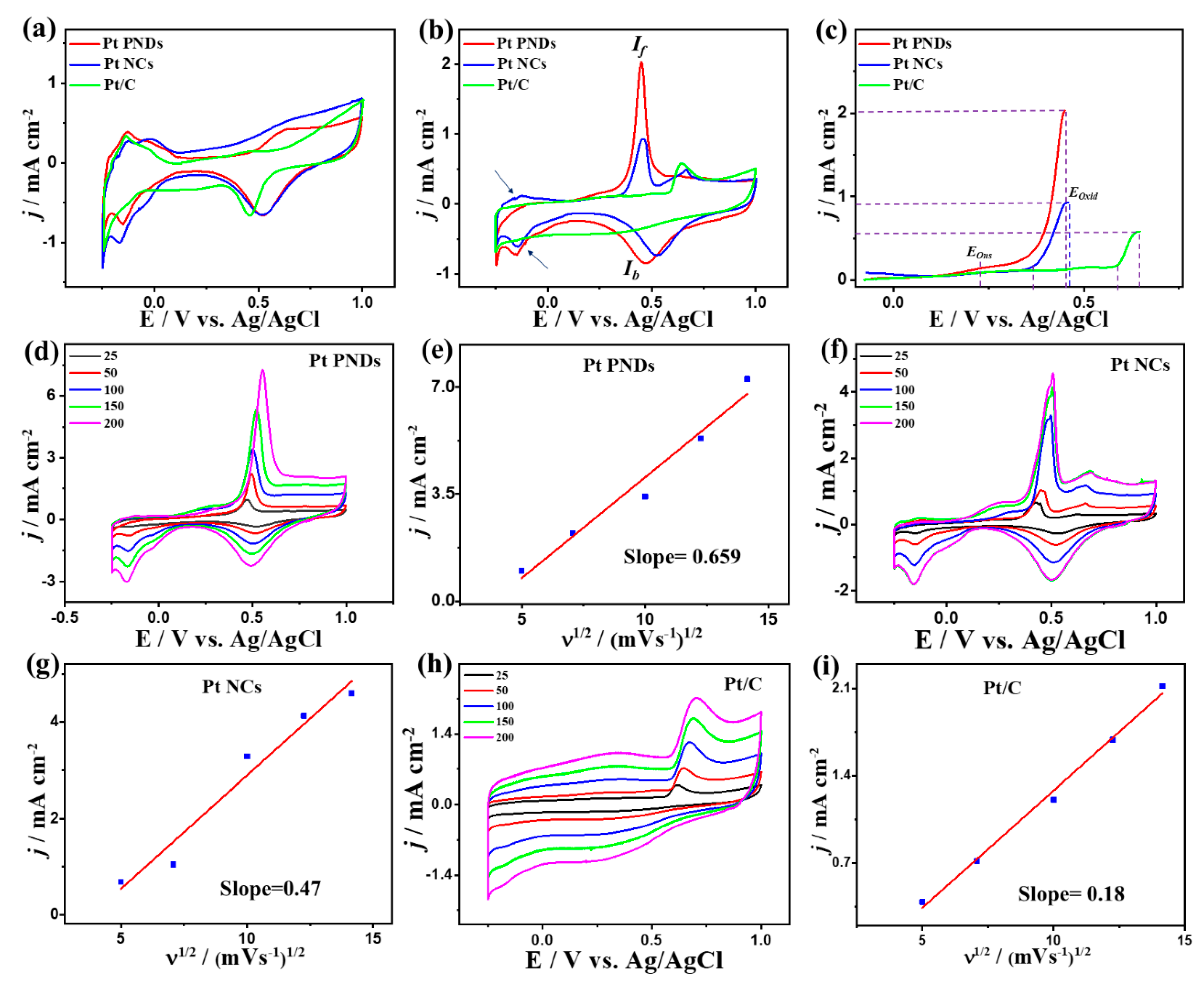
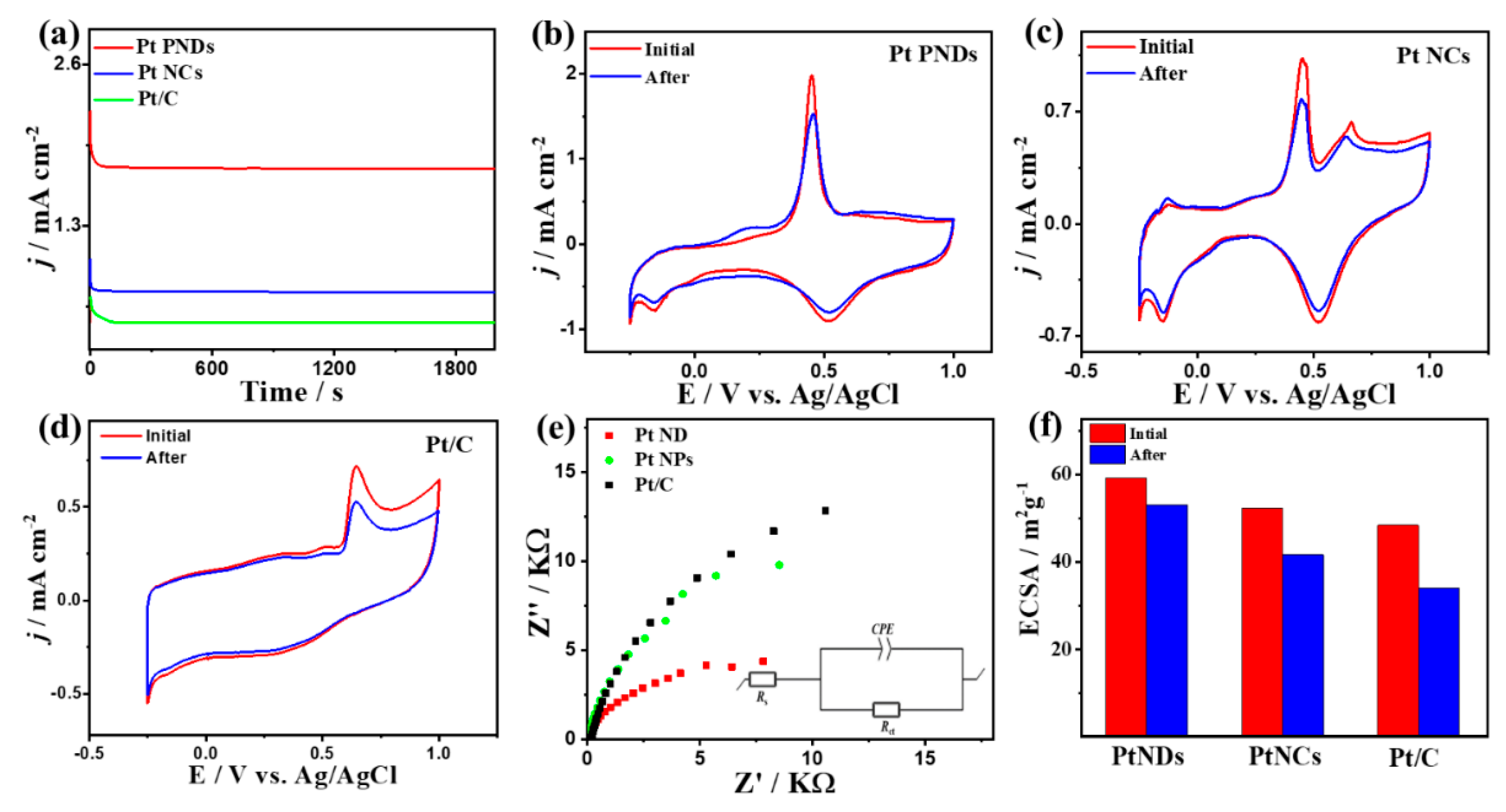
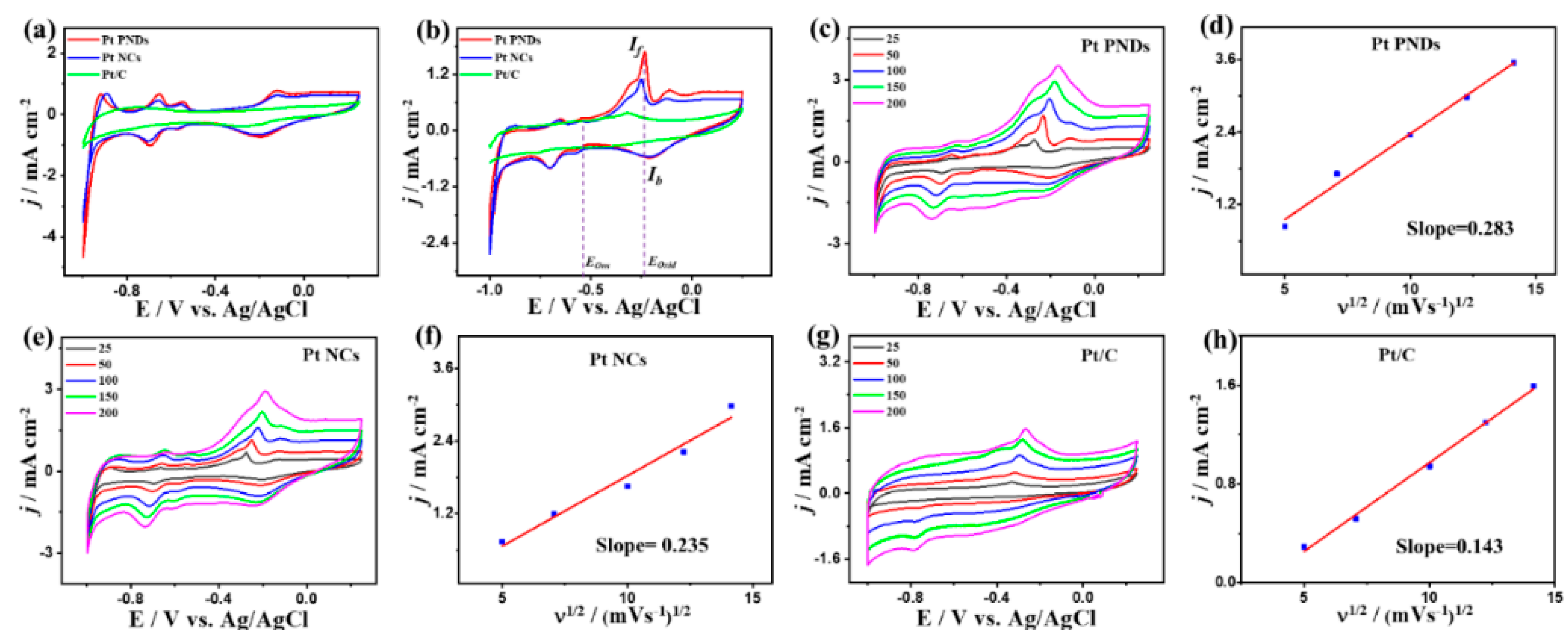
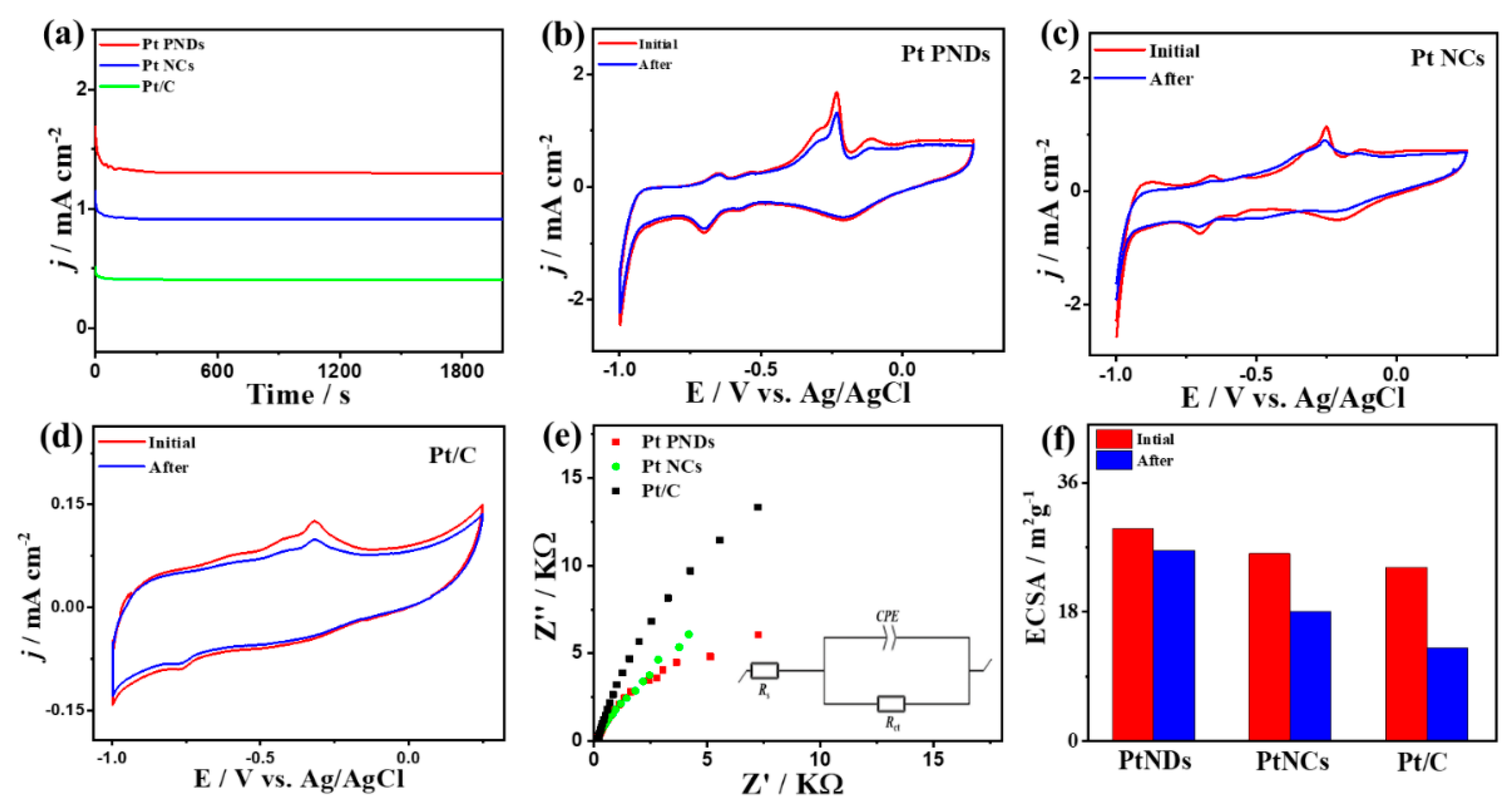
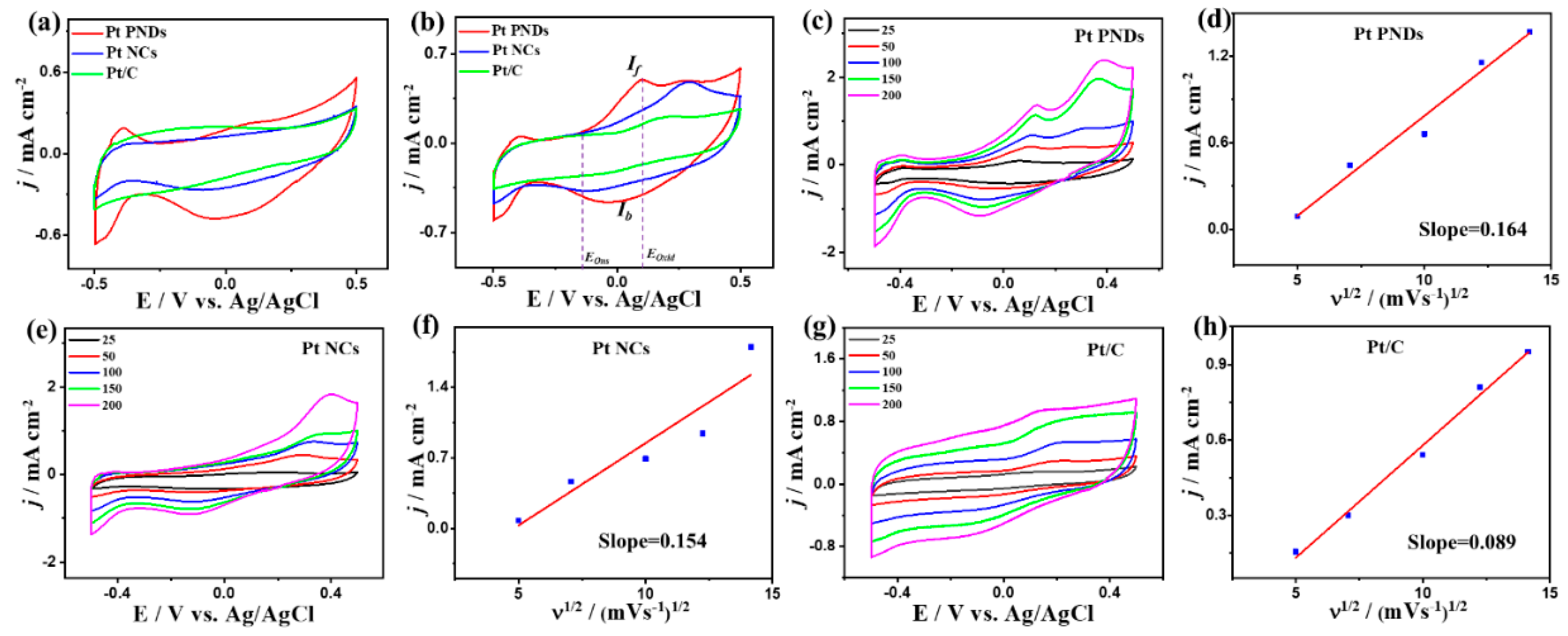
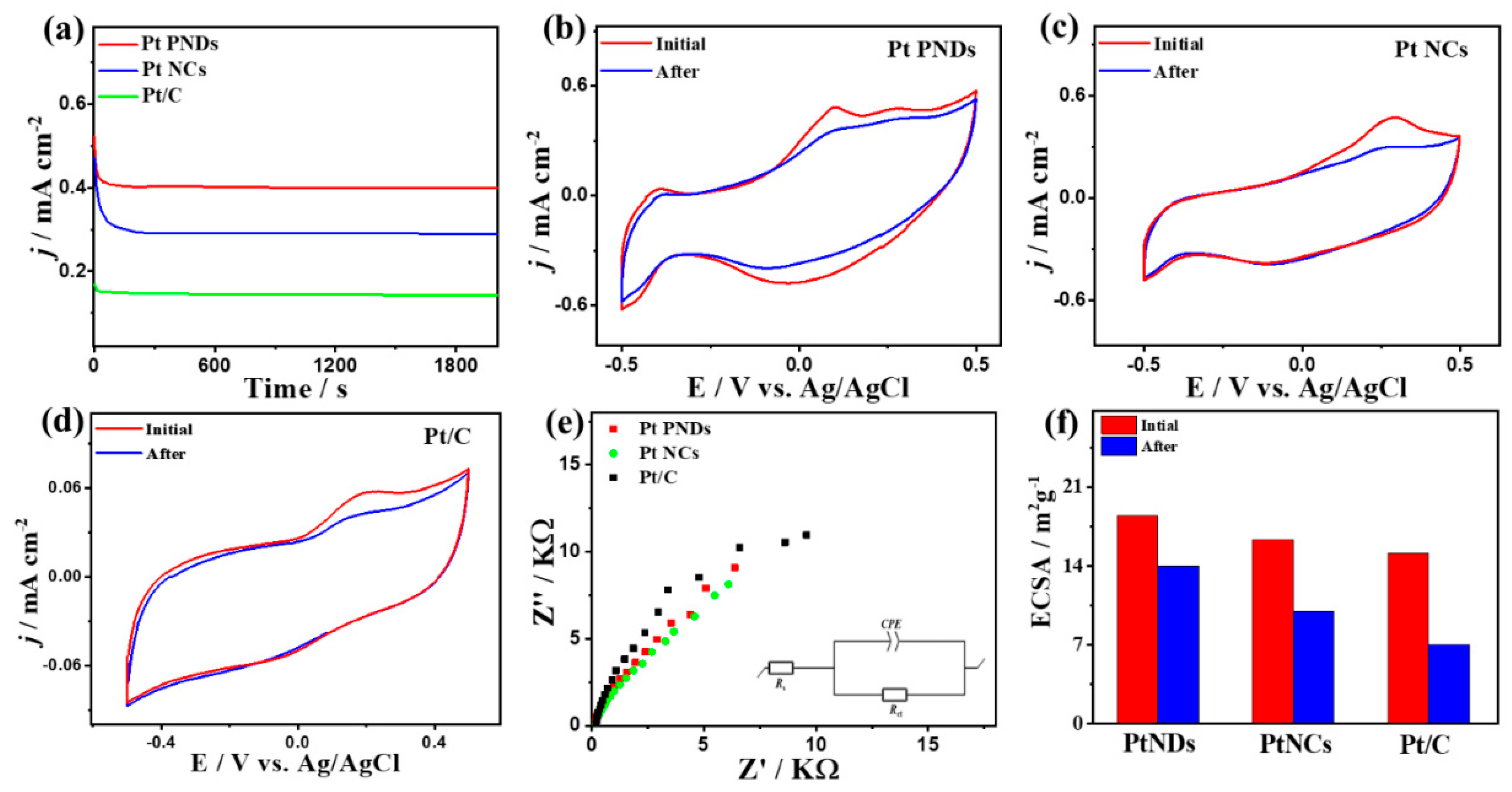
2. Results and Discussion
3. Methods and Materials
3.1. Chemicals and Materials
3.2. Synthesis of porous Pt nanodendrites
3.3. Synthesis of spherical Pt nanoparticles
3.4. Electrochemical COOxid reaction
3.5. Materials Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, Q.; Eid, K.; Li, W.; Abdullah, A.M.; Xu, G.; Varma, R.S. Engineering Graphitic Carbon Nitride (g-C3N4) for Catalytic Reduction of CO2 to Fuels and Chemicals: Strategy and Mechanism. Green Chem. 2021, 23, 5394–5428. [Google Scholar] [CrossRef]
- Eid, K.; Sliem, M.H.; Al-Ejji, M.; Abdullah, A.M.; Harfouche, M.; Varma, R.S. Hierarchical Porous Carbon Nitride-Crumpled Nanosheet-Embedded Copper Single Atoms: An Efficient Catalyst for Carbon Monoxide Oxidation. ACS Appl. Mater. Interfaces 2022, 14, 40749–40760. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Eid, K.; Li, W. Heteroatom-Doped Porous Carbon-Based Nanostructures for Electrochemical CO2 Reduction. Nanomaterials 2022, 12, 2379. [Google Scholar] [CrossRef] [PubMed]
- Gamal, A.; Eid, K.; Abdullah, A.M. Engineering of Pt-based nanostructures for efficient dry (CO2) reforming: Strategy and mechanism for rich-hydrogen production. Int. J. Hydrog. Energy 2021, 47, 5901–5928. [Google Scholar] [CrossRef]
- Eid, K.; Soliman, K.A.; Abdulmalik, D.; Mitoraj, D.; Sleim, M.H.; Liedke, M.O.; El-Sayed, H.A.; AlJaber, A.S.; Al-Qaradawi, I.Y.; Reyes, O.M. Tailored fabrication of iridium nanoparticle-sensitized titanium oxynitride nanotubes for solar-driven water splitting: Experimental insights on the photocatalytic–activity–defects relationship. Catal. Sci. Technol. 2020, 10, 801–809. [Google Scholar] [CrossRef]
- Eid, K.; Sliem, M.H.; Abdullah, A.M. Tailoring the defects of sub-100 nm multipodal titanium nitride/oxynitride nanotubes for efficient water splitting performance. Nanoscale Adv. 2021, 3, 5016–5026. [Google Scholar] [CrossRef]
- Eid, K.; Abdullah, A.M. Porous ternary Pt-based branched nanostructures for electrocatalytic oxygen reduction. Electrochem. Commun. 2022, 136, 107237. [Google Scholar] [CrossRef]
- Lebechi, A.K.; Ipadeola, A.K.; Eid, K.; Abdullah, A.M.; Ozoemena, K.I. Porous spinel-type transition metal oxide nanostructures as emergent electrocatalysts for oxygen reduction reactions. Nanoscale 2022, 14, 10717–10737. [Google Scholar] [CrossRef]
- Lascu, I.; Locovei, C.; Bradu, C.; Gheorghiu, C.; Tanase, A.M.; Dumitru, A. Polyaniline-Derived Nitrogen-Containing Carbon Nanostructures with Different Morphologies as Anode Modifier in Microbial Fuel Cells. Int. J. Mol. Sci. 2022, 23, 11230. [Google Scholar] [CrossRef]
- Guo, X.; Xu, B.; Ma, Z.; Li, Y.; Li, D. Performance Analysis Based on Sustainability Exergy Indicators of High-Temperature Proton Exchange Membrane Fuel Cell. Int. J. Mol. Sci. 2022, 23, 10111. [Google Scholar] [CrossRef]
- Eid, K.; Ahmad, Y.H.; AlQaradawi, S.Y.; Allam, N.K. Rational design of porous binary Pt-based nanodendrites as efficient catalysts for direct glucose fuel cells over a wide pH range. Catal. Sci. Technol. 2017, 7, 2819–2827. [Google Scholar] [CrossRef]
- Lu, S.; Eid, K.; Ge, D.; Guo, J.; Wang, L.; Wang, H.; Gu, H. One-pot synthesis of PtRu nanodendrites as efficient catalysts for methanol oxidation reaction. Nanoscale 2017, 9, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Eid, K.; Lu, Q.; Abdel-Azeim, S.; Soliman, A.; Abdullah, A.M.; Abdelgwad, A.M.; Forbes, R.P.; Ozoemena, K.I.; Varma, R.S.; Shibl, M.F. Highly exfoliated Ti3C2Tx MXene nanosheets atomically doped with Cu for efficient electrochemical CO2 reduction: An experimental and theoretical study. J. Mater. Chem. A 2022, 10, 1965–1975. [Google Scholar] [CrossRef]
- Liu, B.; Wu, H.; Li, S.; Xu, M.; Cao, Y.; Li, Y. Solid-State Construction of CuOx/Cu1.5Mn1.5O4 Nanocomposite with Abundant Surface CuOx Species and Oxygen Vacancies to Promote CO Oxidation Activity. Int. J. Mol. Sci. 2022, 23, 6856. [Google Scholar] [CrossRef] [PubMed]
- Canales, M.; Ramírez-de-Arellano, J.M.; Arellano, J.S.; Magaña, L.F. Ab Initio Study of the Interaction of a Graphene Surface Decorated with a Metal-Doped C30 with Carbon Monoxide, Carbon Dioxide, Methane, and Ozone. Int. J. Mol. Sci. 2022, 23, 4933. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hong, X. PdPt@ Au core@ shell nanoparticles: Alloyed-core manipulation of CO electrocatalytic oxidation properties. Catal. Commun. 2016, 83, 70–73. [Google Scholar] [CrossRef]
- Rosseler, O.; Ulhaq-Bouillet, C.; Bonnefont, A.; Pronkin, S.; Savinova, E.; Louvet, A.; Keller, V.; Keller, N. Structural and electronic effects in bimetallic PdPt nanoparticles on TiO2 for improved photocatalytic oxidation of CO in the presence of humidity. Appl. Catal. B Environ. 2015, 166, 381–392. [Google Scholar] [CrossRef]
- Wu, F.; Eid, K.; Abdullah, A.M.; Niu, W.; Wang, C.; Lan, Y.; Elzatahry, A.A.; Xu, G. Unveiling one-pot template-free fabrication of exquisite multidimensional PtNi multicube nanoarchitectonics for the efficient electrochemical oxidation of ethanol and methanol with a great tolerance for CO. ACS Appl. Mater. Interfaces 2020, 12, 31309–31318. [Google Scholar] [CrossRef]
- Lu, Q.; Li, J.; Eid, K.; Gu, X.; Wan, Z.; Li, W.; Al-Hajri, R.S.; Abdullah, A.M. Facile one-step aqueous-phase synthesis of porous PtBi nanosponges for efficient electrochemical methanol oxidation with a high CO tolerance. J. Electroanal. Chem. 2022, 916, 116361. [Google Scholar] [CrossRef]
- Eid, K.; Sliem, M.H.; Abdullah, A.M. Unraveling template-free fabrication of carbon nitride nanorods codoped with Pt and Pd for efficient electrochemical and photoelectrochemical carbon monoxide oxidation at room temperature. Nanoscale 2019, 11, 11755–11764. [Google Scholar] [CrossRef]
- Eid, K.; Sliem, M.H.; Al-Kandari, H.; Sharaf, M.A.; Abdullah, A.M. Rational synthesis of porous graphitic-like carbon nitride nanotubes codoped with Au and Pd as an efficient catalyst for carbon monoxide oxidation. Langmuir 2019, 35, 3421–3431. [Google Scholar] [CrossRef] [PubMed]
- Eid, K.; Sliem, M.H.; Eldesoky, A.S.; Al-Kandari, H.; Abdullah, A.M. Rational synthesis of one-dimensional carbon nitride-based nanofibers atomically doped with Au/Pd for efficient carbon monoxide oxidation. Int. J. Hydrog. Energy 2019, 44, 17943–17953. [Google Scholar] [CrossRef]
- Eid, K.; Sliem, M.H.; Jlassi, K.; Eldesoky, A.S.; Abdo, G.G.; Al-Qaradawi, S.Y.; Sharaf, M.A.; Abdullah, A.M.; Elzatahry, A.A. Precise fabrication of porous one-dimensional gC3N4 nanotubes doped with Pd and Cu atoms for efficient CO oxidation and CO2 reduction. Inorg. Chem. Commun. 2019, 107, 107460. [Google Scholar] [CrossRef]
- Ipadeola, A.K.; Eid, K.; Abdullah, A.M.; Al-Hajri, R.S.; Ozoemena, K.I. Pd/Ni-Metal-organic Framework-derived Porous Carbon Nanosheets for Efficient CO Oxidation over a Wide pH Range. Nanoscale Adv. 2022. [Google Scholar] [CrossRef]
- Ipadeola, A.K.; Eid, K.; Abdullah, A.M.; Ozoemena, K.I. Pd-Nanoparticles Embedded Metal–Organic Framework-Derived Hier archical Porous Carbon Nanosheets as Efficient Electrocatalysts for Carbon Monoxide Oxidation in Different Electrolytes. Langmuir 2022, 38, 11109–11120. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xu, C.-Q.; Cheng, D.; Li, J. Identification of activity trends for CO oxidation on supported transition-metal single-atom catalysts. Catal. Sci. Technol. 2017, 7, 5860–5871. [Google Scholar] [CrossRef]
- Elias, J.S.; Stoerzinger, K.A.; Hong, W.T.; Risch, M.; Giordano, L.; Mansour, A.N.; Shao-Horn, Y. In situ spectroscopy and mechanistic insights into CO oxidation on transition-metal-substituted ceria nanoparticles. ACS Catal. 2017, 7, 6843–6857. [Google Scholar] [CrossRef]
- Davó-Quiñonero, A.; López-Rodríguez, S.; Bailón-García, E.; Lozano-Castello, D.; Bueno-López, A. Mineral Manganese Oxides as Oxidation Catalysts: Capabilities in the CO-PROX Reaction. ACS Sustain. Chem. Eng. 2021, 9, 6329–6336. [Google Scholar] [CrossRef] [PubMed]
- Eid, K.; Ahmad, Y.H.; Mohamed, A.T.; Elsafy, A.G.; Al-Qaradawi, S.Y. Versatile Synthesis of Pd and Cu Co-Doped Porous Carbon Nitride Nanowires for Catalytic CO Oxidation Reaction. Catalysts 2018, 8, 411. [Google Scholar] [CrossRef]
- Jang, J.-H.; Jeffery, A.A.; Min, J.; Jung, N.; Yoo, S.J. Emerging carbon shell-encapsulated metal nanocatalysts for fuel cells and water electrolysis. Nanoscale 2021, 13, 15116–15141. [Google Scholar] [CrossRef]
- Kim, Y.; Noh, Y.; Lim, E.J.; Lee, S.; Choi, S.M.; Kim, W.B. Star-shaped Pd@ Pt core–shell catalysts supported on reduced graphene oxide with superior electrocatalytic performance. J. Mater. Chem. A 2014, 2, 6976–6986. [Google Scholar] [CrossRef]
- Chen, Z.; Li, X.; Wang, D.; Yang, Q.; Ma, L.; Huang, Z.; Liang, G.; Chen, A.; Guo, Y.; Dong, B. Grafted MXene/polymer electrolyte for high performance solid zinc batteries with enhanced shelf life at low/high temperatures. Energy Environ. Sci. 2021, 14, 3492–3501. [Google Scholar] [CrossRef]
- Choi, H.; Lee, J.; Kim, D.; Kumar, A.; Jeong, B.; Kim, K.-J.; Lee, H.; Park, J.Y. Influence of lattice oxygen on the catalytic activity of blue titania supported Pt catalyst for CO oxidation. Catal. Sci. Technol. 2021, 11, 1698–1708. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Gu, J.; Su, L.; Cheng, L. An overview of metal oxide materials as electrocatalysts and supports for polymer electrolyte fuel cells. Energy Environ. Sci. 2014, 7, 2535–2558. [Google Scholar] [CrossRef]
- Gao, G.; Jiao, Y.; Waclawik, E.R.; Du, A. Single atom (Pd/Pt) supported on graphitic carbon nitride as an efficient photocatalyst for visible-light reduction of carbon dioxide. J. Am. Chem. Soc. 2016, 138, 6292–6297. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-L.; Yin, H.; Kan, X.; Gan, L.-Y.; Schwingenschlögl, U.; Zhao, Y. Potential of transition metal atoms embedded in buckled monolayer gC 3 N 4 as single-atom catalysts. Phys. Chem. Chem. Phys. 2017, 19, 30069–30077. [Google Scholar] [CrossRef]
- Chen, Z.; Mitchell, S.; Vorobyeva, E.; Leary, R.K.; Hauert, R.; Furnival, T.; Ramasse, Q.M.; Thomas, J.M.; Midgley, P.A.; Dontsova, D. Stabilization of single metal atoms on graphitic carbon nitride. Adv. Funct. Mater. 2017, 27, 1605785. [Google Scholar] [CrossRef]
- Kumar, R.; Oh, J.-H.; Kim, H.-J.; Jung, J.-H.; Jung, C.-H.; Hong, W.G.; Kim, H.-J.; Park, J.-Y.; Oh, I.-K. Nanohole-structured and palladium-embedded 3D porous graphene for ultrahigh hydrogen storage and CO oxidation multifunctionalities. ACS Nano 2015, 9, 7343–7351. [Google Scholar] [CrossRef]
- Mukri, B.D.; Waghmare, U.V.; Hegde, M. Platinum Ion-Doped TiO2: High Catalytic Activity of Pt2+ with Oxide Ion Vacancy in Ti4+1–xPt2+xO2–x Compared to Pt4+ without Oxide Ion Vacancy in Ti4+1-xPt4+xO2. Chem. Mater. 2013, 25, 3822–3833. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Yang, Z. Recent progress of MXenes as the support of catalysts for the CO oxidation and oxygen reduction reaction. Chin. Chem. Lett. 2020, 31, 931–936. [Google Scholar] [CrossRef]
- Tahini, H.A.; Tan, X.; Smith, S.C. Facile CO Oxidation on Oxygen-functionalized MXenes via the Mars-van Krevelen Mechanism. ChemCatChem 2020, 12, 1007–1012. [Google Scholar] [CrossRef]
- Cheng, C.; Zhang, X.; Yang, Z.; Hermansson, K. Identification of High-Performance Single-Atom MXenes Catalysts for Low-Temperature CO Oxidation. Adv. Theory Simul. 2019, 2, 1900006. [Google Scholar] [CrossRef]
- Morales-Garciía, A.n.; Calle-Vallejo, F.; Illas, F. MXenes: New Horizons in Catalysis. ACS Catal. 2020, 10, 13487–13503. [Google Scholar] [CrossRef]
- Alhabeb, M.; Maleski, K.; Anasori, B.; Lelyukh, P.; Clark, L.; Sin, S.; Gogotsi, Y. Guidelines for synthesis and processing of two-dimensional titanium carbide (Ti3C2Tx MXene). Chem. Mater. 2017, 29, 7633–7644. [Google Scholar] [CrossRef]
- Salah, B.; Eid, K.; Abdelgwad, A.M.; Ibrahim, Y.; Abdullah, A.M.; Hassan, M.K.; Ozoemena, K.I. Titanium Carbide (Ti3C2Tx) MXene Ornamented with Pallidum Nanoparticles for Electrochemical CO Oxidation. Electroanalysis 2022, 34, 677–683. [Google Scholar] [CrossRef]
- Eid, K.; Wang, H.; Malgras, V.; Alothman, Z.A.; Yamauchi, Y.; Wang, L. Trimetallic PtPdRu dendritic nanocages with three-dimensional electrocatalytic surfaces. J. Phys. Chem. C 2015, 119, 19947–19953. [Google Scholar] [CrossRef]
- Guo, Z.; Dai, X.; Yang, Y.; Zhang, Z.; Zhang, X.; Mi, S.; Xu, K.; Li, Y. Highly stable and active PtNiFe dandelion-like alloys for methanol electrooxidation. J. Mater. Chem. A 2013, 1, 13252–13260. [Google Scholar] [CrossRef]
- Lu, W.; Xia, X.; Wei, X.; Li, M.; Zeng, M.; Guo, J.; Cheng, S. Nanoengineering 2D Dendritic PdAgPt Nanoalloys with Edge-Enriched Active Sites for Enhanced Alcohol Electroxidation and Electrocatalytic Hydrogen Evolution. ACS Appl. Mater. Interfaces 2020, 12, 21569–21578. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Chen, Z.; Liu, Y.; Zhang, M.; Han, X.; Zhong, C.; Hu, W.; Deng, Y. One-step synthesis of the PdPt bimetallic nanodendrites with controllable composition for methanol oxidation reaction. Sci. China Mater. 2018, 61, 697–706. [Google Scholar] [CrossRef]
- Eid, K.; Ahmad, Y.H.; Yu, H.; Li, Y.; Li, X.; AlQaradawi, S.Y.; Wang, H.; Wang, L. Rational one-step synthesis of porous PtPdRu nanodendrites for ethanol oxidation reaction with a superior tolerance for CO-poisoning. Nanoscale 2017, 9, 18881–18889. [Google Scholar] [CrossRef]
- Eid, K.; Wang, H.; Malgras, V.; Alshehri, S.M.; Ahamad, T.; Yamauchi, Y.; Wang, L. One-step solution-phase synthesis of bimetallic PtCo nanodendrites with high electrocatalytic activity for oxygen reduction reaction. J. Electroanal. Chem. 2016, 779, 250–255. [Google Scholar] [CrossRef]
- Eid, K.; Wang, H.; He, P.; Wang, K.; Ahamad, T.; Alshehri, S.M.; Yamauchi, Y.; Wang, L. One-step synthesis of porous bimetallic PtCu nanocrystals with high electrocatalytic activity for methanol oxidation reaction. Nanoscale 2015, 7, 16860–16866. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xia, W.-Y.; Xu, C.-W.; Chen, S. Pt/C and Pd/C catalysts promoted by Au for glycerol and CO electrooxidation in alkaline medium. J. Energy Inst. 2017, 90, 725–733. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, L.; Duan, Z.; Lapp, A.S.; Henkelman, G.; Crooks, R.M. Efficient CO oxidation using dendrimer-encapsulated Pt nanoparticles activated with < 2% Cu surface atoms. ACS nano 2016, 10, 8760–8769. [Google Scholar]
- Sun, M.; Lv, Y.; Song, Y.; Wu, H.; Wang, G.; Zhang, H.; Chen, M.; Fu, Q.; Bao, X. CO-tolerant PtRu@ h-BN/C core–shell electrocatalysts for proton exchange membrane fuel cells. Appl. Surf. Sci. 2018, 450, 244–250. [Google Scholar] [CrossRef]
- Weir, M.G.; Myers, V.S.; Frenkel, A.I.; Crooks, R.M. In situ X-ray Absorption Analysis of ∼1.8 nm Dendrimer-Encapsulated Pt Nanoparticles during Electrochemical CO Oxidation. ChemPhysChem 2010, 11, 2942–2950. [Google Scholar] [CrossRef]
- Ueda, A.; Yamada, Y.; Ioroi, T.; Fujiwara, N.; Yasuda, K.; Miyazaki, Y.; Kobayashi, T.J.C.t. Electrochemical oxidation of CO in sulfuric acid solution over Pt and PtRu catalysts modified with TaOx and NbOx. Catal. Today 2003, 84, 223–229. [Google Scholar] [CrossRef]
- McPherson, I.J.; Ash, P.A.; Jones, L.; Varambhia, A.; Jacobs, R.M.; Vincent, K.A. Electrochemical CO oxidation at platinum on carbon studied through analysis of anomalous in situ IR spectra. J. Phys. Chem. C 2017, 121, 17176–17187. [Google Scholar] [CrossRef]
- Spendelow, J.; Goodpaster, J.; Kenis, P.; Wieckowski, A. Mechanism of CO oxidation on Pt (111) in alkaline media. J. Phys. Chem. B 2006, 110, 9545–9555. [Google Scholar] [CrossRef]
- Davies, J.C.; Hayden, B.E.; Pegg, D.J. The electrooxidation of carbon monoxide on ruthenium modified Pt (110). Electrochim. Acta 1998, 44, 1181–1190. [Google Scholar] [CrossRef]
- Matsui, T.; Fujiwara, K.; Okanishi, T.; Kikuchi, R.; Takeguchi, T.; Eguchi, K. Electrochemical oxidation of CO over tin oxide supported platinum catalysts. J. Power Source 2006, 155, 152–156. [Google Scholar] [CrossRef]
- Ciapina, E.G.; Santos, S.F.; Gonzalez, E.R. The electrooxidation of carbon monoxide on unsupported Pt agglomerates. J. Electroanal. Chem. 2010, 644, 132–143. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Rs (Ω) | Rct (kΩ) | CPE (μF.s(1−a)) | α |
|---|---|---|---|---|
| Pt NDs | 41.20 | 6.837 | 104.9 | 0.9 |
| Pt NCs | 50.21 | 24.60 | 69.77 | 0.914 |
| Pt/C(20 wt.%) | 95.51 | 55.67 | 27.46 | 0.92 |
| Catalyst | Rs (Ω) | Rct (kΩ) | CPE (μF.s(1−a)) | α |
|---|---|---|---|---|
| Pt NDs | 81.86 | 18.92 | 173.5 | 0.769 |
| Pt NCs | 89.39 | 25.17 | 124.9 | 0.768 |
| Pt/C (20 wt.%) | 149.40 | 52.20 | 89.39 | 0.87 |
| Catalyst | Rs (Ω) | Rct (kΩ) | CPE (μF.s(1−a)) | α |
|---|---|---|---|---|
| Pt NDs | 87.18 | 25.65 | 112.4 | 0.832 |
| Pt NCs | 105.9 | 25.91 | 108.2 | 0.789 |
| Pt/C (20 wt.%) | 160.60 | 33.28 | 47.78 | 0.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelgawad, A.; Salah, B.; Eid, K.; Abdullah, A.M.; Al-Hajri, R.S.; Al-Abri, M.; Hassan, M.K.; Al-Sulaiti, L.A.; Ahmadaliev, D.; Ozoemena, K.I. Pt-Based Nanostructures for Electrochemical Oxidation of CO: Unveiling the Effect of Shapes and Electrolytes. Int. J. Mol. Sci. 2022, 23, 15034. https://doi.org/10.3390/ijms232315034
Abdelgawad A, Salah B, Eid K, Abdullah AM, Al-Hajri RS, Al-Abri M, Hassan MK, Al-Sulaiti LA, Ahmadaliev D, Ozoemena KI. Pt-Based Nanostructures for Electrochemical Oxidation of CO: Unveiling the Effect of Shapes and Electrolytes. International Journal of Molecular Sciences. 2022; 23(23):15034. https://doi.org/10.3390/ijms232315034
Chicago/Turabian StyleAbdelgawad, Ahmed, Belal Salah, Kamel Eid, Aboubakr M. Abdullah, Rashid S. Al-Hajri, Mohammed Al-Abri, Mohammad K. Hassan, Leena A. Al-Sulaiti, Doniyorbek Ahmadaliev, and Kenneth I. Ozoemena. 2022. "Pt-Based Nanostructures for Electrochemical Oxidation of CO: Unveiling the Effect of Shapes and Electrolytes" International Journal of Molecular Sciences 23, no. 23: 15034. https://doi.org/10.3390/ijms232315034
APA StyleAbdelgawad, A., Salah, B., Eid, K., Abdullah, A. M., Al-Hajri, R. S., Al-Abri, M., Hassan, M. K., Al-Sulaiti, L. A., Ahmadaliev, D., & Ozoemena, K. I. (2022). Pt-Based Nanostructures for Electrochemical Oxidation of CO: Unveiling the Effect of Shapes and Electrolytes. International Journal of Molecular Sciences, 23(23), 15034. https://doi.org/10.3390/ijms232315034